
Identification of Small Molecule Inhibitors against Staphylococcus aureus Dihydroorotase via HTS

 , and
, and
Abstract
:
1. Introduction
2. Results
2.1. Preparation, Purification and Activity of SaDHOase
2.2. High-Throughput Screening and Secondary Confirmation of SaDHOase Inhibitors
2.3. Binding Affinity of Selected Hits and Catalytic Site Binders
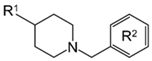
2.4. Preliminary Structure–Activity Relationship (SAR)
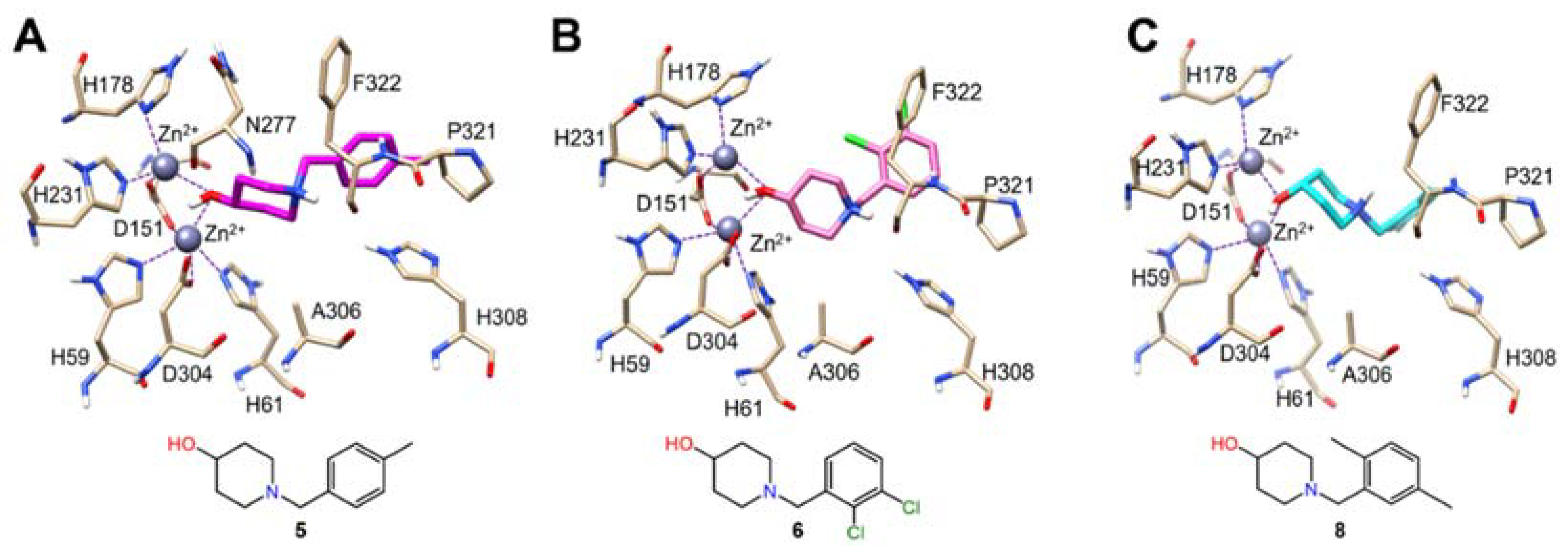
2.5. Docking Studies
3. Materials and Methods
3.1. Preparation and Purification of SaDHOase
3.2. Primary High-Throughput Screen
3.3. Secondary Counter-Screen by Surface Plasmon Resonance
3.4. Competition SPR and Analogs
3.5. Molecular Docking for Compounds 5, 6, 8
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ong, P.Y.; Leung, D.Y. Bacterial and Viral Infections in Atopic Dermatitis: A Comprehensive Review. Clin. Rev. Allergy Immunol. 2016, 51, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Hatzenbuehler, J.; Pulling, T.J. Diagnosis and management of osteomyelitis. Am. Fam. Physician 2011, 84, 1027–1033. [Google Scholar] [PubMed]
- McGuinness, W.A.; Malachowa, N.; DeLeo, F.R. Vancomycin Resistance in Staphylococcus aureus. Yale J. Biol. Med. 2017, 90, 269–281. [Google Scholar] [PubMed]
- Lakhundi, S.; Zhang, K. Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin. Microbiol. Rev. 2018, 31, e00020-18. [Google Scholar] [CrossRef] [Green Version]
- Holland, T.L.; Arnold, C.; Fowler, V.G., Jr. Clinical management of Staphylococcus aureus bacteremia: A review. JAMA 2014, 312, 1330–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, S.S.; Hung, D.T. Persistent bacterial infections, antibiotic tolerance, and the oxidative stress response. Virulence 2013, 4, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Saeed, K.; Bal, A.; Gould, I.M.; David, M.Z.; Dryden, M.; Giannitsioti, E.; Hijazi, K.; Meisner, J.A.; Esposito, S.; Scaglione, F.; et al. An update on Staphylococcus aureus infective endocarditis from the International Society of Antimicrobial Chemotherapy (ISAC). Int. J. Antimicrob. Agents. 2019, 53, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, A.; Gaca, J.G.; Chu, V.H. Management Considerations in Infective Endocarditis: A Review. JAMA 2018, 320, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.H.; Huang, Y.H.; Lin, E.S.; Chen, C.J.; Huang, C.Y. Structural basis for the interaction modes of dihydroorotase with the anticancer drugs 5-fluorouracil and 5-aminouracil. Biochem. Biophys. Res. Commun. 2021, 551, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.J.; Lei, H.; Santarsiero, B.D.; Lee, H.; Johnson, M.E. Ca-asp bound X-ray structure and inhibition of Bacillus anthracis dihydroorotase (DHOase). Bioorg. Med. Chem. 2016, 24, 4536–4543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Kolli, M.; Coulibaly, A.; Chevalier, J.; Barbe, J.; Cremieux, A. Antibacterial activity of 5-aminoorotic acid derivatives. Curr. Microbiol. 1998, 36, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Truong, L.; Hevener, K.E.; Rice, A.J.; Patel, K.; Johnson, M.E.; Lee, H. High-level expression, purification, and characterization of Staphylococcus aureus dihydroorotase (PyrC) as a cleavable His-SUMO fusion. Protein Expr. Purif. 2013, 88, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, A.J.; Truong, L.; Johnson, M.E.; Lee, H. A colorimetric assay optimization for high-throughput screening of dihydroorotase by detecting ureido groups. Anal. Biochem. 2013, 441, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Sander, E.G.; Wright, L.D.; McCormick, D.B. Evidence for function of a metal ion in the activity of dihydroorotase from Zymobacterium oroticum. J. Biol. Chem. 1965, 240, 3628–3630. [Google Scholar] [CrossRef]
- Storr, T.; Barnard, P.J. Ligand Design in Medicinal Inorganic Chemistry; Wiley: Chichester, UK, 2014. [Google Scholar]
- Schrödinger Release 2016-1: Schrödinger Suite 2016-1 Protein Preparation Wizard, Epik version 3.5, Impact version 7.0, Prime version 4.3; Schrödinger, LLC: New York, NY, USA, 2016.
- Jorgensen, W.L.; Maxwell, D.S.; TiradoRives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Schrödinger Release 2016-1: Ligprep, version 3.7; Schrödinger, LLC: New York, NY, USA, 2016.
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Compound | R1 | R2 | KD (µM) |
| 5 | -OH (p) | -4-CH3 | 48 ± 21 |
| 6 | -OH (p) | -2,3-Cl | 90 ± 4 |
| 7 | -OH (p) | -2-CH3 | 41 ± 18 |
| 8 | -OH (p) | -2,5-CH3 | 11 ± 5 |
| 9 | -OH (p) | -4-CH2CH3 | 125 ± 20 |
| 10 | -OH (p) | -3-CH3 | NB a |
| 11 | -OH (p) | -3-CF3 | NB a |
| 12 | -OH (p) | -4-CF3 | 48 ± 24 |
| 13 | -OH (m) | -2,4-CH3 | 77 ± 15 |
| 14 | -CH2OH (m) | -3-CH3 | 38 ± 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rice, A.J.; Pesavento, R.P.; Ren, J.; Youn, I.; Kwon, Y.; Ellepola, K.; Che, C.-T.; Johnson, M.E.; Lee, H. Identification of Small Molecule Inhibitors against Staphylococcus aureus Dihydroorotase via HTS. Int. J. Mol. Sci. 2021, 22, 9984. https://doi.org/10.3390/ijms22189984
Rice AJ, Pesavento RP, Ren J, Youn I, Kwon Y, Ellepola K, Che C-T, Johnson ME, Lee H. Identification of Small Molecule Inhibitors against Staphylococcus aureus Dihydroorotase via HTS. International Journal of Molecular Sciences. 2021; 22(18):9984. https://doi.org/10.3390/ijms22189984
Chicago/Turabian StyleRice, Amy J., Russell P. Pesavento, Jinhong Ren, Isoo Youn, Youngjin Kwon, Kassapa Ellepola, Chun-Tao Che, Michael E. Johnson, and Hyun Lee. 2021. "Identification of Small Molecule Inhibitors against Staphylococcus aureus Dihydroorotase via HTS" International Journal of Molecular Sciences 22, no. 18: 9984. https://doi.org/10.3390/ijms22189984