
Celiac Disease Defined by Over-Sensitivity to Gliadin Activation and Superior Antigen Presentation of Dendritic Cells

 , ,
, ,  and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
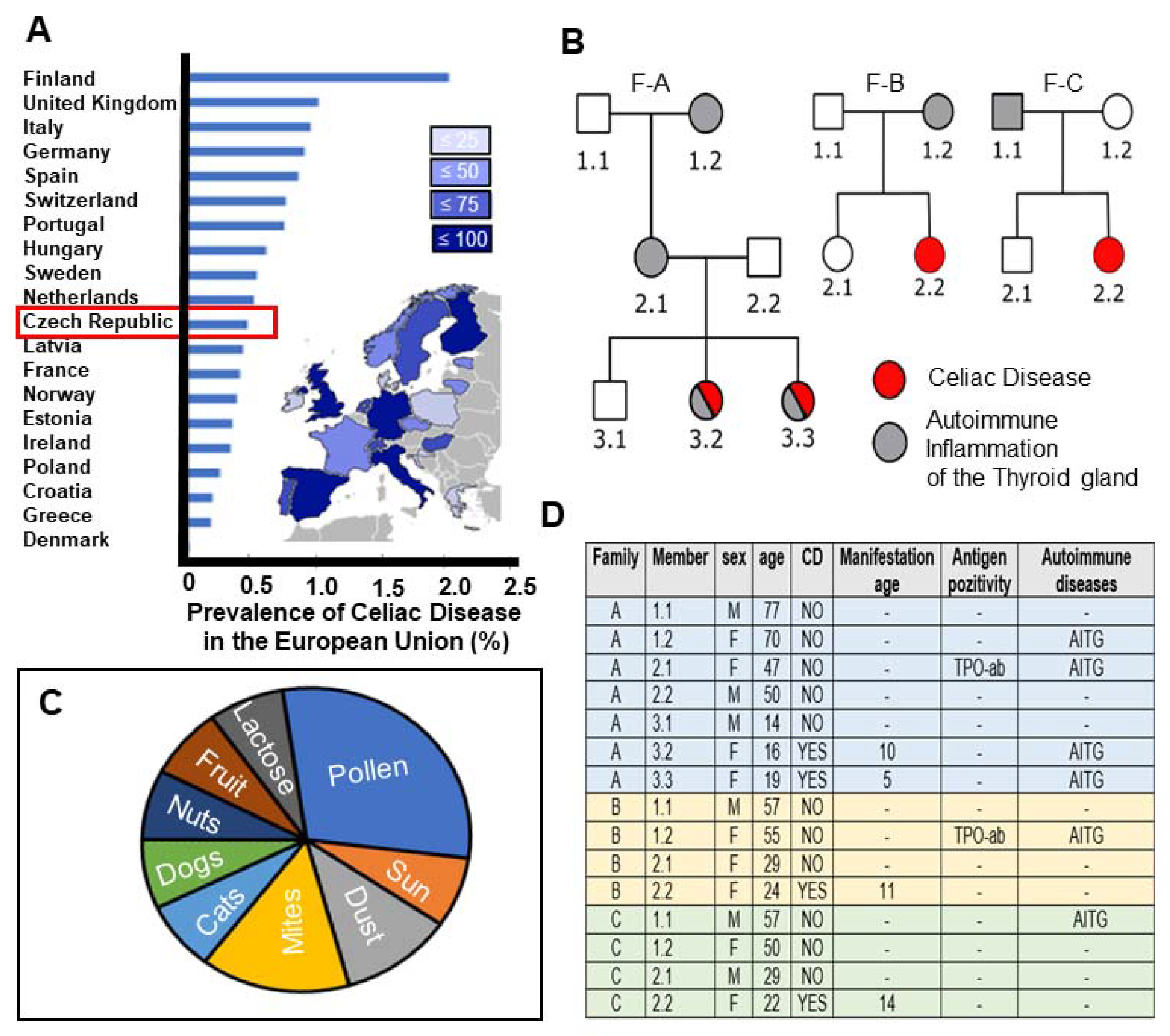
2.1. High Prevalence of CeD in the Czech Republic with Study Participants Representing Autoimmune Disease Susceptibility across Familial Generations
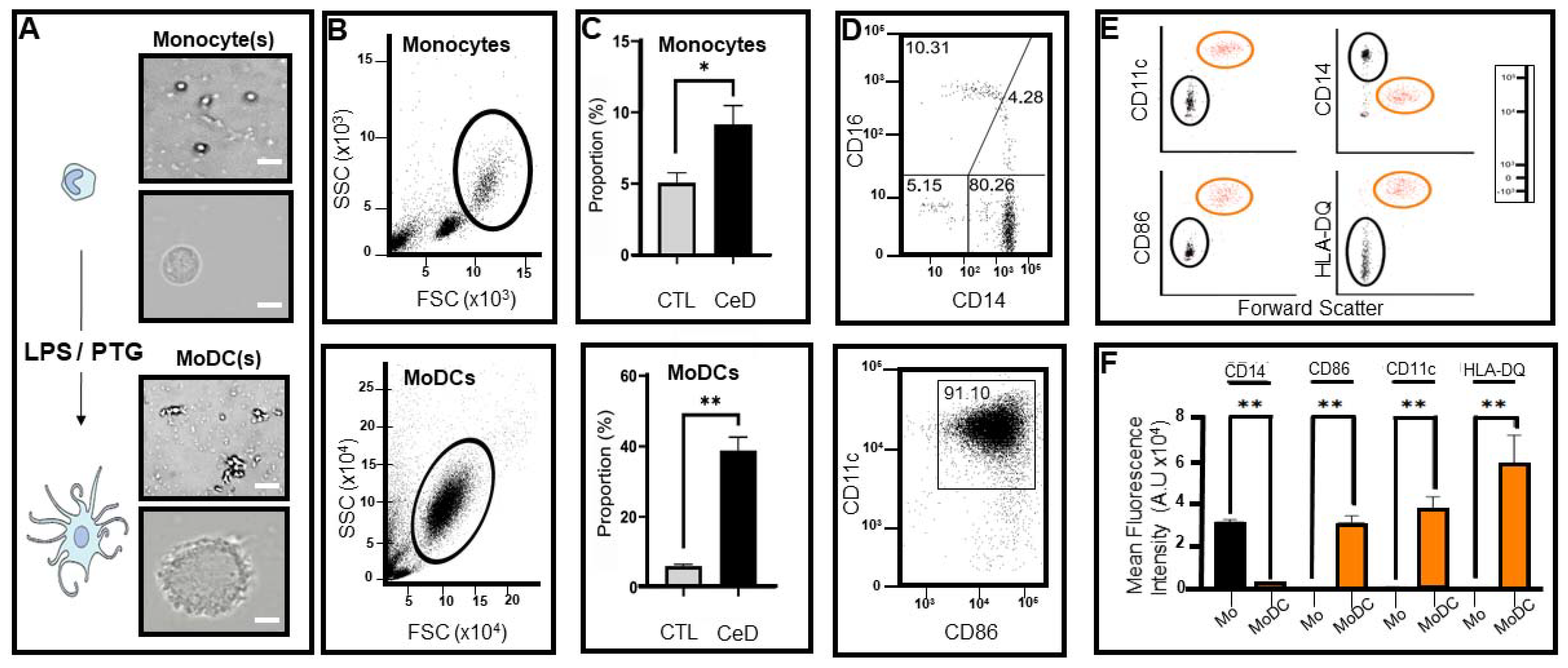
2.2. Differential Expression of Transmembrane Markers Indicates Monocytes Extracted from CeD Patients Have the Ability to Transform into Dendritic Cells after Exposure to LPS/PTG
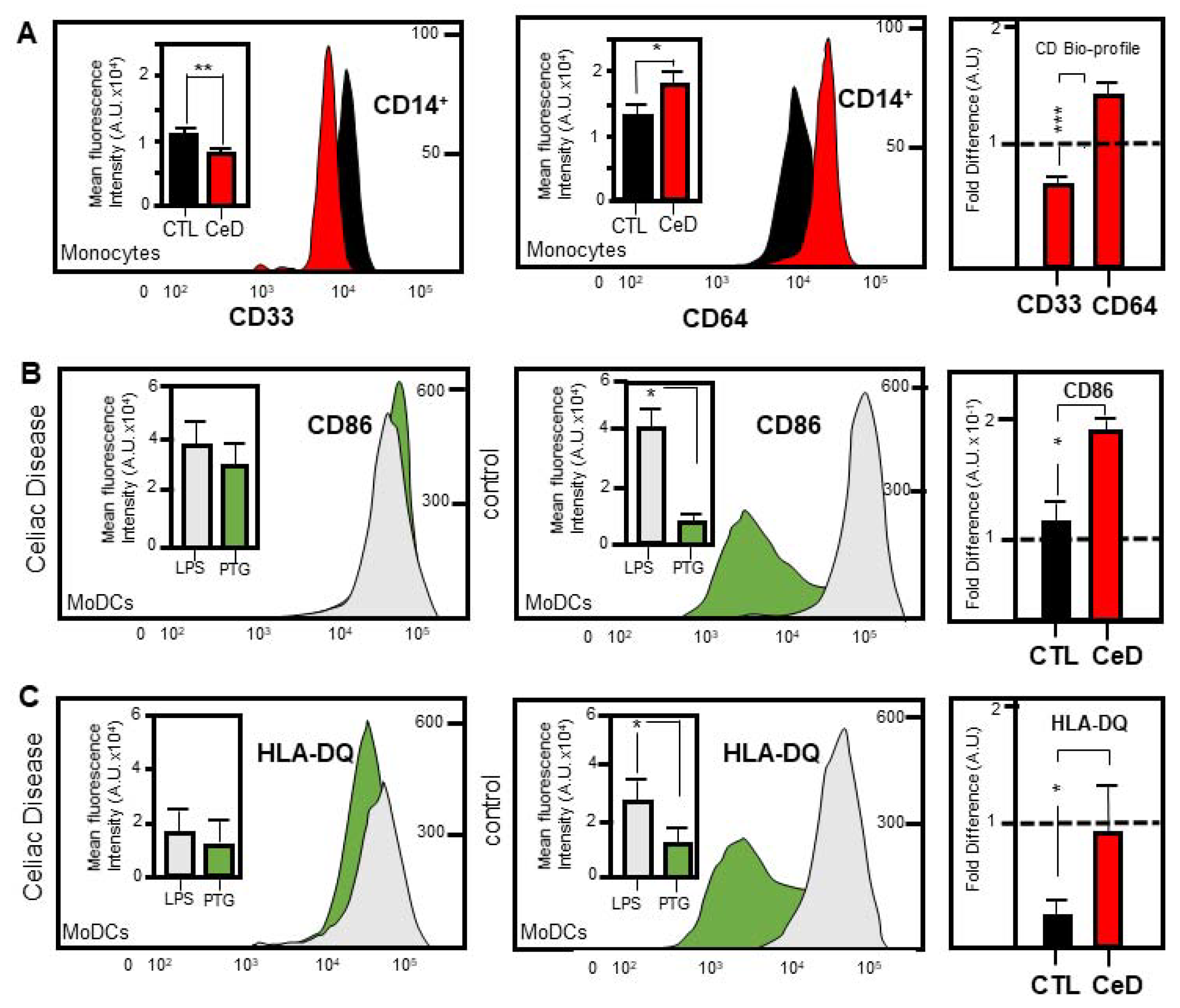
2.3. Identification of a Novel CD33:CD64 Bio-Profile Marker for CeD Patients
2.4. MoDCs from CeD Patients Exhibit Hypersensitivity to Gliadin Activation
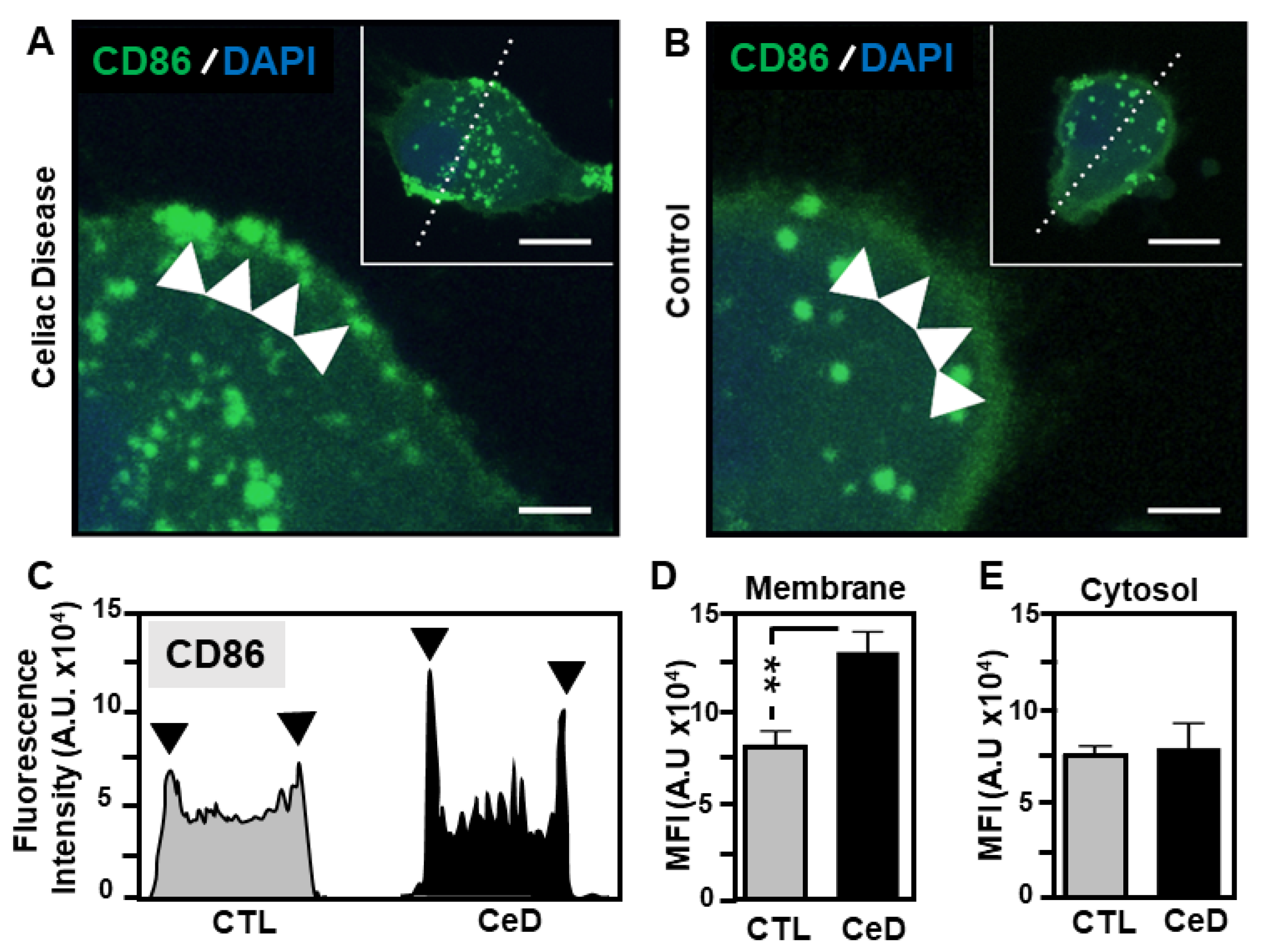
2.5. Superior CD86 Presentation in MoDCs from CeD Patients Compared to CTL
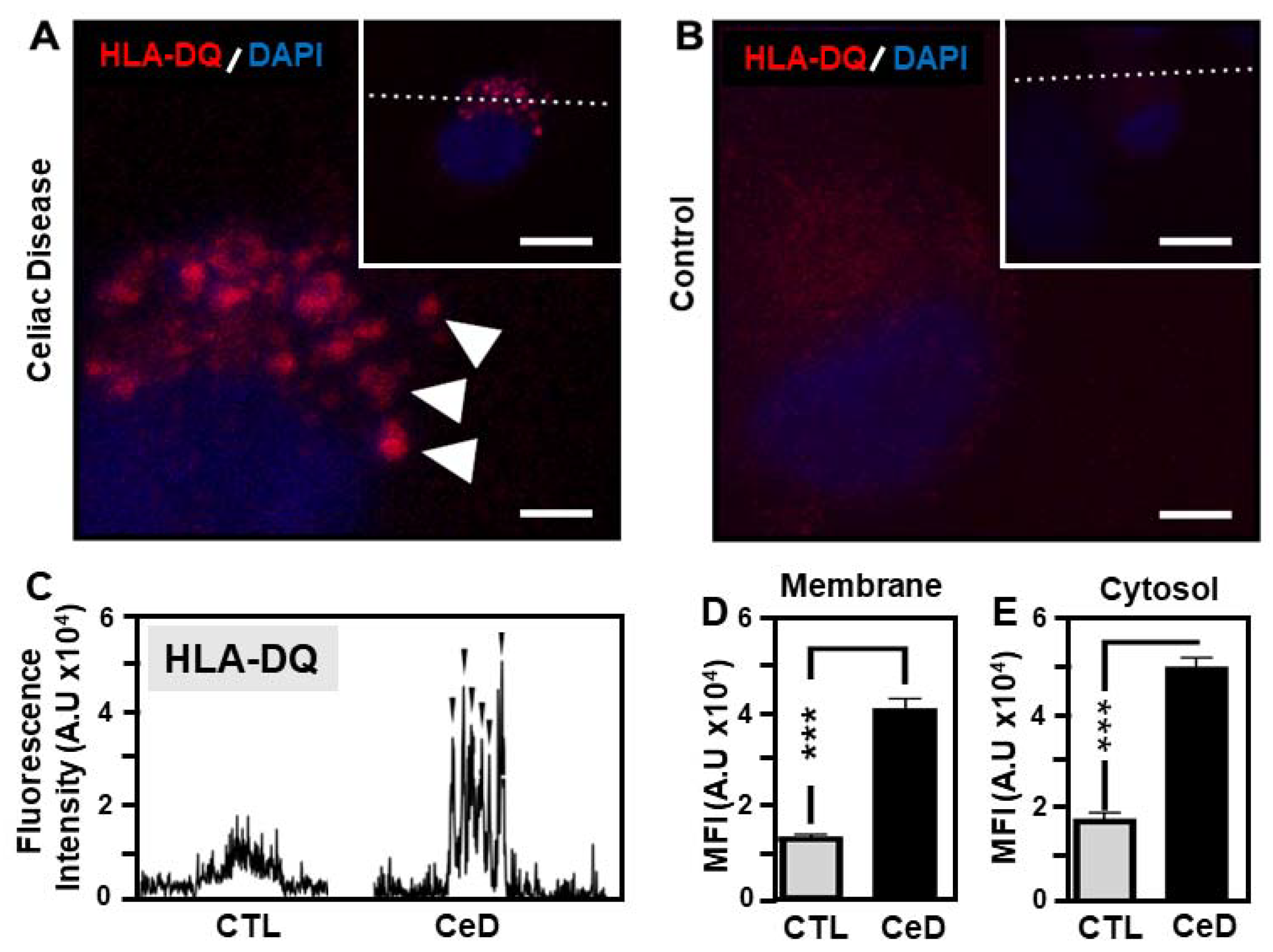
2.6. Predominant Membrane-Associated HLA-DQ Expression in MoDCs Originating from CeD Patients Compared to CTL
3. Discussion
4. Materials and Methods
4.1. Clinical Diagnosis of CeD Patients and Pedigree Overview
4.2. Monocyte Isolation from Peripheral Blood Mononuclear Cells
4.3. Generation of Monocyte Derived Dendritic Cells (MoDCs)
4.4. Flow Cytometry and FACS Staining
4.5. Immunohistochemical and Confocal Microscopy Detection of MoDC Surface Antigens
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lundin, K.E.; Wijmenga, C. Coeliac disease and autoimmune disease-genetic overlap and screening. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 507–515. [Google Scholar] [CrossRef]
- Shiina, T.; Hosomichi, K.; Inoko, H.; Kulski, J.K. The HLA genomic loci map: Expression, interaction, diversity and disease. J. Hum. Genet. 2009, 54, 15–39. [Google Scholar] [CrossRef] [Green Version]
- Zajacova, M.; Kotrbova-Kozak, A.; Cerna, M. HLA-DRB1, -DQA1 and -DQB1 genotyping of 180 Czech individuals from the Czech Republic pop 3. Hum. Immunol. 2016, 77, 365–366. [Google Scholar] [CrossRef]
- Lebwohl, B.; Sanders, D.S.; Green, P.H.R. Coeliac disease. Lancet 2018, 391, 70–81. [Google Scholar] [CrossRef]
- Maki, M.; Collin, P. Coeliac disease. Lancet 1997, 349, 1755–1759. [Google Scholar] [CrossRef]
- Marsh, M.N. Gluten, major histocompatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity (‘celiac sprue’). Gastroenterology 1992, 102, 330–354. [Google Scholar] [CrossRef]
- Sollid, L.M. Coeliac disease: Dissecting a complex inflammatory disorder. Nat. Rev. Immunol. 2002, 2, 647–655. [Google Scholar] [CrossRef]
- Shan, L.; Molberg, O.; Parrot, I.; Hausch, F.; Filiz, F.; Gray, G.M.; Sollid, L.M.; Khosla, C. Structural basis for gluten intolerance in celiac sprue. Science 2002, 297, 2275–2279. [Google Scholar] [CrossRef] [Green Version]
- Matysiak-Budnik, T.; Moura, I.C.; Arcos-Fajardo, M.; Lebreton, C.; Menard, S.; Candalh, C.; Ben-Khalifa, K.; Dugave, C.; Tamouza, H.; Van Niel, G.; et al. Secretory IgA mediates retrotranscytosis of intact gliadin peptides via the transferrin receptor in celiac disease. J. Exp. Med. 2007, 205, 143–154. [Google Scholar] [CrossRef]
- Visser, J.; Rozing, J.; Sapone, A.; Lammers, K.; Fasano, A. Tight junctions, intestinal permeability, and autoimmunity: Celiac disease and type 1 diabetes paradigms. Ann. Acad. Sci. 2009, 1165, 195–205. [Google Scholar] [CrossRef] [Green Version]
- Schumann, M.; Siegmund, B.; Schulzke, J.D.; Fromm, M. Celiac Disease: Role of the Epithelial Barrier. Cell. Mol. Gastroenterol. Hepatol. 2017, 3, 150–162. [Google Scholar] [CrossRef] [Green Version]
- Banchereau, J.; Briere, F.; Caux, C.; Davoust, J.; Lebecque, S.; Liu, Y.-J.; Pulendran, B.; Palucka, K. Immunobiology of Dendritic Cells. Annu. Rev. Immunol. 2000, 18, 767–811. [Google Scholar] [CrossRef] [PubMed]
- Palová-Jelínková, L.; Rožková, D.; Pecharová, B.; Bártová, J.; Šedivá, A.; Tlaskalová-Hogenová, H.; Spíšek, R.; Tučková, L. Gliadin Fragments Induce Phenotypic and Functional Maturation of Human Dendritic Cells. J. Immunol. 2005, 175, 7038–7045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varol, C.; Landsman, L.; Fogg, D.K.; Greenshtein, L.; Gildor, B.; Margalit, R.; Kalchenko, V.; Geissmann, F.; Jung, S. Monocytes give rise to mucosal, but not splenic, conventional dendritic cells. J. Exp. Med. 2006, 204, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.-W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate Mapping Reveals Origins and Dynamics of Monocytes and Tissue Macrophages under Homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, K.L.; Tai, J.J.-Y.; Wong, W.-C.; Han, H.; Sem, X.; Yeap, W.-H.; Kourilsky, P.; Wong, S.-C. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011, 118, e16–e31. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.L.; Yeap, W.H.; Tai, J.J.Y.; Ong, S.M.; Dang, T.M.; Wong, S.C. The three human monocyte subsets: Implications for health and disease. Immunol. Res. 2012, 53, 41–57. [Google Scholar] [CrossRef]
- Zawada, A.M.; Rogacev, K.S.; Rotter, B.; Winter, P.; Marell, R.-R.; Fliser, D.; Heine, G.H. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood 2011, 118, e50–e61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinova, J.; Palová-Jelínková, L.; Smythies, L.E.; Černá, M.; Pecharová, B.; Dvorak, M.; Fruhauf, P.; Tlaskalová-Hogenová, H.; Smith, P.D.; Tučková, L. Gliadin Peptides Activate Blood Monocytes from Patients with Celiac Disease. J. Clin. Immunol. 2007, 27, 201–209. [Google Scholar] [CrossRef]
- Bai, J.C.; Ciacci, C. World Gastroenterology Organisation Global Guidelines. J. Clin. Gastroenterol. 2017, 51, 755–768. [Google Scholar] [CrossRef]
- Kleiveland, C.R. Peripheral Blood Mononuclear Cells. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Springer: Cham, Switzerland, 2015; pp. 161–167. [Google Scholar]
- Gren, S.T.; Rasmussen, T.B.; Janciauskiene, S.; Håkansson, K.; Gerwien, J.G.; Grip, O. A Single-Cell Gene-Expression Profile Reveals Inter-Cellular Heterogeneity within Human Monocyte Subsets. PLoS ONE 2015, 10, e0144351. [Google Scholar] [CrossRef] [Green Version]
- Kenderian, S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Aikawa, V.; Morrissette, J.J.D.; Scholler, J.; Song, D.; Porter, D.L.; Carroll, M.C.; et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leuk. 2015, 29, 1637–1647. [Google Scholar] [CrossRef]
- Akinrinmade, O.A.; Chetty, S.; Daramola, A.K.; Islam, M.-U.; Thepen, T.; Barth, S. CD64: An Attractive Immunotherapeutic Target for M1-type Macrophage Mediated Chronic Inflammatory Diseases. Biomed. 2017, 5, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbí, A.L.; Rodríguez, C.L. CD11c Integrin Gene Promoter Activity during Myeloid Differentiation. Leuk. Lymphoma 1997, 25, 415–425. [Google Scholar] [CrossRef]
- Mbongue, J.C.; Nieves, H.A.; Torrez, T.W.; Langridge, W.H.R. The Role of Dendritic Cell Maturation in the Induction of Insulin-Dependent Diabetes Mellitus. Front. Immunol. 2017, 8, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapa, B.R.; Rawal, P.; Sapra, B.; Vaiphei, K.; Nain, C.K.; Singh, K. Familial Prevalence of Celiac Disease. J. Trop. Pediatr. 2010, 57, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Title Page/List of Contents/Preface; S. Karger AG: Basel, Switzerland, 2009; Volume 26.
- Taneja, V.; David, C.S. HLA class II transgenic mice as models of human diseases. Immunol. Rev. 1999, 169, 67–79. [Google Scholar] [CrossRef]
- Mustalahti, K.; Catassi, C.; Reunanen, A.; Fabiani, E.; Heier, M.; McMillan, S.; Murray, L.; Metzger, M.H.; Gasparin, M.; Bravi, E.; et al. The prevalence of celiac disease in Europe: Results of a centralized, international mass screening project. Ann. Med. 2010, 42, 587–595. [Google Scholar] [CrossRef]
- Vilppula, A.; Collin, P.; Mäki, M.; Valve, R.; Luostarinen, M.; Krekelä, I.; Patrikainen, H.; Kaukinen, K. Undetected coeliac disease in the elderly: A biopsy-proven population-based study. Dig. Liver Dis. 2008, 40, 809–813. [Google Scholar] [CrossRef]
- Sumník, Z.; Kolousková, S.; Cinek, O.; Kotalová, R.; Vavrinec, J.; Snajderová, M. HLA—DQA1*05-DQB1*0201 positivity predisposes to coeliac disease in Czech diabetic children. Acta Paediatr. 2007, 89, 1426–1430. [Google Scholar] [CrossRef]
- Lebwohl, B.; Ludvigsson, J.F.; Green, P.H.R. Celiac disease and non-celiac gluten sensitivity. BMJ 2015, 351, h4347. [Google Scholar] [CrossRef]
- Fasano, A.; Araya, M.; Bhatnagar, S.; Cameron, D.; Catassi, C.; Dirks, M.; Mearin, M.; Ortigosa, L.; Phillips, A. Federation of International Societies of Pediatric Gastroenterology, Hepatology, and Nutrition Consensus Report on Celiac Disease. J. Pediatr. Gastroenterol. Nutr. 2008, 47, 214–219. [Google Scholar] [CrossRef] [Green Version]
- Biagi, F.; Campanella, J.; Bianchi, P.; Zanellati, G.; Capriglione, I.; Klersy, C.; Corazza, G.R. The incidence of coeliac disease in adult first degree relatives. Dig. Liver Dis. 2008, 40, 97–100. [Google Scholar] [CrossRef]
- Lionetti, E.; Castellaneta, S.; Francavilla, R.; Pulvirenti, A.; Tonutti, E.; Amarri, S.; Barbato, M.; Barbera, C.; Barera, G.; Bellantoni, A.; et al. Introduction of Gluten, HLA Status, and the Risk of Celiac Disease in Children. N. Engl. J. Med. 2014, 371, 1295–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasano, A.; Attwood, S.E.A.; Lewis, C.J.; Bronder, C.S.; Morris, C.D.; Armstrong, G.R.; Whittam, J. European and North American populations should be screened for coeliac disease. Gut 2003, 52, 168–169. [Google Scholar] [CrossRef] [Green Version]
- Moerkens, R.; Mooiweer, J.; Withoff, S.; Wijmenga, C. Celiac disease-on-chip: Modeling a multifactorial disease in vitro. United Eur. Gastroenterol. J. 2019, 7, 467–476. [Google Scholar] [CrossRef] [Green Version]
- Kurki, A.; Kemppainen, E.; Laurikka, P.; Kaukinen, K.; Lindfors, K. The use of peripheral blood mononuclear cells in celiac disease diagnosis and treatment. Expert Rev. Gastroenterol. Hepatol. 2021, 15, 305–316. [Google Scholar] [CrossRef]
- du Pré, F.M.; van Berkel, L.A.; Ráki, M.; van Leeuwen, M.A.; de Ruiter, L.F.; Broere, F.; ter Borg, M.N.D.; Lund, F.E.; Escher, J.C.; Lundin, K.E.A.; et al. CD62LnegCD38+ Expression on Circulating CD4+ T Cells Identifies Mucosally Differentiated Cells in Protein Fed Mice and in Human Celiac Disease Patients and Controls. Am. J. Gastroenterol. 2011, 106, 1147–1159. [Google Scholar] [CrossRef] [Green Version]
- Butler, C.A.; Popescu, A.S.; Kitchener, E.J.A.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021, 158, 621–639. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Jung, J.; Zia, S.; Ho, M.; Eskandari-Sedighi, G.; Laurent, C.D.S.; McCord, K.A.; Bains, A.; Sidhu, G.; Sarkar, S.; et al. The CD33 short isoform is a gain-of-function variant that enhances Aβ1–42 phagocytosis in microglia. Mol. Neurodegener. 2021, 16, 1–22. [Google Scholar] [CrossRef]
- Ng, P.C.; Li, G.; Chui, K.M.; Chu, W.C.W.; Li, K.; Wong, R.P.O.; Chik, K.W.; Wong, E.; Fok, T.F. Neutrophil CD64 Is a Sensitive Diagnostic Marker for Early-Onset Neonatal Infection. Pediatr. Res. 2004, 56, 796–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azeem, W.; Bakke, R.M.; Appel, S.; Øyan, A.M.; Kalland, K.-H. Dual Pro- and Anti-Inflammatory Features of Monocyte-Derived Dendritic Cells. Front. Immunol. 2020, 11, 438. [Google Scholar] [CrossRef]
- Ráki, M.; Tollefsen, S.; Molberg, Ø.; Lundin, K.E.; Sollid, L.M.; Jahnsen, F.L. A Unique Dendritic Cell Subset Accumulates in the Celiac Lesion and Efficiently Activates Gluten-Reactive T Cells. Gastroenterol. 2006, 131, 428–438. [Google Scholar] [CrossRef]
- Choi, I.; Cho, B.; Kim, S.D.; Park, D.; Kim, J.Y.; Park, C.-G.; Chung, O.H.; Hwang, W.S.; Lee, J.S.; Ahn, C. Molecular cloning, expression and functional characterization of miniature swine CD86. Mol. Immunol. 2006, 43, 480–486. [Google Scholar] [CrossRef]
- Thorsby, E.; Lie, B.A. HLA associated genetic predisposition to autoimmune diseases: Genes involved and possible mechanisms. Transpl. Immunol. 2005, 14, 175–182. [Google Scholar] [CrossRef]
- Jeannin, P.; Magistrelli, G.; Aubry, J.-P.; Caron, G.; Gauchat, J.-F.; Renno, T.; Herbault, N.; Goetsch, L.; Blaecke, A.; Dietrich, P.-Y.; et al. Soluble CD86 Is a Costimulatory Molecule for Human T Lymphocytes. Immunity 2000, 13, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Husby, S.; Koletzko, S.; Korponay-Szabó, I.; Kurppa, K.; Mearin, M.L.; Ribes-Koninckx, C.; Shamir, R.; Troncone, R.; Auricchio, R.; Castillejo, G.; et al. European Society Paediatric Gastroenterology, Hepatology and Nutrition Guidelines for Diagnosing Coeliac Disease 2020. J. Pediatr. Gastroenterol. Nutr. 2020, 70, 141–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hudec, M.; Riegerová, K.; Pala, J.; Kútna, V.; Černá, M.; O´Leary, V.B. Celiac Disease Defined by Over-Sensitivity to Gliadin Activation and Superior Antigen Presentation of Dendritic Cells. Int. J. Mol. Sci. 2021, 22, 9982. https://doi.org/10.3390/ijms22189982
Hudec M, Riegerová K, Pala J, Kútna V, Černá M, O´Leary VB. Celiac Disease Defined by Over-Sensitivity to Gliadin Activation and Superior Antigen Presentation of Dendritic Cells. International Journal of Molecular Sciences. 2021; 22(18):9982. https://doi.org/10.3390/ijms22189982
Chicago/Turabian StyleHudec, Michael, Kamila Riegerová, Jan Pala, Viera Kútna, Marie Černá, and Valerie Bríd O´Leary. 2021. "Celiac Disease Defined by Over-Sensitivity to Gliadin Activation and Superior Antigen Presentation of Dendritic Cells" International Journal of Molecular Sciences 22, no. 18: 9982. https://doi.org/10.3390/ijms22189982