
Highlights on the Role of KRAS Mutations in Reshaping the Microenvironment of Pancreatic Adenocarcinoma


Abstract
:
1. Introduction
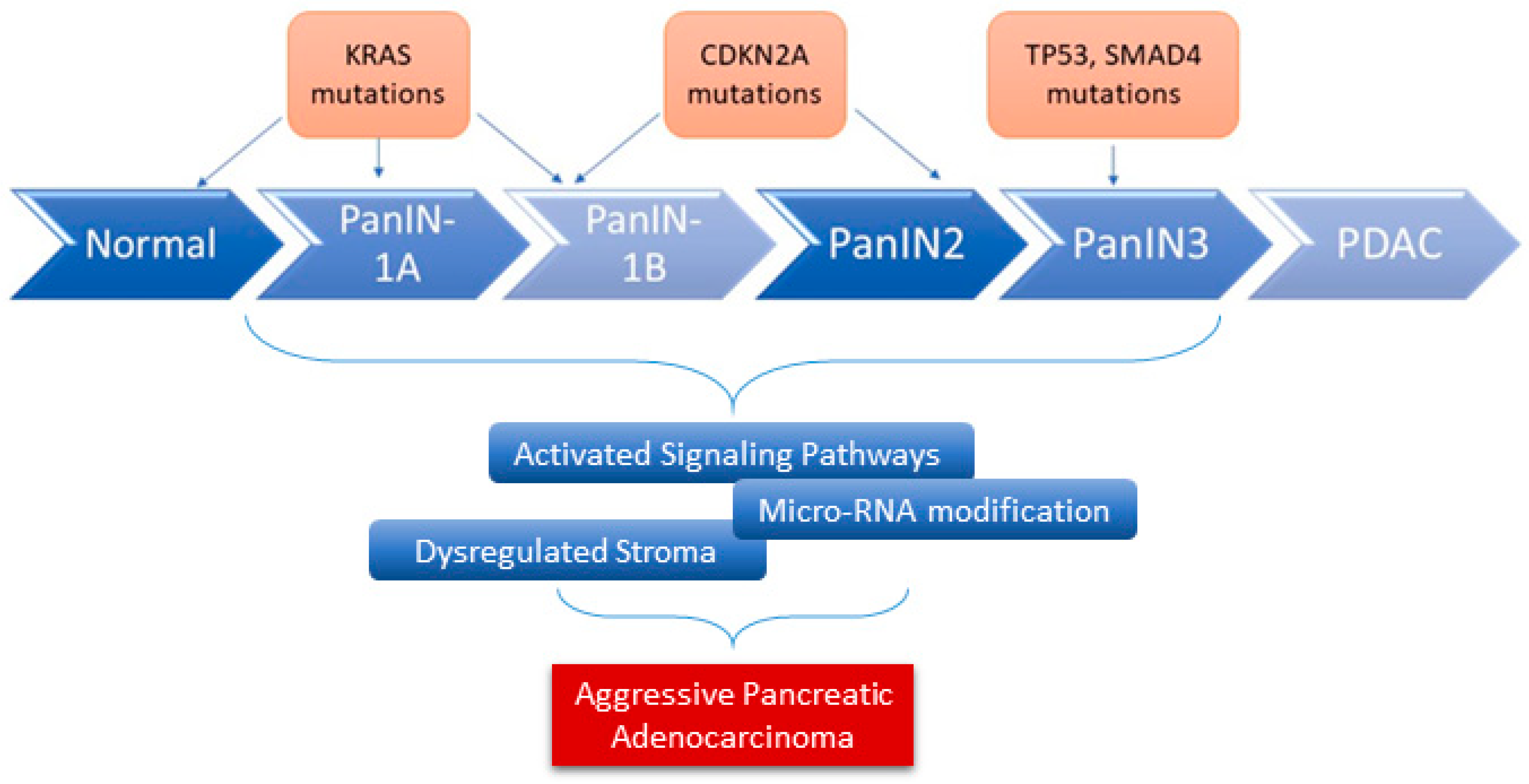
2. Pathobiology of Pancreatic Ductal Adenocarcinoma
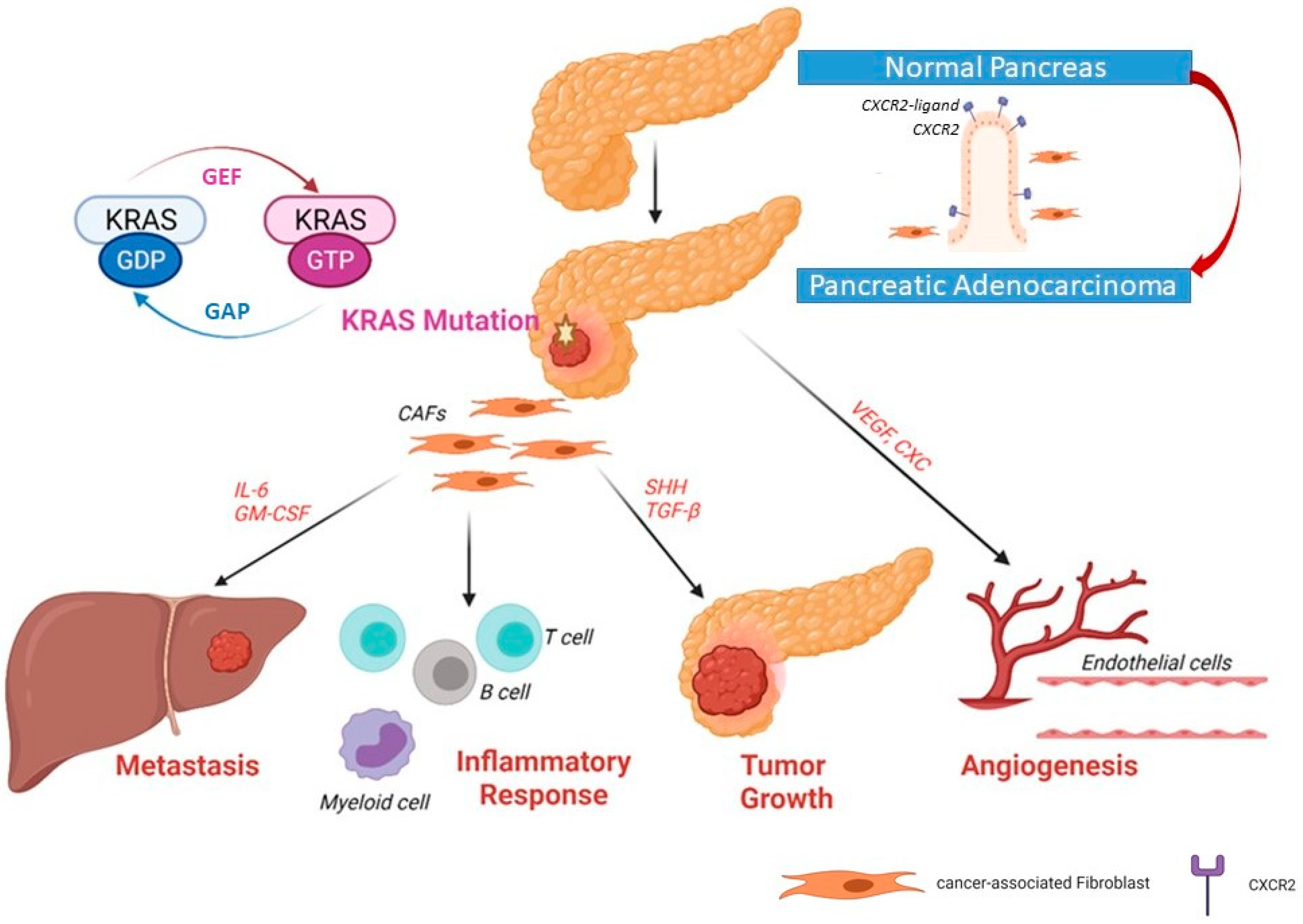
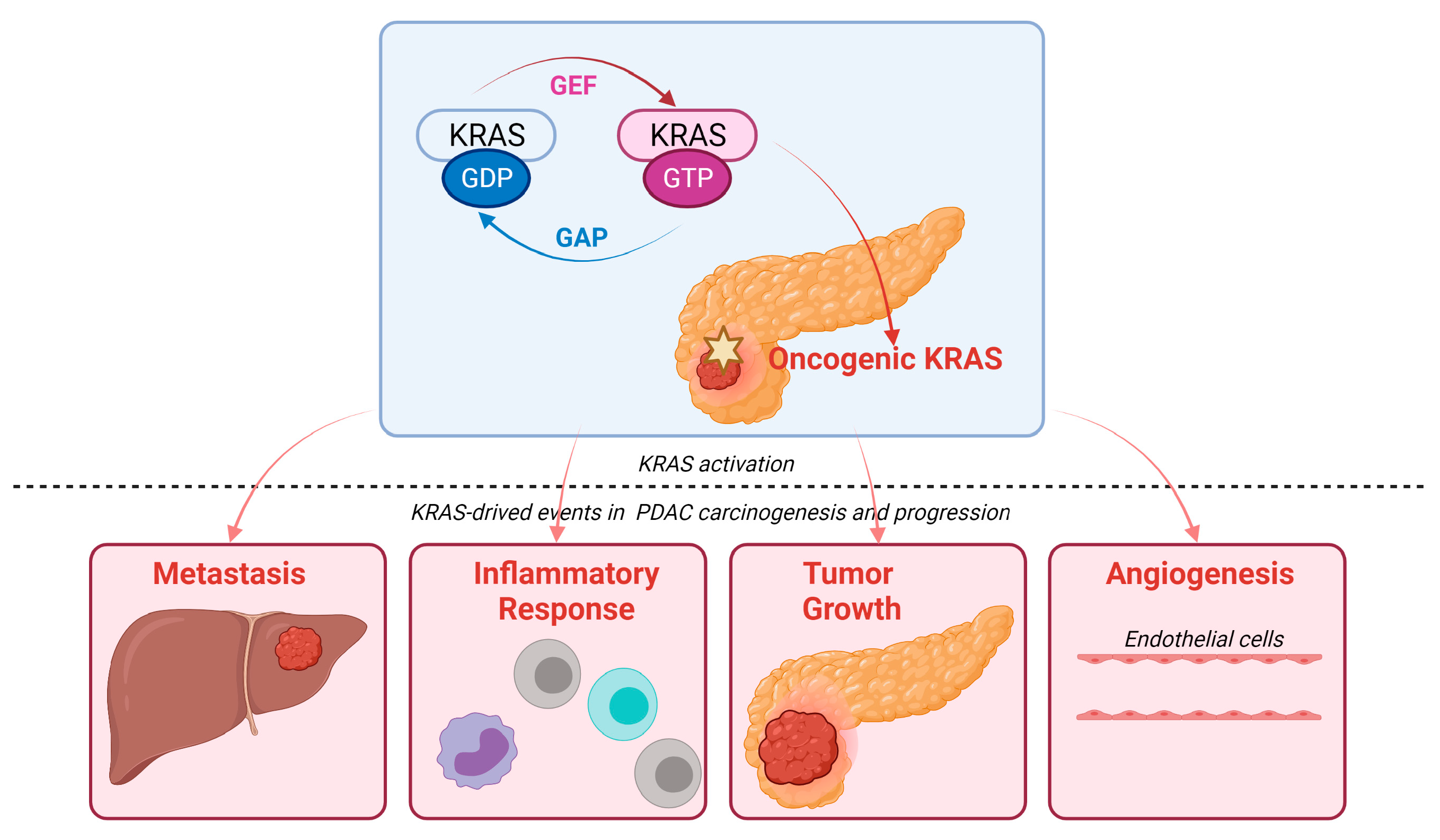
3. KRAS Signaling Pathways in PDAC
4. Mutated KRAS and the Tumor Microenvironment
4.1. Inflammatory Chemokines, Cytokines, and Interleukin 6
4.2. Mutated KRAS Effect on the Surrounding Stromal Cells
4.3. Mutated KRAS Interaction with the Immune Cells
5. KRAS Mutation and Metabolic Reprogramming
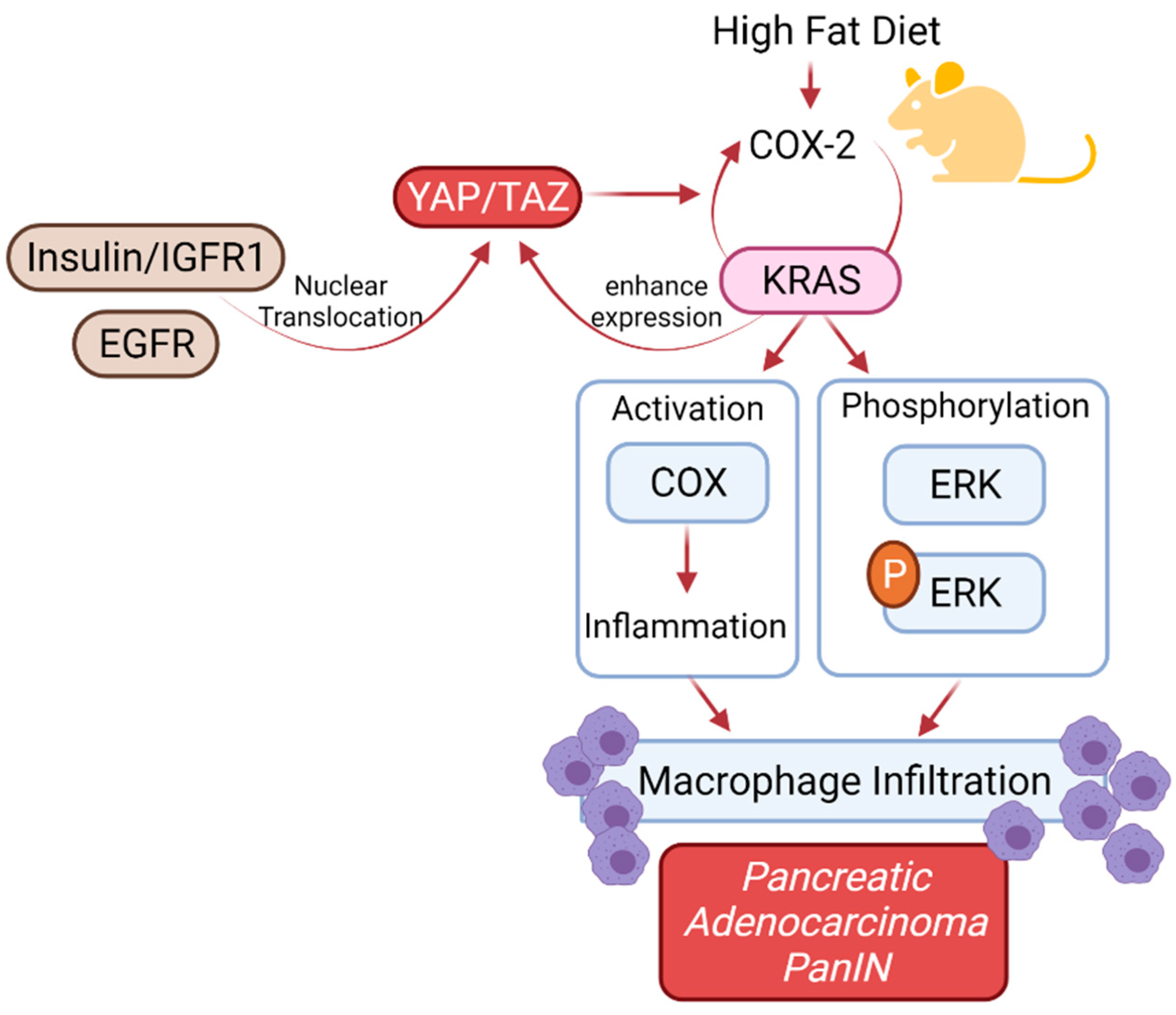
6. KRAS Mutation in Patients with Diabetes Mellitus
6.1. Therapeutic Targets in KRAS-Mutated Pancreatic Cancer
6.2. Therapeutic Targets of KRAS Mutation in CAFs and Importance of Vitamin D Therapy
6.3. Modulating the Immune Status of PDAC Microenvironment
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Glossary and Abbreviations
| AXL | Is a member of the TAM (Tyro3, Axl, MerTK) receptor tyrosine kinase family. It has been found to drive metastasis and cause immune suppression in different cancers including PDAC. |
| CAFs | Cancer-associated fibroblasts. It can promote tumorigenic features by initiating the remodeling of the extracellular matrix by secreting cytokines. |
| ERK | MEK–extracellular signal-regulated kinase. |
| FoxM1 | Fork-head box M1. |
| GAS6 | Growth arrest-specific gene 6 (GAS6). It has an important role in the stimulation of cell proliferation. |
| IGF-1 | Insulin-like growth factor 1 (IGF1), is a hormone that plays an important role in childhood growth. Through inhibition of apoptosis, IGF-1 has been shown to promote cancer development. |
| INK4a–ARF | Inhibitors of CDK4 (INK4). The INK4a–ARF locus on chromosome 9 is one of the sites mutated most frequently in human cancer. Two genes comprising over-lapping reading frames encoding p16 (INK4a) and p19 (ARF) have been discovered at this locus, and remarkably, both play an important role in regulating cell growth, survival, and senescence. |
| KRAS | Kirsten rat sarcoma viral oncogene homolog (KRAS) gene: A gene that makes a protein that is involved in cell signaling pathways that control cell growth, cell maturation, and cell death. The natural, unchanged form of the gene is called wild-type KRAS. Mutated (changed) forms of the KRAS gene have been found in some types of cancer, including non-small-cell lung cancer, colorectal cancer, and pancreatic cancer. These changes may cause cancer cells to grow and spread in the body. |
| MAPK | mitogen-activated protein kinase. |
| MEK | MAPK kinase. |
| MHC | The major histocompatibility complex (MHC) is a large locus on vertebrate DNA containing a set of closely linked polymorphic genes that code for cell surface proteins essential for the adaptive immune system. PDAC cells show a reduced expression of MHC-Class1 on their cell surface. |
| mTOR | mechanistic target of rapamycin. |
| MUC4 | Mucin4 is a large membrane-anchored glycoprotein that belongs to the mucin family. They play an important role in the protection of epithelial cells. Its overexpression has been seen in many types of carcinomas. |
| NF-κB | Nuclear factor- κB. |
| PI3K | phosphoinositide 3-kinase. |
| PPP | pentose phosphate pathway. |
| RAF | rapidly accelerated fibrosarcoma. |
| SHH | Sonic Hedgehog (SHH), is one of the hedgehog pathways that play an important role in the regulation of embryonic development. It has been found to play an important role in tumor initiation and invasiveness. |
| SMAD4 | Mothers against decapentaplegic homolog4 (SMAD4), also called DPC4, is an Intra-cellular messenger of TGF-β and shows an anti-tumor effect by inhibiting the cell growth. |
| TCA | tricarboxylic acid. |
| VDR | Vitamin D receptor. |
References
- Bournet, B.; Buscail, C.; Muscari, F.; Cordelier, P.; Buscail, L. Targeting KRAS for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur. J. Cancer 2016, 54, 75–83. [Google Scholar] [CrossRef] [PubMed]
- di Magliano, M.P.; Logsdon, C.D. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology 2013, 144, 1220–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indini, A.; Rijavec, E.; Ghidini, M.; Cortellini, A.; Grossi, F. Targeting KRAS in Solid Tumors: Current Challenges and Future Op-portunities of Novel KRAS Inhibitors. Pharmaceutics 2021, 13, 653. [Google Scholar] [CrossRef] [PubMed]
- Waters, A.M.; Ozkan-Dagliyan, I.; Vaseva, A.V.; Fer, N.; Strathern, L.A.; Hobbs, G.A.; Tessier-Cloutier, B.; Gillette, W.K.; Bagni, R.; Whiteley, G.R.; et al. Evaluation of the selectivity and sensitivity of isoform- and mutation-specific RAS antibodies. Sci. Signal. 2017, 10, eaao3332. [Google Scholar] [CrossRef] [Green Version]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathology. 2008, 3, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhang, B.; Li, H. The Roles of Frequently Mutated Genes of Pancreatic Cancer in Regulation of Tumor Microenvi-ronment. Technol. Cancer Res. Treat. 2020, 19, 1533033820920969. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The Pancreas Cancer Microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Electronic address aadhe, Cancer Genome Atlas Research, N. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017, 32, 185–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.-C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.-M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Masetti, M.; Acquaviva, G.; Visani, M.; Tallini, G.; Fornelli, A.; Ragazzi, M.; Vasuri, F.; Grifoni, D.; Di Giacomo, S.; Fiorino, S.; et al. Long-term survivors of pancreatic adenocarcinoma show low rates of genetic alterations in KRAS, TP53 and SMAD4. Cancer Biomark. 2018, 21, 323–334. [Google Scholar] [CrossRef] [PubMed]
- Visani, M.; Acquaviva, G.; De Leo, A.; Sanza, V.; Merlo, L.; Maloberti, T.; Brandes, A.A.; Franceschi, E.; Di Battista, M.; Masetti, M.; et al. Molecular alterations in pancreatic tumors. World J. Gastroenterol. 2021, 27, 2710–2726. [Google Scholar] [CrossRef] [PubMed]
- Tanizaki, J.; Okamoto, I.; Sakai, K.; Nakagawa, K. Differential roles of trans-phosphorylated EGFR, HER2, HER3, and RET as heterodimerisation partners of MET in lung cancer with MET amplification. Br. J. Cancer 2011, 105, 807–813. [Google Scholar] [CrossRef]
- Jahng, A.W.; Reicher, S.; Chung, D.; Varela, D.; Chhablani, R.; Dev, A.; Pham, B.; Nieto, J.; Venegas, R.J.; French, S.W.; et al. Staining for p53 and Ki-67 increases the sensitivity of EUS-FNA to detect pancreatic malignancy. World J. Gastrointest. Endosc. 2010, 2, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.-Y.; Liu, T.-P.; Chou, C.-J.; Chen, H.-Y.; Lee, K.-H.; Yang, P.-M. Integration of Bioinformatics Resources Reveals the Therapeutic Benefits of Gemcitabine and Cell Cycle Intervention in SMAD4-Deleted Pancreatic Ductal Adenocarcinoma. Genes 2019, 10, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartsch, D.; Shevlin, D.W.; Tung, W.S.; Kisker, O.; Wells, S.A., Jr.; Goodfellow, P.J. Frequent mutations ofCDKN2 in primary pancreatic adenocarcinomas. Genes Chromosom. Cancer 1995, 14, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Rozenblum, E.; Schutte, M.; Goggins, M.; Hahn, S.; Panzer, S.; Zahurak, M.; Goodman, S.N.; Sohn, T.A.; Hruban, R.H.; Yeo, C.J.; et al. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res. 1997, 57, 1731–1734. [Google Scholar]
- Oshima, M.; Okano, K.; Muraki, S.; Haba, R.; Maeba, T.; Suzuki, Y.; Yachida, S. Immunohistochemically Detected Expression of 3 Major Genes (CDKN2A/p16, TP53, and SMAD4/DPC4) Strongly Predicts Survival in Patients with Resectable Pancreatic Cancer. Ann. Surg. 2013, 258, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Gong, Y.; Fan, Z.; Luo, G.; Huang, Q.; Deng, S.; Cheng, H.; Jin, K.; Ni, Q.; Yu, X.; et al. Molecular alterations and targeted therapy in pancreatic ductal ade-nocarcinoma. J. Hematol. Oncol. 2020, 13, 130. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.; Raufi, A.G.; Safyan, R.A.; Bates, S.E.; Manji, G.A. BRCA Mutations in Pancreas Cancer: Spectrum, Current Management, Challenges and Future Prospects. Cancer Manag. Res. 2020, 12, 2731–2742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Azim, S.; Zubair, H.; Bhardwaj, A.; Patel, G.K.; Khushman, M.; Singh, S.; Singh, A.P. Molecular Drivers of Pancreatic Cancer Patho-genesis: Looking Inward to Move Forward. Int. J. Mol. Sci. 2017, 18, 779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleeff, J.; Korc, M.; Apte, M.; Vecchia, C.L.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef]
- Consortium TAPG. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, S.; Jang, H.; Nussinov, R.; Zhang, J. The Structural Basis of Oncogenic Mutations G12, G13 and Q61 in Small GTPase K-Ras4B. Sci. Rep. 2016, 6, 21949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicenas, J.; Kvederaviciute, K.; Meskinyte, I.; Meskinyte-Kausiliene, E.; Skeberdyte, A. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 Mutations in Pancreatic Cancer. Cancers 2017, 9, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeitouni, D.; Pylayeva-Gupta, Y.; Der, C.J.; Bryant, K.L. KRAS Mutant Pancreatic Cancer: No Lone Path to an Effective Treatment. Cancers 2016, 8, 45. [Google Scholar] [CrossRef] [PubMed]
- Pylayeva-Gupta, Y.; Grabocka, E.; Bar-Sagi, D. RAS oncogenes: Weaving a tumorigenic web. Nat. Rev. Cancer 2011, 11, 761–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonckheere, N.; Vasseur, R.; Van Seuningen, I. The cornerstone K-RAS mutation in pancreatic adenocarcinoma: From cell sig-naling network, target genes, biological processes to therapeutic targeting. Crit. Rev. Oncol. Hematology 2017, 111, 7–19. [Google Scholar] [CrossRef]
- Dey, P.; Li, J.; Zhang, J.; Chaurasiya, S.; Strom, A.; Wang, H.; Liao, W.-T.; Cavallaro, F.; Denz, P.; Bernard, V.; et al. Oncogenic KRAS-Driven Metabolic Reprogramming in Pancreatic Cancer Cells Utilizes Cytokines from the Tumor Microenvironment. Cancer Discov. 2020, 10, 608–625. [Google Scholar] [CrossRef]
- Buscail, L.; Bournet, B.; Cordelier, P. Role of oncogenic KRAS in the diagnosis, prognosis and treatment of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 153–168. [Google Scholar] [CrossRef]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef]
- Waters, A.M.; Der, C.J. KRAS: The Critical Driver and Therapeutic Target for Pancreatic Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a03145. [Google Scholar] [CrossRef]
- Preis, M.; Korc, M. Signaling pathways in pancreatic cancer. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Wang, Y.; Cao, L.; Ouellette, M.M.; Freeman, J.W. Expression of oncogenic K-ras and loss of Smad4 cooperate to induce the expression of EGFR and to promote invasion of immortalized human pancreas ductal cells. Int. J. Cancer 2010, 127, 2076–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.; Merza, M.; Luo, L.; Thorlacius, H. Inhibition of Ras signalling reduces neutrophil infiltration and tissue damage in severe acute pancreatitis. Eur. J. Pharmacol. 2015, 746, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, Y.; Campbell, P.M.; Brekken, R.A.; Sung, B.; Ouellette, M.M.; Fleming, J.B.; Aggarwal, B.B.; Der, C.; Guha, S. K-Ras Promotes Angiogenesis Mediated by Immortalized Human Pancreatic Epithelial Cells through Mitogen-Activated Protein Kinase Signaling Pathways. Mol. Cancer Res. 2009, 7, 799–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitajima, S.; Thummalapalli, R.; Barbie, D.A. Inflammation as a driver and vulnerability of KRAS mediated oncogenesis. Semin. Cell Dev. Biol. 2016, 58, 127–135. [Google Scholar] [CrossRef] [Green Version]
- Awaji, M.; Saxena, S.; Wu, L.; Prajapati, D.R.; Purohit, A.; Varney, M.L.; Kumar, S.; Rachagani, S.; Ly, Q.P.; Jain, M.; et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J. 2020, 34, 9405–9418. [Google Scholar] [CrossRef]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Monteran, L.; Erez, N. The Dark Side of Fibroblasts: Cancer-Associated Fibroblasts as Mediators of Immunosuppression in the Tumor Microenvironment. Front. Immunol. 2019, 10, 1835. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, M.F.B.; Mortensen, M.B.; Detlefsen, S. Key players in pancreatic cancer-stroma interaction: Cancer-associated fibroblasts, endothelial and inflammatory cells. World J. Gastroenterol. 2016, 22, 2678–2700. [Google Scholar] [CrossRef]
- Mei, L.; Du, W.; Ma, W.W. Targeting stromal microenvironment in pancreatic ductal adenocarcinoma: Controversies and promises. J. Gastrointest. Oncol. 2016, 7, 487–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Notta, F.; Navab, R.; Joseph, J.; Ibrahimov, E.; Xu, J.; Zhu, C.-Q.; Borgida, A.; Gallinger, S.; Tsao, M.-S. Senescent Carcinoma-associated Fibroblasts Upregulate IL8 to Enhance Pro-metastatic Phenotypes. Mol. Cancer Res. 2016, 15, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldinucci, D.; Borghese, C.; Casagrande, N. The CCL5/CCR5 Axis in Cancer Progression. Cancers 2020, 12, 1765. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Mishra, M.K.; Eltoum, I.-E.A.; Bae, S.; Lillard, J.W., Jr.; Singh, R. CCR5/CCL5 axis interaction promotes migratory and invasiveness of pancreatic cancer cells. Sci. Rep. 2018, 8, 1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huffman, A.P.; Lin, J.H.; Kim, S.I.; Byrne, K.T.; Vonderheide, R.H. CCL5 mediates CD40-driven CD4+ T cell tumor infiltration and immunity. JCI Insight 2020, 5, e137263. [Google Scholar] [CrossRef] [PubMed]
- Veatch, J.R.; Jesernig, B.L.; Kargl, J.; Fitzgibbon, M.; Lee, S.M.; Baik, C.; Martins, R.; Hughton, M.; Riddell, S.R. Endogenous CD4(+) T Cells Recognize Neoantigens in Lung Cancer Patients, Including Recurrent Oncogenic KRAS and ERBB2 (Her2) Driver Mutations. Cancer Immunol. Res. 2019, 7, 910–922. [Google Scholar] [CrossRef] [Green Version]
- Ryan, D.P.; Hong, T.S.; Bardeesy, N. Pancreatic adenocarcinoma. N. Engl. J. Med. 2014, 371, 2140–2141. [Google Scholar] [CrossRef] [PubMed]
- Lesina, M.; Kurkowski, M.U.; Ludes, K.; Rose-John, S.; Treiber, M.; Klöppel, G.; Yoshimura, A.; Reindl, W.; Sipos, B.; Akira, S.; et al. Stat3/Socs3 Activation by IL-6 Transsignaling Promotes Progression of Pancreatic Intraepithelial Neoplasia and Development of Pancreatic Cancer. Cancer Cell 2011, 19, 456–469. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yan, W.; Collins, M.A.; Bednar, F.; Rakshit, S.; Zetter, B.R.; Stanger, B.Z.; Chung, I.; Rhim, A.D.; Di Magliano, M.P. Interleukin-6 Is Required for Pancreatic Cancer Progression by Promoting MAPK Signaling Activation and Oxidative Stress Resistance. Cancer Res. 2013, 73, 6359–6374. [Google Scholar] [CrossRef] [Green Version]
- Tape, C.J.; Ling, S.; Dimitriadi, M.; McMahon, K.M.; Worboys, J.D.; Leong, H.S.; Norre, I.C.; Miller, C.J.; Poulogiannis, G.; Laffenburger, D.A.; et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell 2016, 165, 1818. [Google Scholar] [CrossRef] [PubMed]
- Gu, D.; Schlotman, K.E.; Xie, J. Deciphering the role of hedgehog signaling in pancreatic cancer. J. Biomed. Res. 2016, 30, 353–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, P. Pancreatic stellate cells and fibrosis. In Pancreatic Cancer and Tumor Microenvironment; Grippo, P.J., Munshi, H.G., Eds.; Transworld Research Network: Trivandrum, India, 2012; Chapter 3. [Google Scholar]
- Jonckheere, N.; Van Seuningen, I. The membrane-bound mucins: From cell signalling to transcriptional regulation and expression in epithelial cancers. Biochimie 2010, 92, 1–11. [Google Scholar] [CrossRef]
- Vasseur, R.; Skrypek, N.; Duchêne, B.; Renaud, F.; Martínez-Maqueda, D.; Vincent, A.; Porchet, N.; Van Seuningen, I.; Jonckheere, N. The mucin MUC4 is a transcriptional and post-transcriptional target of K-ras oncogene in pancreatic cancer. Implication of MAPK/AP-1, NF-κB and RalB signaling pathways. Biochim. Biophys. Acta Bioenerg. 2015, 1849, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Pommier, A.; Anaparthy, N.; Memos, N.; Kelley, Z.L.; Gouronnec, A.; Yan, R.; Auffray, C.; Albrengues, J.; Egeblad, M.; Iacobuzio-Donahue, C.A.; et al. Unresolved endoplasmic reticulum stress engenders immune-resistant, latent pancreatic cancer metastases. Science 2018, 360, eaao4908. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy Promotes Immune Evasion of Pancreatic Cancer by Degrading MHC-I; Springer: Berlin/Heidelberg, Germany, 2020; Volume 581, ISBN 0000007587. [Google Scholar]
- Kaur, K.; Chang, H.-H.; Topchyan, P.; Cook, J.M.; Barkhordarian, A.; Eibl, G.; Jewett, A. Deficiencies in Natural Killer Cell Numbers, Expansion, and Function at the Pre-Neoplastic Stage of Pancreatic Cancer by KRAS Mutation in the Pancreas of Obese Mice. Front. Immunol. 2018, 9, 1229. [Google Scholar] [CrossRef] [Green Version]
- Nowak, N.; Kulma, A.; Gutowicz, J. Up-regulation of key glycolysis proteins in cancer development. Open Life Sci. 2018, 13, 569–581. [Google Scholar] [CrossRef]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras Maintains Pancreatic Tumors through Regulation of Anabolic Glucose Metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [Green Version]
- Elia, I.; Doglioni, G.; Fendt, S.-M. Metabolic Hallmarks of Metastasis Formation. Trends Cell Biol. 2018, 28, 673–684. [Google Scholar] [CrossRef]
- Rozeveld, C.N.; Johnson, K.M.; Zhang, L.; Razidlo, G.L. KRAS Controls Pancreatic Cancer Cell Lipid Metabolism and Invasive Potential through the Lipase HSL. Cancer Res. 2020, 80, 4932–4945. [Google Scholar] [CrossRef]
- Badea, L.; Herlea, V.; Dima, S.O.; Dumitrascu, T.; Popescu, I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology 2009, 55, 2016–2027. [Google Scholar]
- Logsdon, C.D.; Simeone, D.M.; Binkley, C.; Arumugam, T.; Greenson, J.K.; Giordano, T.J.; Misek, D.E.; Kuick, R.; Hanash, S. Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res. 2003, 63, 2649–2657. [Google Scholar] [PubMed]
- Pei, H.; Li, L.; Fridley, B.L.; Jenkins, G.D.; Kalari, K.R.; Lingle, W.; Petersen, G.; Lou, Z.; Wang, L. FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell 2009, 16, 259–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segara, D.; Biankin, A.; Kench, J.; Langusch, C.C.; Dawson, A.C.; Skalicky, D.; Gotley, D.; Coleman, M.J.; Sutherland, R.L.; Henshall, S.M. Expression of HOXB2, a Retinoic Acid Signaling Target in Pancreatic Cancer and Pancreatic Intraepithelial Neoplasia. Clin. Cancer Res. 2005, 11, 3587–3596. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.V. MYC, Metabolism, Cell Growth, and Tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef]
- Wang, H.W.; Joyce, J.A. Alternative activation of tumor-associated macrophages by IL-4: Priming for protumoral functions. Cell Cycle 2010, 9, 4824–4835. [Google Scholar] [CrossRef] [Green Version]
- Vaseva, A.V.; Blake, D.R.; Gilbert, T.; Ng, S.; Hostetter, G.; Azam, S.H.; Ozkan-Dagliyan, I.; Gautam, P.; Bryant, K.L.; Pearce, K.H.; et al. KRAS Suppression-Induced Degradation of MYC Is Antagonized by a MEK5-ERK5 Compensatory Mechanism. Cancer Cell 2018, 34, 807–822. [Google Scholar] [CrossRef] [Green Version]
- Eibl, G.; Rozengurt, E. KRAS, YAP, and obesity in pancreatic cancer: A signaling network with multiple loops. Semin. Cancer Biol. 2019, 54, 50–62. [Google Scholar] [CrossRef]
- Philip, B.; Roland, C.L.; Daniluk, J.; Liu, Y.; Chatterjee, D.; Gomez, S.B.; Ji, B.; Huang, H.; Wang, H.; Fleming, J.B.; et al. A High-Fat Diet Activates Oncogenic Kras and COX2 to Induce Development of Pancreatic Ductal Adenocarcinoma in Mice. Gastroenterology 2013, 145, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Burke, J.P.; Phan, J.; Burns, M.C.; Olejniczak, E.T.; Waterson, A.G.; Lee, T.; Rossanese, O.W.; Fesik, S.W. Discovery of Small Molecules that Bind to K-Ras and Inhibit Sos-Mediated Activation. Angew. Chem. Int. Ed. 2012, 51, 6140–6143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torphy, R.J.; Zhu, Y.; Schulick, R.D. Immunotherapy for pancreatic cancer: Barriers and breakthroughs. Ann. Gastroenterol. Surg. 2018, 2, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shima, F.; Yoshikawa, Y.; Ye, M.; Araki, M.; Matsumoto, S.; Liao, J.; Hu, L.; Sugimoto, T.; Ijiri, Y.; Takeda, A.; et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc. Natl. Acad. Sci. USA 2013, 110, 8182–8187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission Possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, S.M.; Westover, K.; Ficarro, S.B.; Harrison, R.A.; Choi, H.G.; Pacold, M.E.; Carrasco, M.; Hunter, J.; Kim, N.D.; Xie, T.; et al. Therapeutic Targeting of Oncogenic K-Ras by a Covalent Catalytic Site Inhibitor. Angew. Chem. Int. Ed. 2014, 53, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nat. Cell Biol. 2013, 503, 548–551. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, G.; Papke, B.; Ismail, S.; Vartak, N.; Chandra, A.; Hoffmann, M.; Hahn, S.A.; Triola, G.; Wittinghofer, A.; Bastiaens, P.I.H.; et al. Small molecule inhibition of the KRAS-PDEdelta interaction impairs oncogenic KRAS signalling. Nature 2013, 497, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Rhim, A.D.; Oberstein, P.E.; Thomas, D.H.; Mirek, E.T.; Palermo, C.F.; Sastra, S.A.; Dekleva, E.N.; Saunders, T.; Becerra, C.P.; Tattersall, I.; et al. Stromal Elements Act to Restrain, Rather Than Support, Pancreatic Ductal Adenocarcinoma. Cancer Cell 2014, 25, 735–747. [Google Scholar] [CrossRef] [Green Version]
- Beg, M.S.; Lowy, A.M.; O’Dwyer, P.J.; Jameson, G.S.; Borazanci, E.H.; Patel, H.; Massey, C.; Schoelermann, J.; Lorens, J.; Fattah, F.; et al. A randomized clinical trial of chemotherapy with gemcitabine/cisplatin/nabpaclitaxel with or without the AXL inhibitor bemcentinib (BGB324) for patients with advanced pancreatic cancer. J. Clin. Oncol. 2019, 37, TPS473. [Google Scholar] [CrossRef]
- Sarantopoulos, J.; Beg, M.S.; Fotopoulos, G.; Taverna, J.A.; Anthony, S.P.; Weitman, S.D.; Warner, S.L.; Mouritsen, L.; Bearss, D.; Smith, S.; et al. A phase 1a / 1b first-in-human, open-label, dose-escalation, safety, pharmacokinetic, and pharmacodynamic study of oral TP-0903, a potent inhibitor of AXL kinase, administered daily for 21 days to patients with advanced solid tumors. J. Clin. Oncol. 2018, 36, TPS2612. [Google Scholar] [CrossRef]
- Pan, B.; Liao, Q.; Niu, Z.; Zhou, L.; Zhao, Y. Cancer-associated fibroblasts in pancreatic adenocarcinoma. Futur. Oncol. 2015, 11, 2603–2610. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.D.; Jiang, X.; Sullivan, K.M.; Jalikis, F.G.; Smythe, K.S.; Abbasi, A.; Vignali, M.; Park, J.O.; Daniel, S.K.; Pollack, S.M.; et al. Mobilization of CD8+ T Cells via CXCR4 Blockade Facilitates PD-1 Checkpoint Therapy in Human Pancreatic Cancer. Clin. Cancer Res. 2019, 25, 3934–3945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.; Pereg, Y.; Bulvik, B.; Klein, S.; Mishalian, I.; Wald, H.; Eizenberg, O.; Beider, K.; Nagler, A.; Golan, R.; et al. Single Dose of the CXCR4 Antagonist BL-8040 Induces Rapid Mobilization for the Collection of Human CD34+ Cells in Healthy Volunteers. Clin. Cancer Res. 2017, 23, 6790–6801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maehira, H.; Miyake, T.; Iida, H.; Tokuda, A.; Mori, H.; Yasukawa, D.; Mukaisho, K.-I.; Shimizu, T.; Tani, M. Vimentin Expression in Tumor Microenvironment Predicts Survival in Pancreatic Ductal Adenocarcinoma: Heterogeneity in Fibroblast Population. Ann. Surg. Oncol. 2019, 26, 4791–4804. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Jia, Z.; Gao, Y.; Xie, D.; Wei, D.; Cui, J.; Mishra, L.; Huang, S.; Zhang, Y.; Xie, K. Activation of Vitamin D Receptor Signaling Downregulates the Expression of Nuclear FOXM1 Protein and Suppresses Pancreatic Cancer Cell Stemness. Clin. Cancer Res. 2015, 21, 844–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorchs, L.; Ahmed, S.; Mayer, C.; Knauf, A.; Moro, C.F.; Svensson, M.; Heuchel, R.; Rangelova, E.; Bergman, P.; Kaipe, H. The vitamin D analogue calcipotriol promotes an anti-tumorigenic phenotype of human pancreatic CAFs but reduces T cell mediated immunity. Sci. Rep. 2020, 10, 17444. [Google Scholar] [CrossRef]
- Garg, B.; Giri, B.; Modi, S.; Sethi, V.; Castro, I.; Umland, O.; Ban, Y.; Lavania, S.; Dawra, R.; Banerjee, R.; et al. NFkappaB in Pancreatic Stellate Cells Reduces Infiltration of Tumors by Cytotoxic T Cells and Killing of Cancer Cells, via Up-regulation of CXCL12. Gastroenterology 2018, 155, 880–891. [Google Scholar] [CrossRef]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 antagonist, in com-bination with pembrolizumab and chemotherapy for pancreatic cancer: The COMBAT trial. Nat Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef] [Green Version]
- Kim, V.M.; Blair, A.; Lauer, P.; Foley, K.; Che, X.; Soares, K.; Xia, T.; Muth, S.T.; Kleponis, J.; Armstrong, T.D.; et al. Anti-pancreatic tumor efficacy of a Listeria-based, Annexin A2-targeting immunotherapy in combination with anti-PD-1 antibodies. J. Immunother. Cancer 2019, 7, 132. [Google Scholar] [CrossRef] [PubMed]
- Christensen, M.V.; Hogdall, C.K.; Jochumsen, K.M.; Hogdall, E.V.S. Annexin A2 and cancer: A systematic review. Int. J. Oncology. 2018, 52, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.C. Annexin A2 (ANX A2): An emerging biomarker and potential therapeutic target for aggressive cancers. Int. J. Cancer 2019, 144, 2074–2081. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, J.S.; Kim, W.K.; Oh, K.J.; Kim, J.M.; Lee, H.J.; Han, B.S.; Kim, D.S.; Seo, Y.S.; Lee, S.C.; et al. Intracellular annexin A2 regulates NF-kappaB signaling by binding to the p50 subunit: Implications for gemcitabine resistance in pancreatic cancer. Cell Death Disease 2015, 6, e1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Wang, J.; Zhang, H.; Li, C.; Chen, C.; Zhu, T. Transcription factor Sp1 is upregulated by PKCiota to drive the expression of YAP1 during pancreatic carcinogenesis. Carcinogenesis 2021, 42, 344–356. [Google Scholar] [CrossRef]
- Kaczynski, J.; Cook, T.; Urrutia, R. Sp1- and Krüppel-like transcription factors. Genome Biol. 2003, 4, 206. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets/Diseases | Drugs | Phase of Trial | Patients/ In Vivo/In Vitro | Outcomes | References |
|---|---|---|---|---|---|
| Targeting CXCR4 in PDAC | BL-8040 (CXCR4 inhibitor) plus pembrolizumab with or without 5-FU and liposomal irinotecan | Phase 2 | 80 Patients | Objective response rate | [90] NCT02826486 [91] |
| Targeting AXL | (Nab-paclitaxel, Gemcitabine, Cisplatin) with or without BGB324 (Axl inhibitor) TP-0903 | Phase 1 and 2 Phase 1 | 74 Patients 177 Patients | Decreased tumor volume and increased cancer cell apoptosis | NCT03649321 [81] NCT02729298 [82] |
| Metabolism in RAS-driven Pancreatic cancer. Stage II, III, IV pancreatic cancer | Trametinib, hydroxychloroquine | Phase 1 | 33 participants | Ongoing Results are not yet available | NCT03825289 |
| Targeting autophagy/Metabolism in RAS-driven Pancreatic cancer. Metastatic pancreatic adenocarcinoma, stage IV pancreatic cancer | Hydroxychloroquine, binimetinib | Phase 1 | 39 participants | Ongoing Results are not yet available | NCT04132505 |
| KRAS p.G12C Mutant Advanced Solid Tumors | AMG 510 (Sotorasib) Anti PD-1/L1 Midazolam | Phase 1 and 2 | 733 participants | Partial responses in two of four NSCLC patients, with stable disease achieved in the remaining two | NCT03600883 |
| Multiple clinical trials are underway to assess the benefit of vitamin D treatment in PDAC | |||||
| Multiple clinical trials are underway to assess the benefit of vitamin D treatment in PDAC | calcipotriol (a synthetic form of vitamin D) Combined Calcipotriol and gemcitabine treatment | In vivo In vivo | Reduced markers of inflammation and fibrosis in pancreatitis and human tumor strom aEnhanced the survival of the KPC (KRASLSL-G12D/+; Trp53LSL-R172H/+; Pdx-1-Cre) mouse model, ultimately increasing median animal survival by 57%. | NCT03472833 NCT03300921 NCT02754726 [87] | |
| Targeting vitamin D receptor (VDR) /PDAC | Vitamin D receptor agonist paricalcitol plus gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer | Phase 2 | 112 Patients | Ongoing Results are not yet available | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hafezi, S.; Saber-Ayad, M.; Abdel-Rahman, W.M. Highlights on the Role of KRAS Mutations in Reshaping the Microenvironment of Pancreatic Adenocarcinoma. Int. J. Mol. Sci. 2021, 22, 10219. https://doi.org/10.3390/ijms221910219
Hafezi S, Saber-Ayad M, Abdel-Rahman WM. Highlights on the Role of KRAS Mutations in Reshaping the Microenvironment of Pancreatic Adenocarcinoma. International Journal of Molecular Sciences. 2021; 22(19):10219. https://doi.org/10.3390/ijms221910219
Chicago/Turabian StyleHafezi, Shirin, Maha Saber-Ayad, and Wael M. Abdel-Rahman. 2021. "Highlights on the Role of KRAS Mutations in Reshaping the Microenvironment of Pancreatic Adenocarcinoma" International Journal of Molecular Sciences 22, no. 19: 10219. https://doi.org/10.3390/ijms221910219
APA StyleHafezi, S., Saber-Ayad, M., & Abdel-Rahman, W. M. (2021). Highlights on the Role of KRAS Mutations in Reshaping the Microenvironment of Pancreatic Adenocarcinoma. International Journal of Molecular Sciences, 22(19), 10219. https://doi.org/10.3390/ijms221910219