
In Silico Identification and Evaluation of Natural Products as Potential Tumor Necrosis Factor Function Inhibitors Using Advanced Enalos Asclepios KNIME Nodes


 , ,
, , 
Abstract
:1. Introduction
2. Results
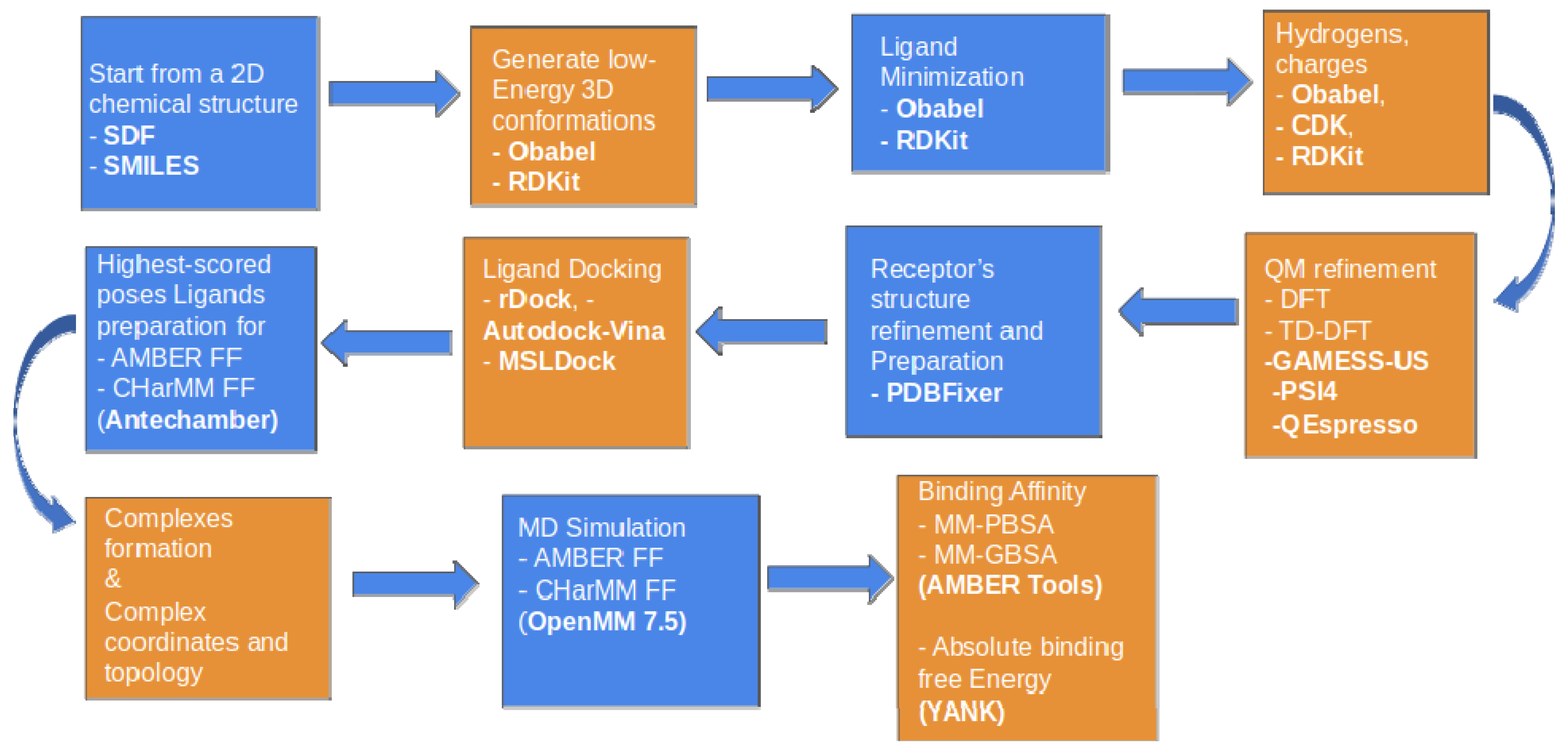
2.1. Computer-Aided Drug Design (CADD)
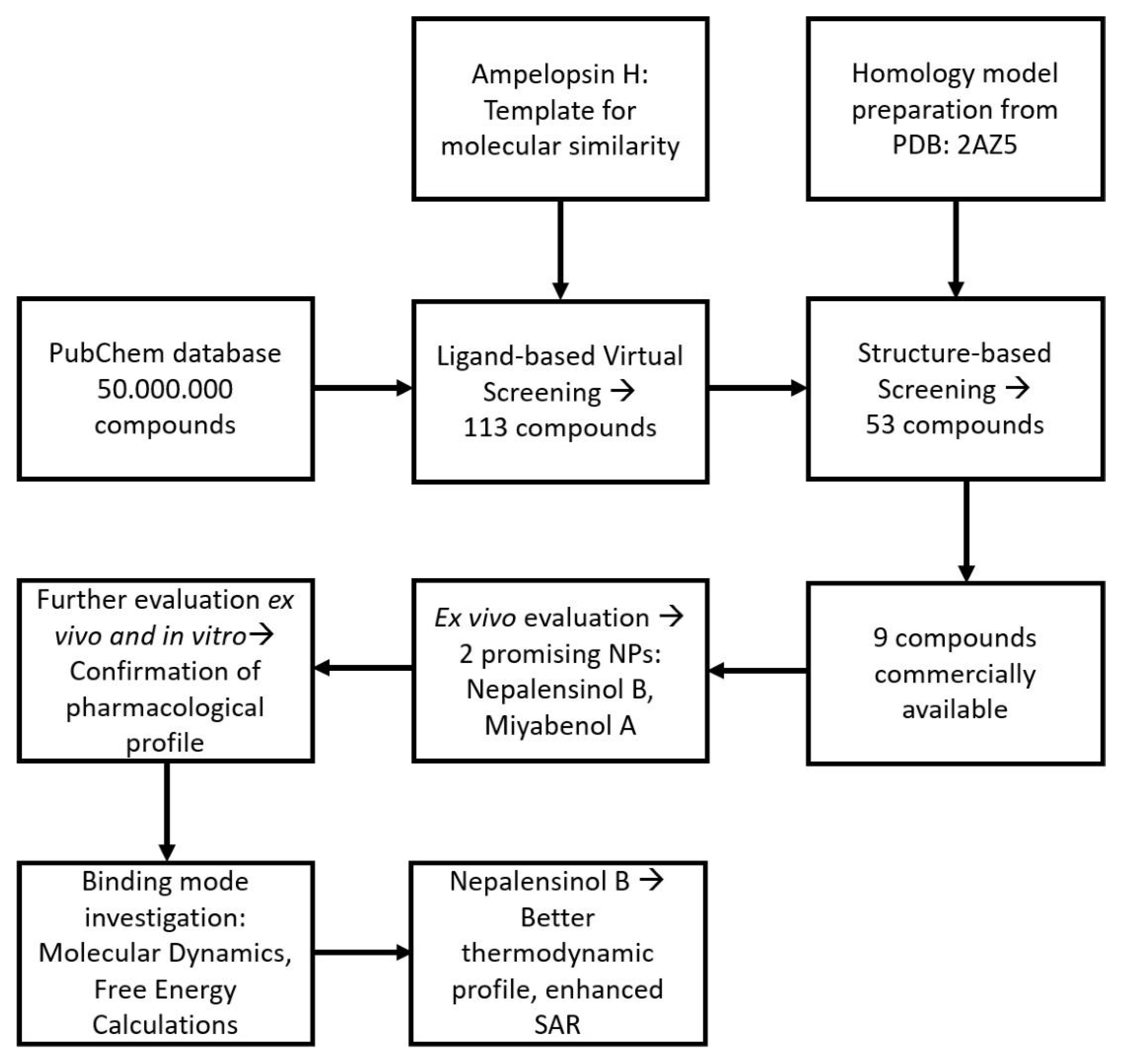
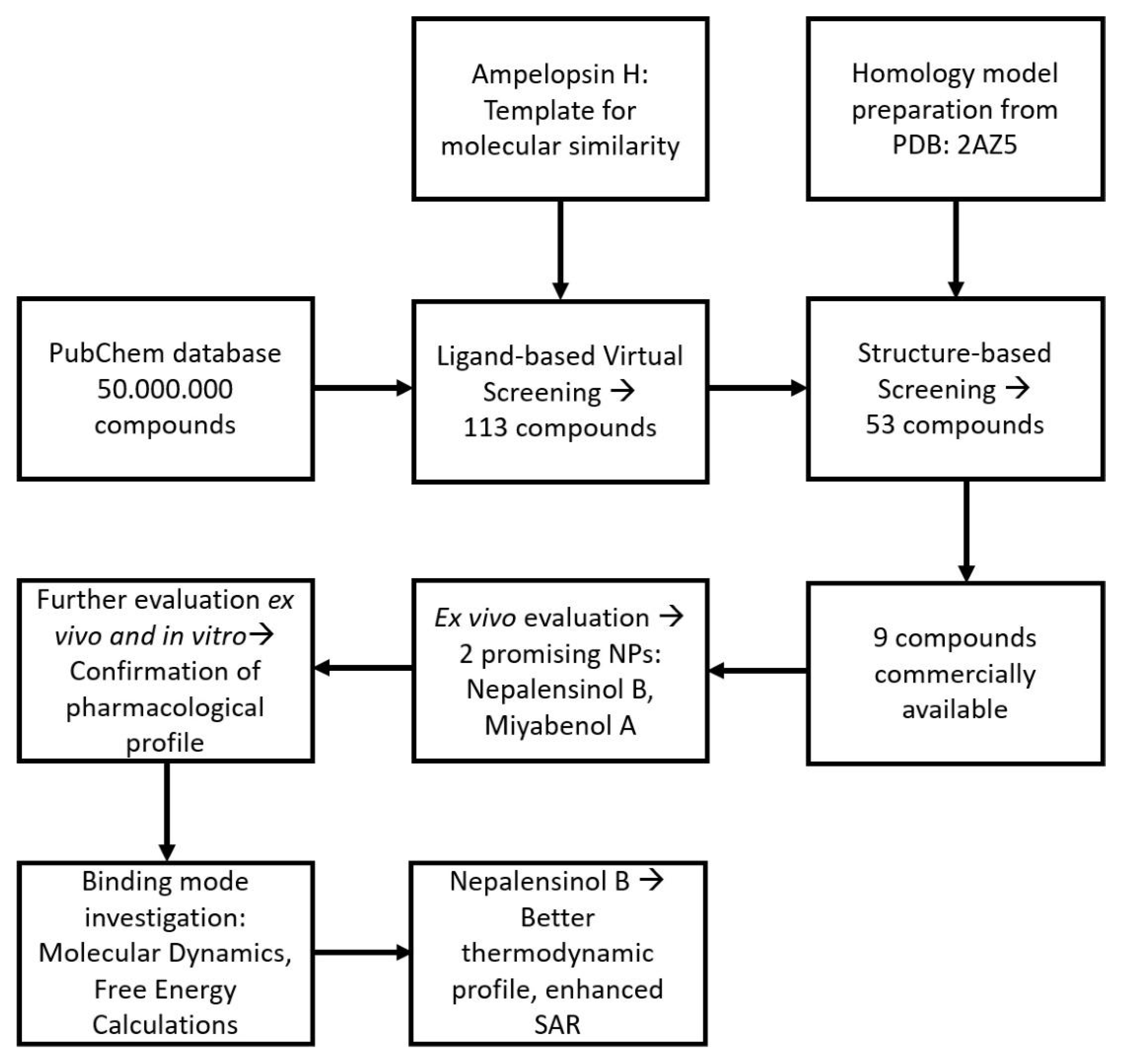
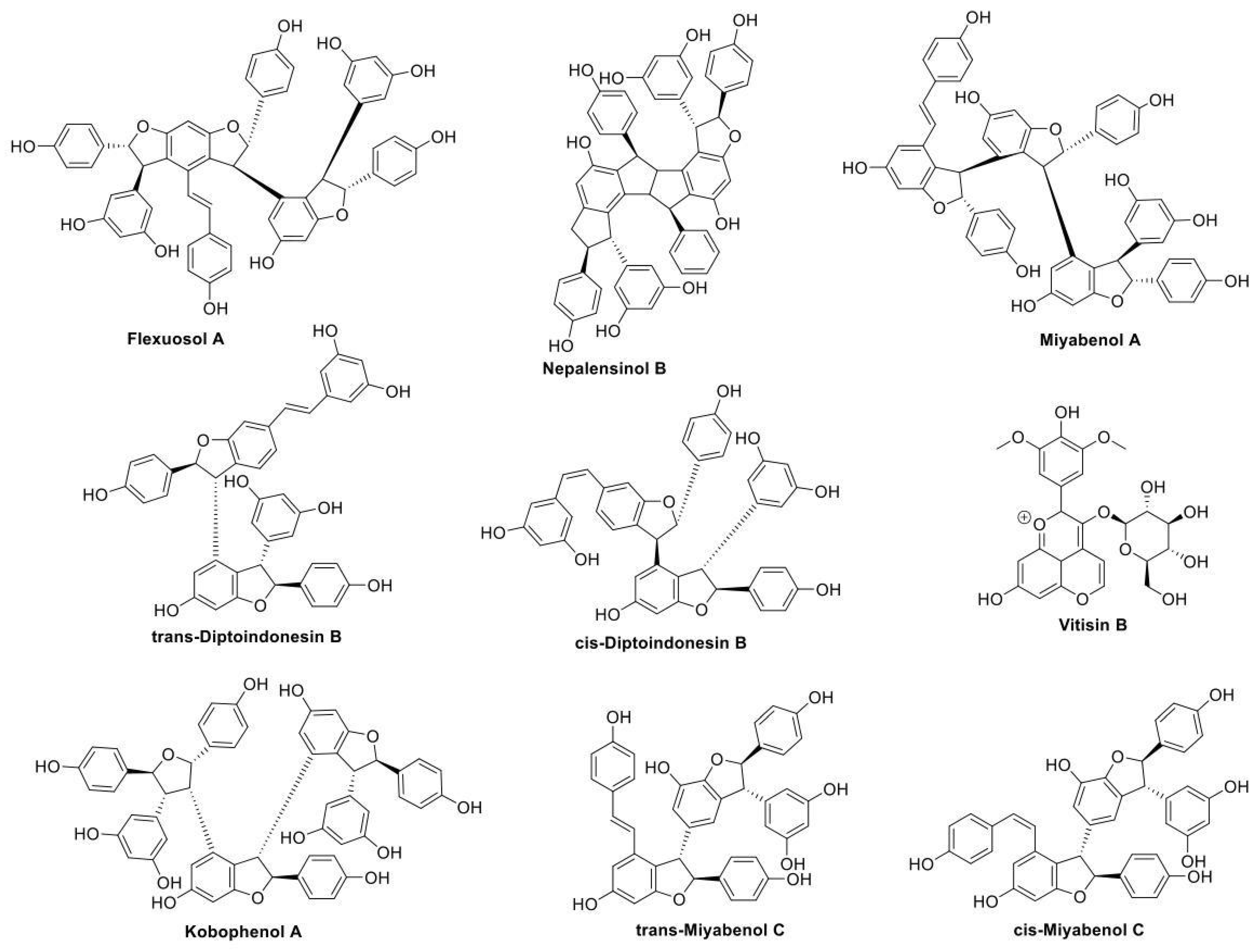
2.1.1. Initial Search and Filtering
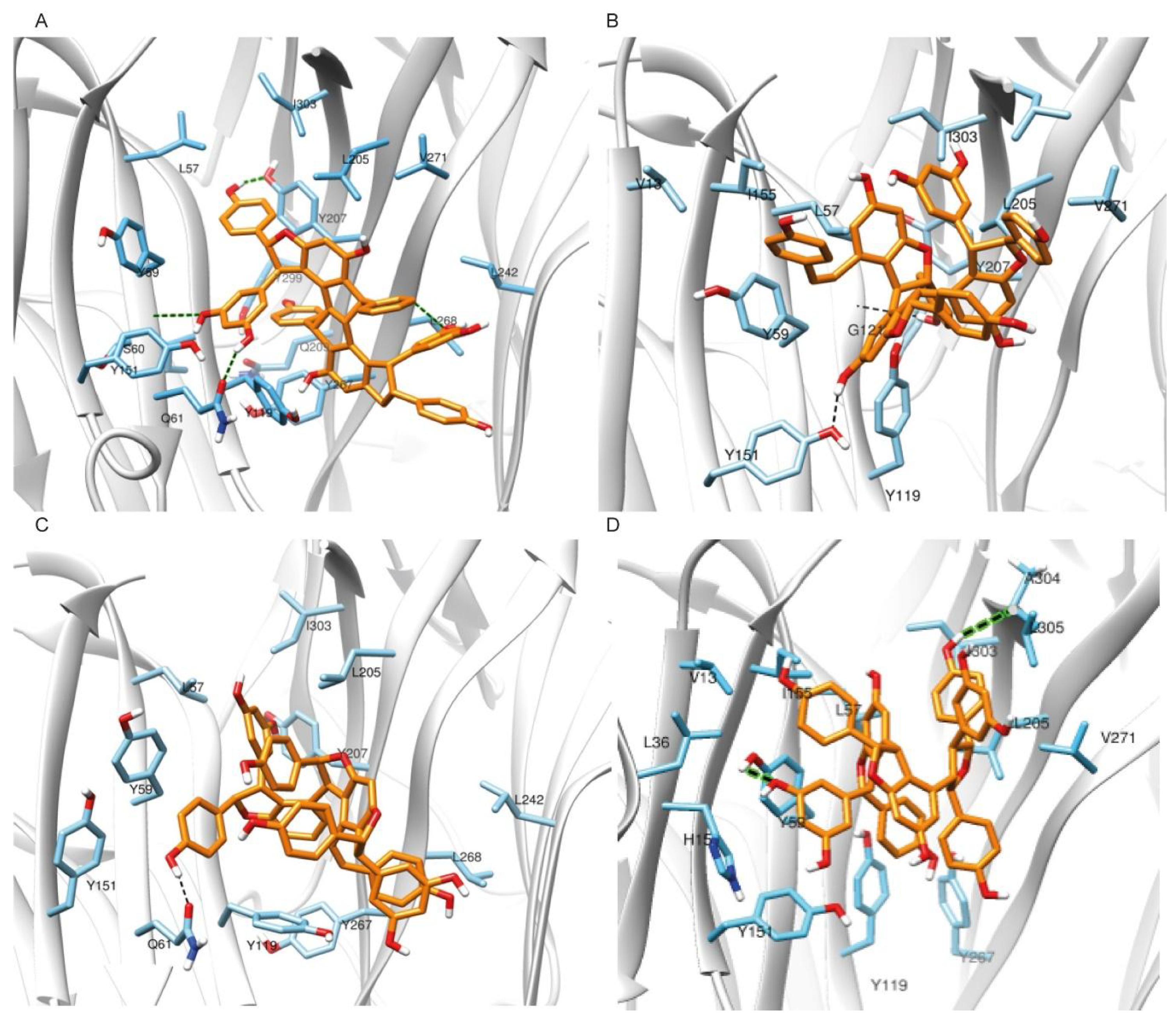
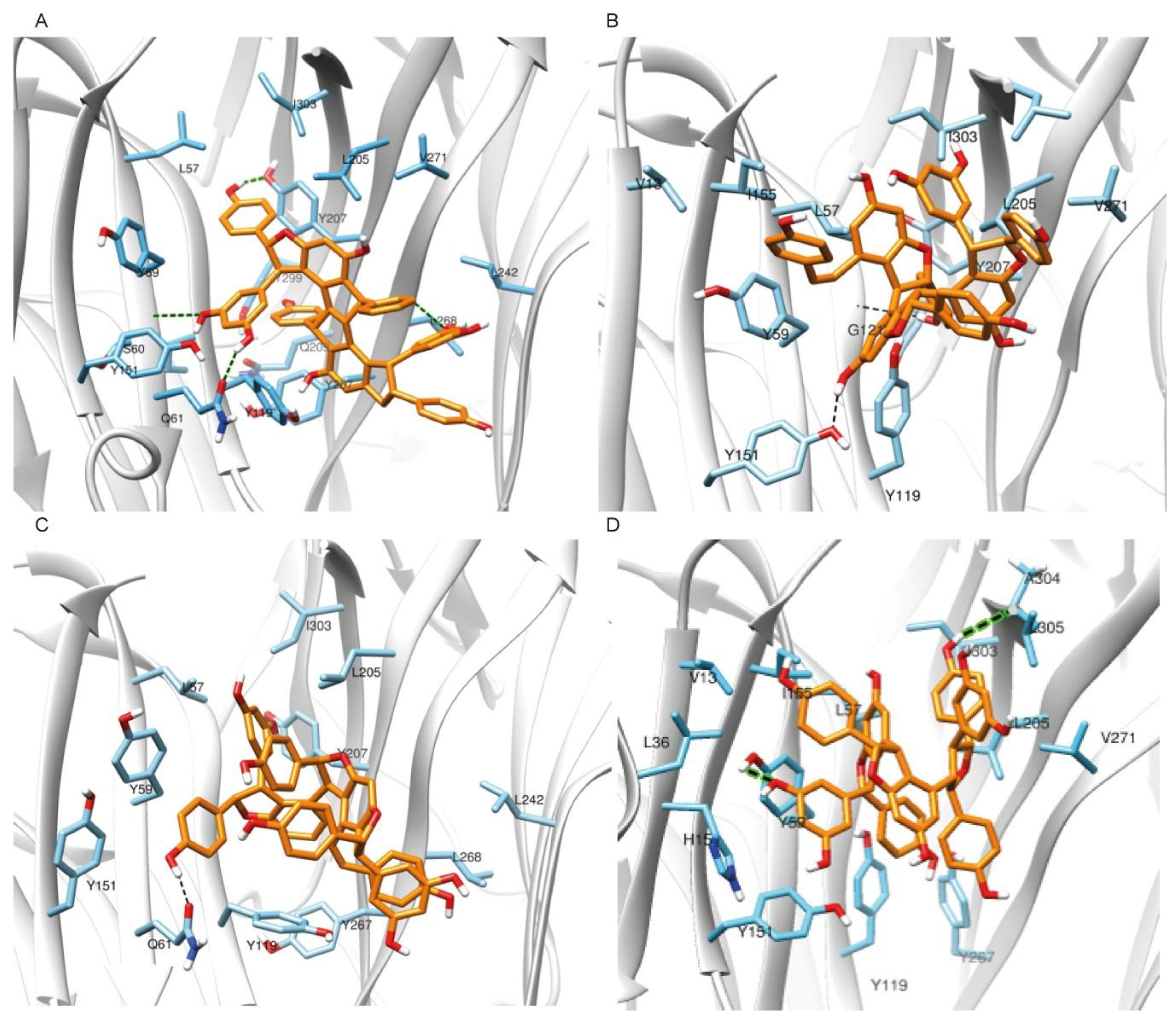
2.1.2. Molecular Docking Simulations Using Enalos Asclepios KNIME rxDock Node
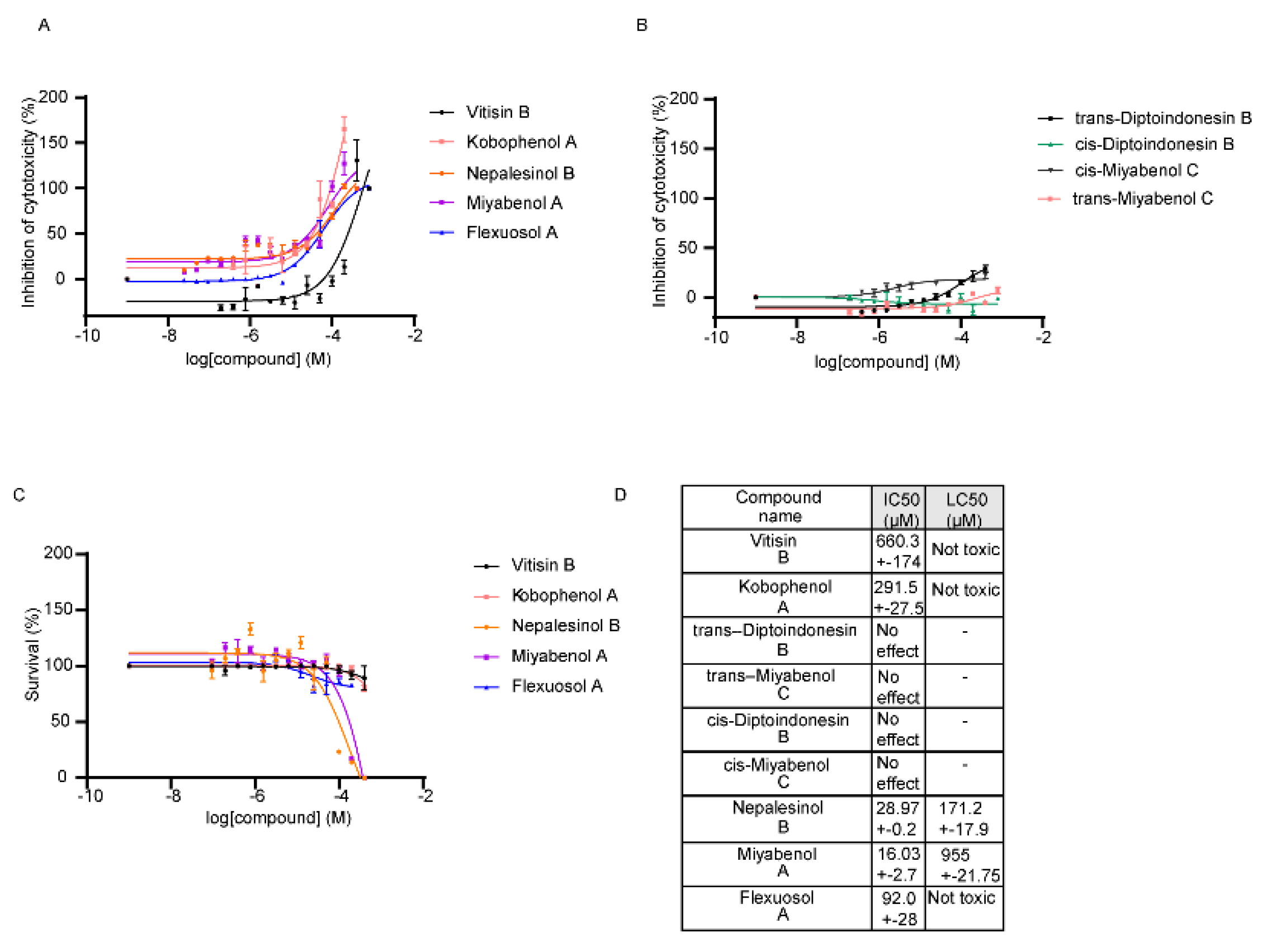
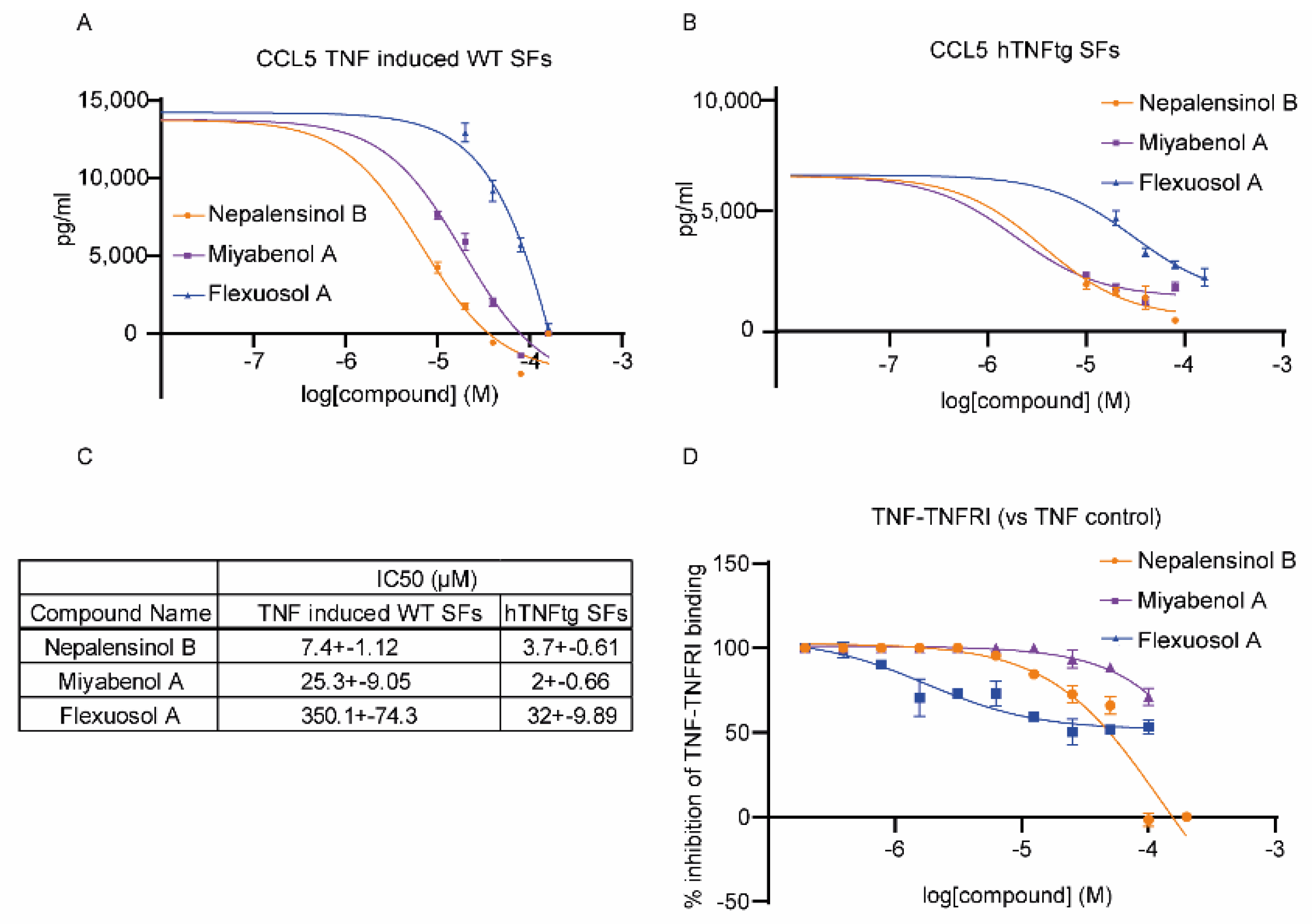
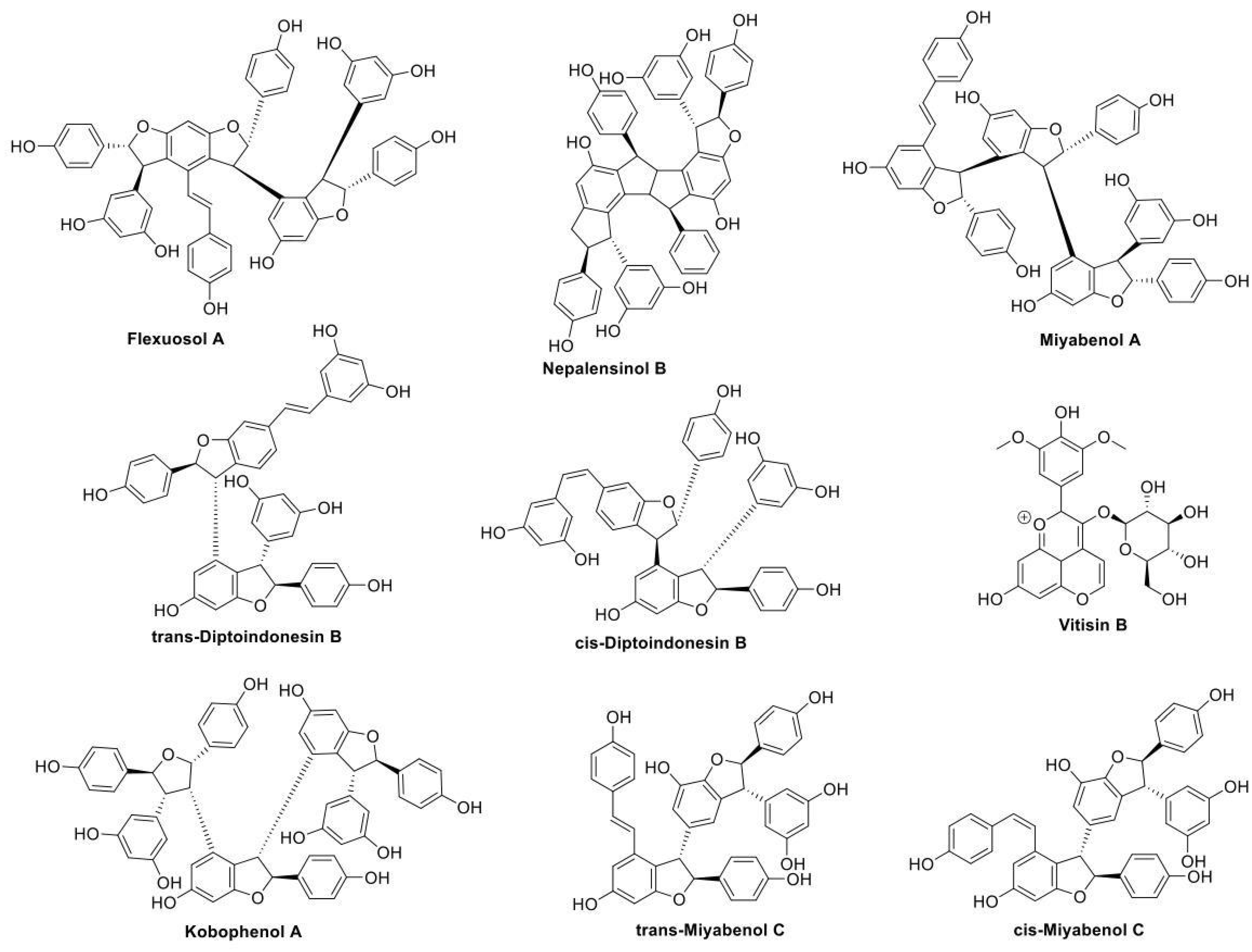
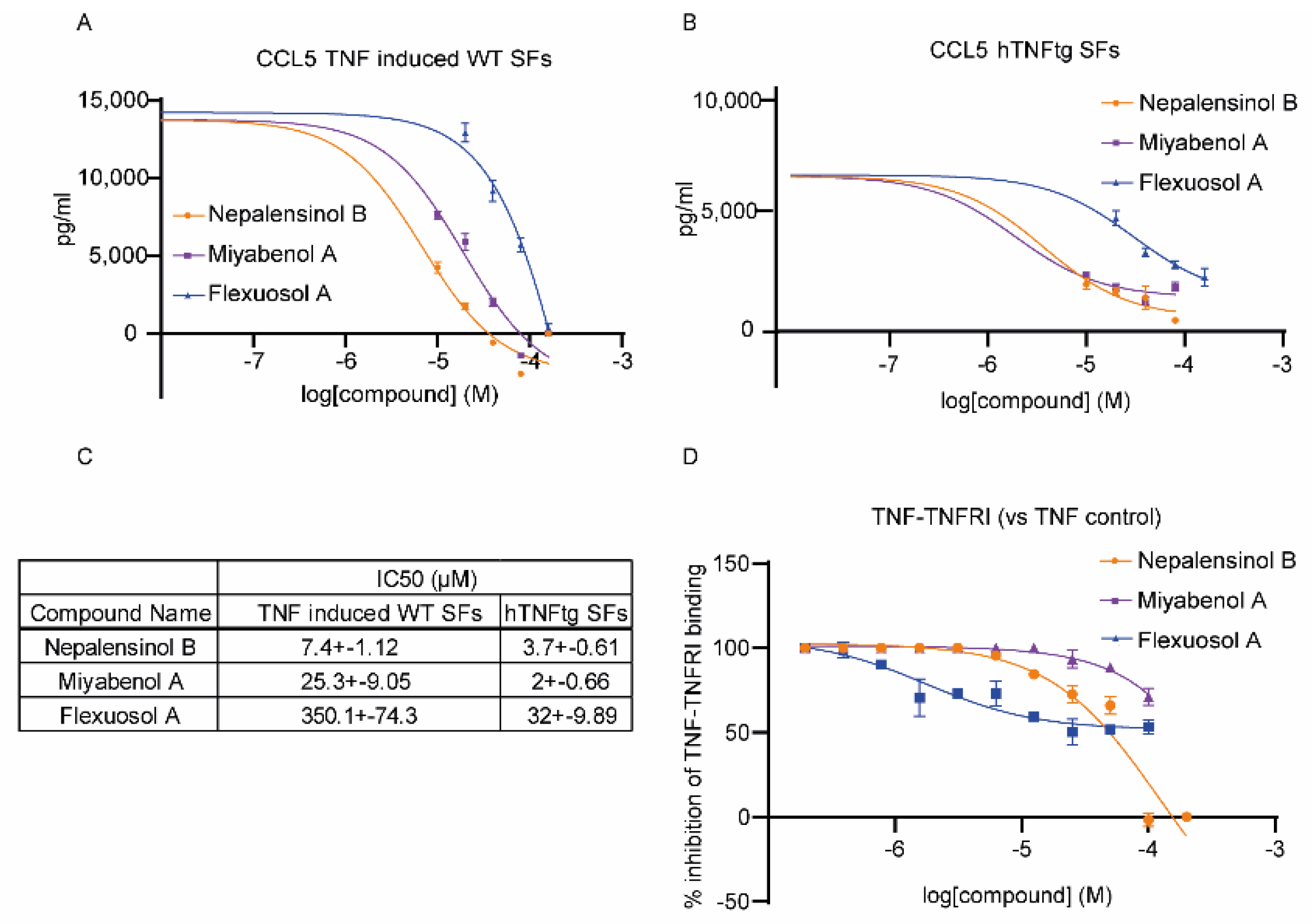
2.2. Pharmacological Testing
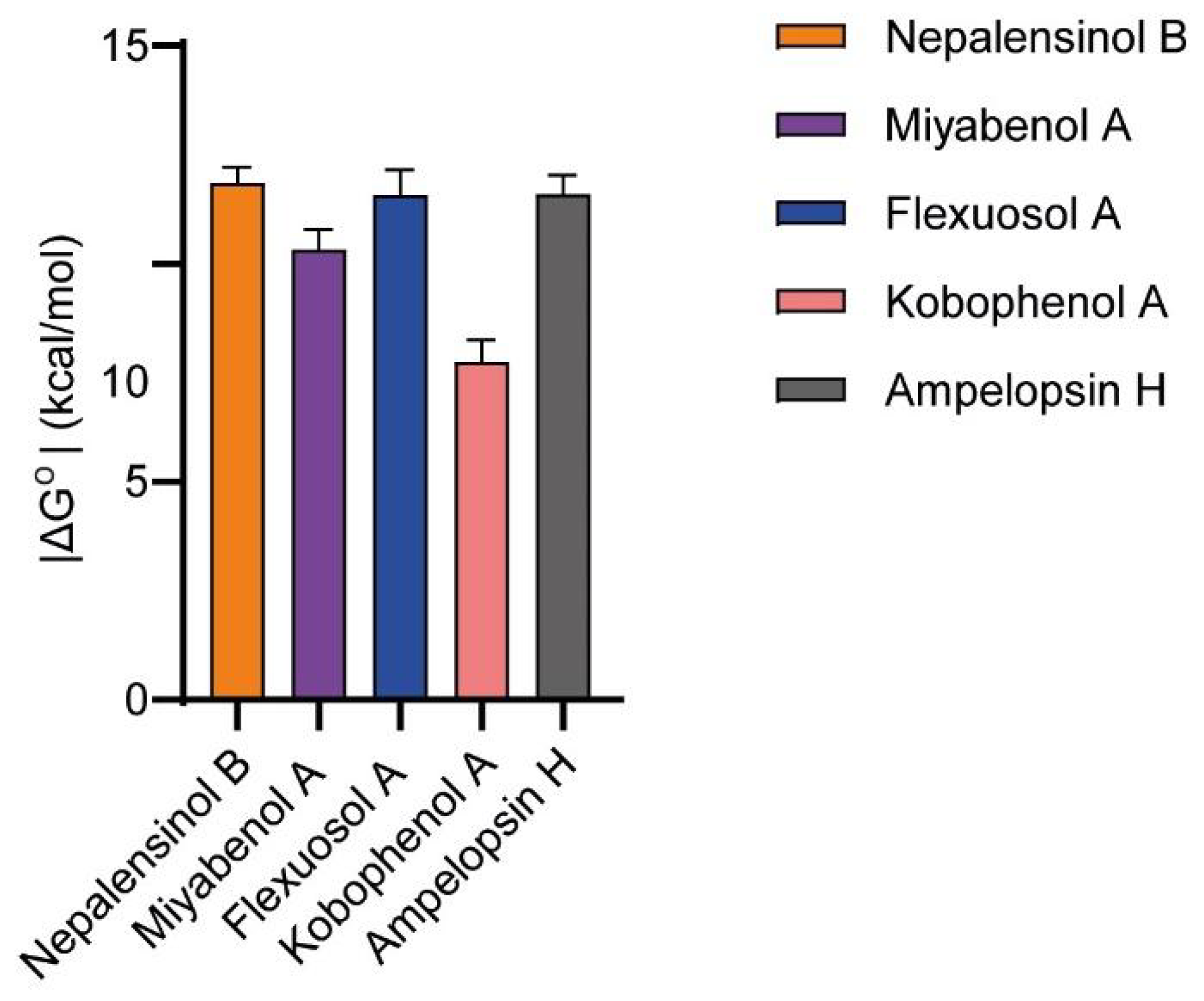
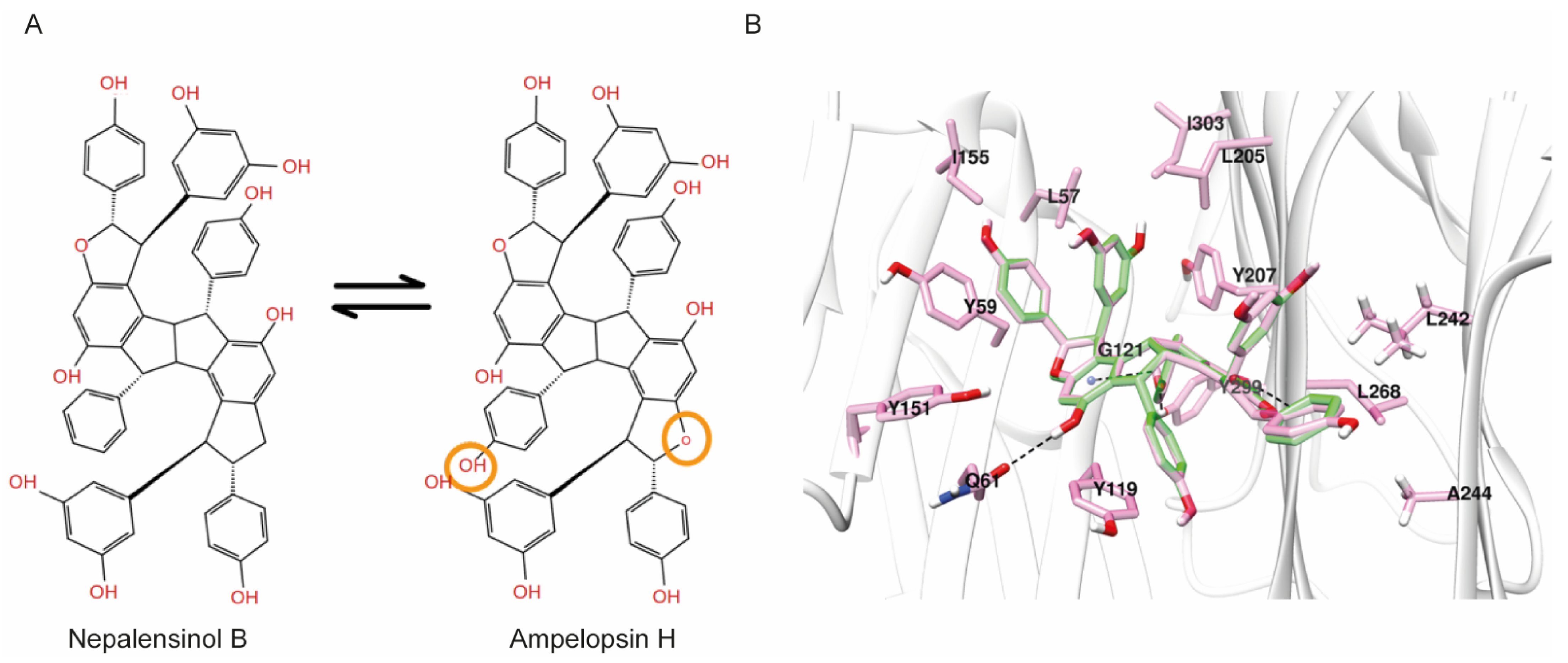
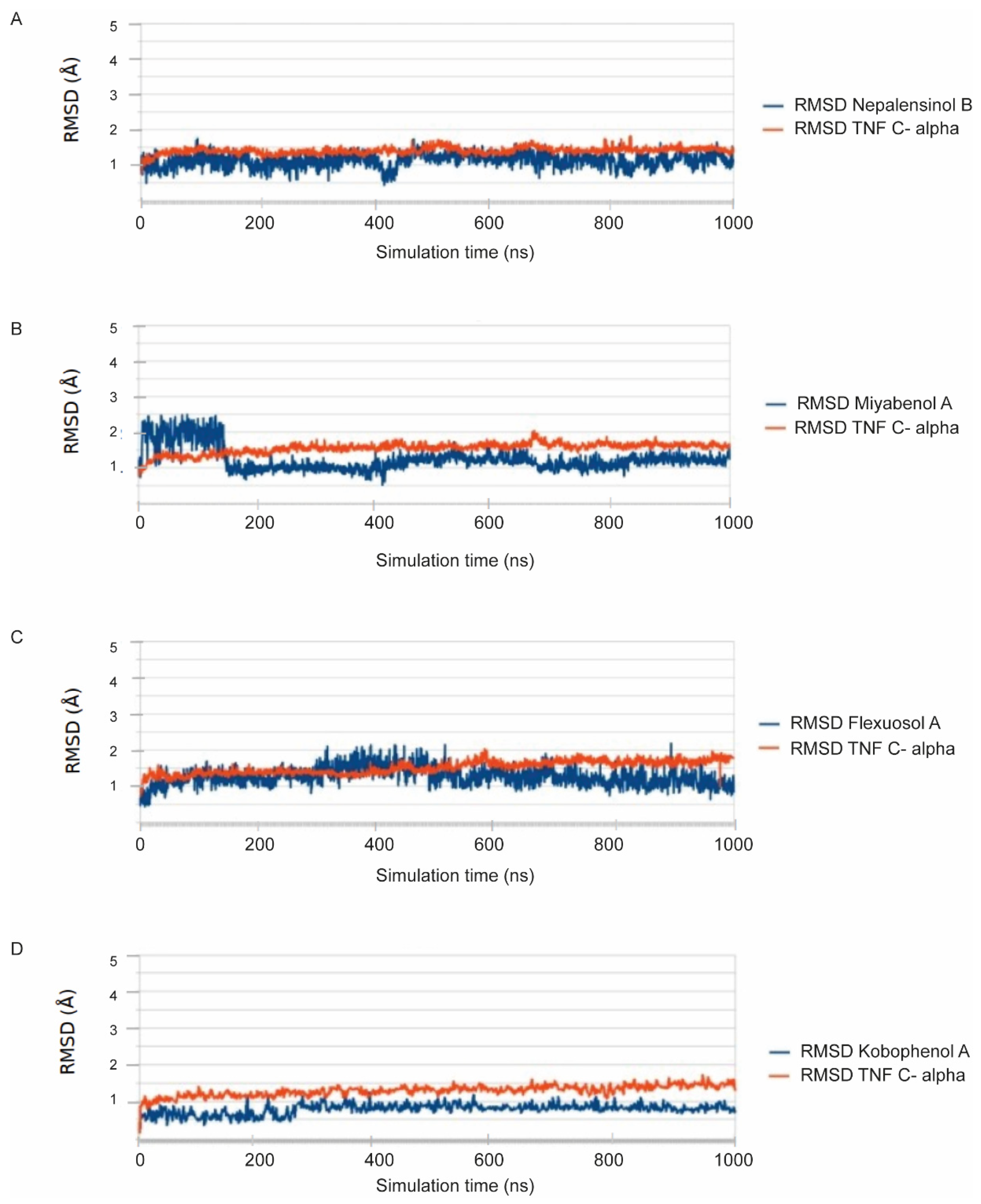
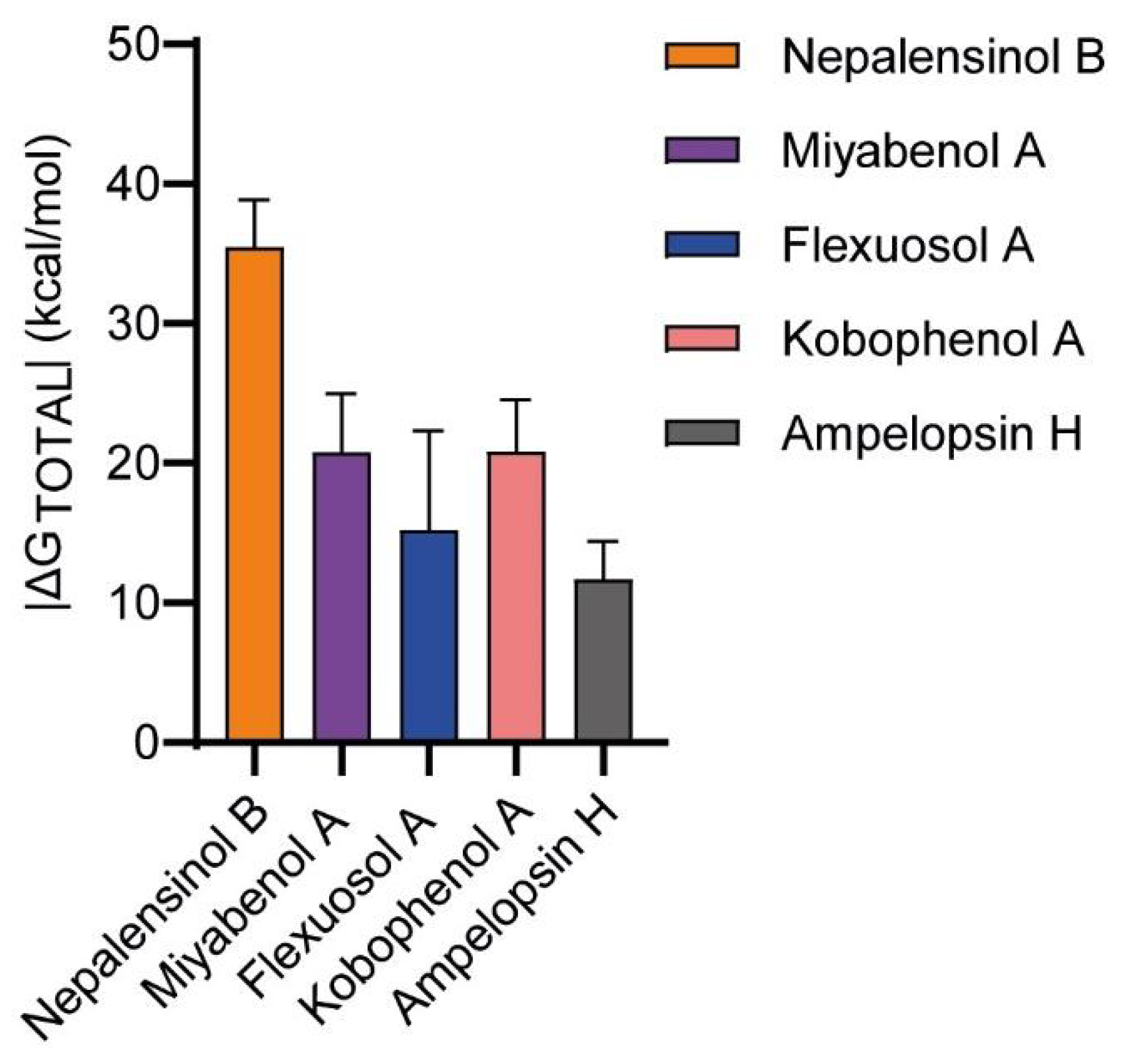
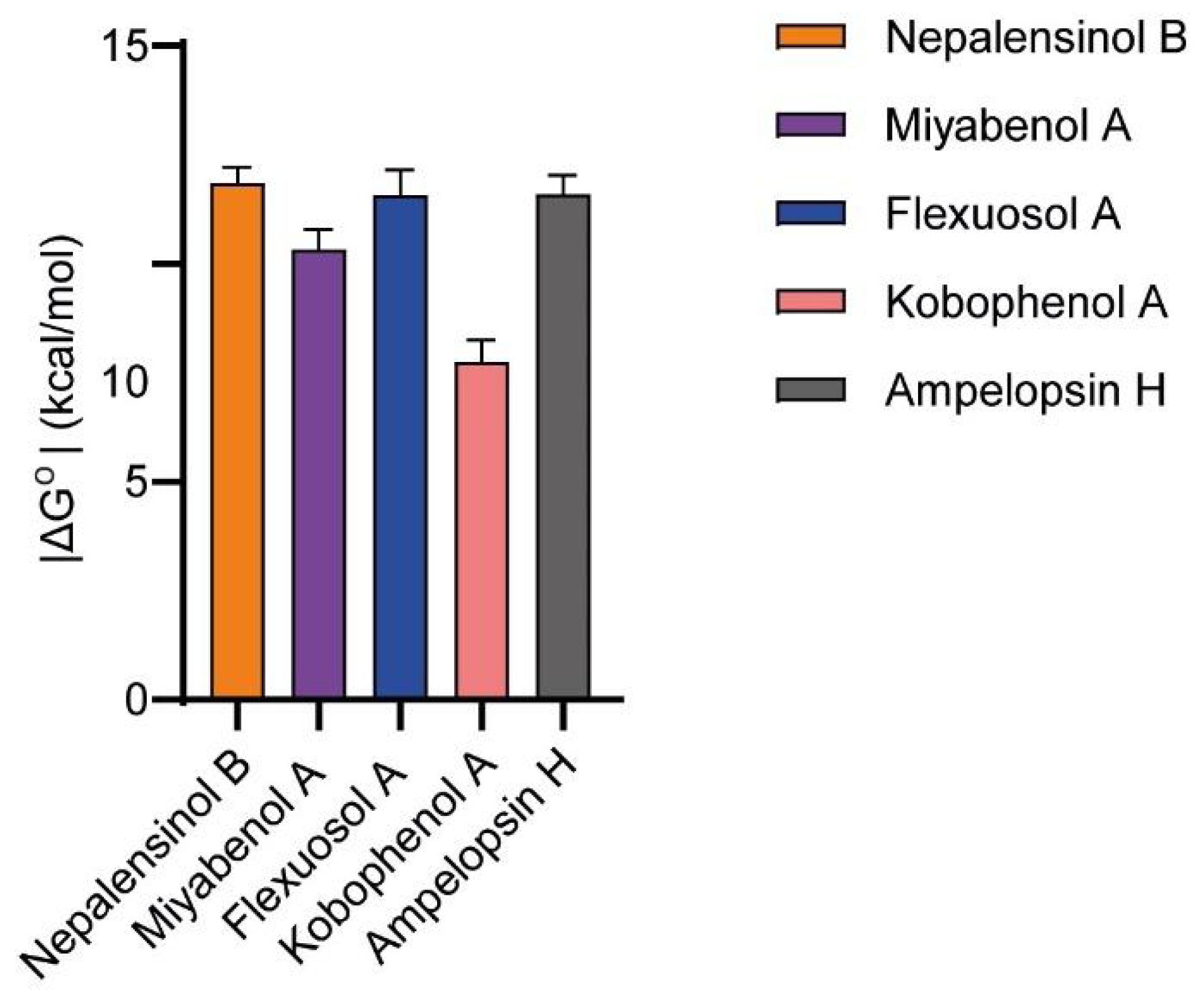
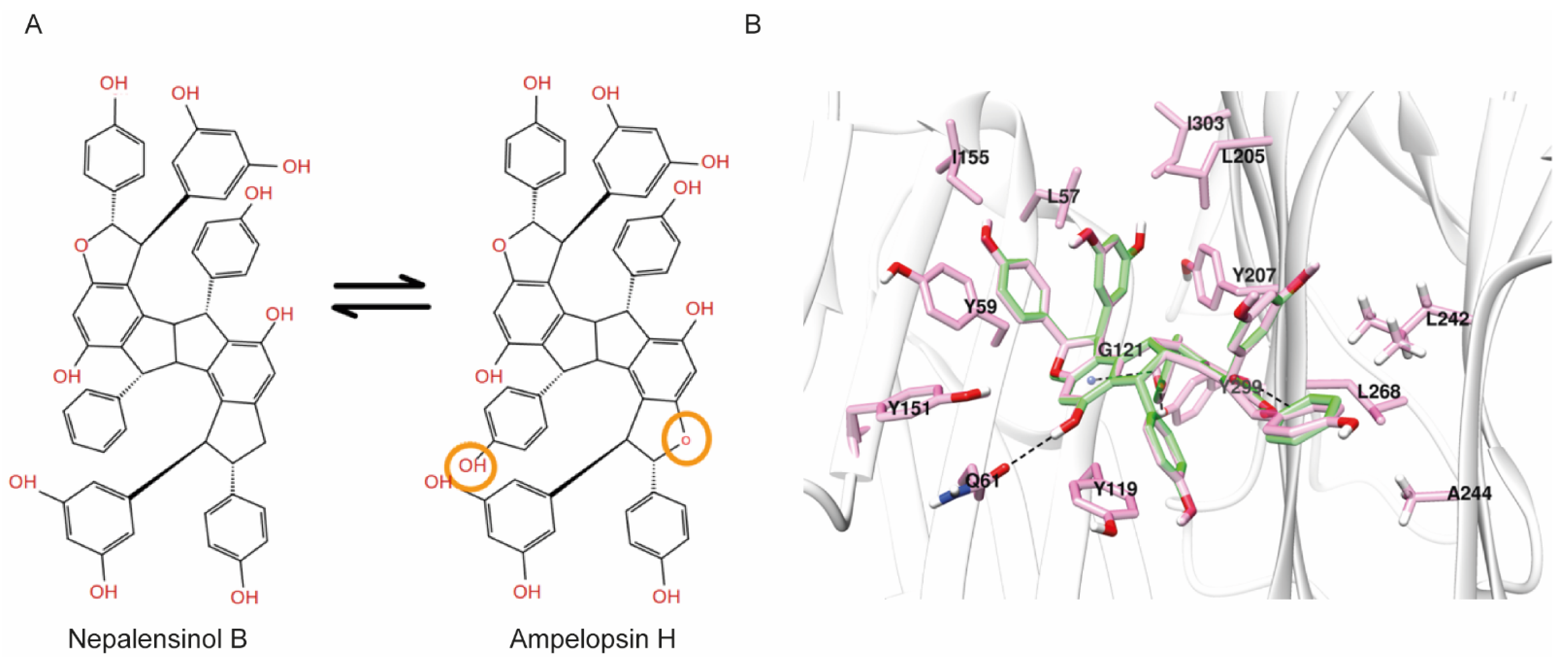
2.3. Molecular Dynamics and Free Energy Calculations
Molecular Dynamics Simulations with Enalos Asclepios KNIME MD-Simulation Node, Free Energy Calculations with MM-GBSA Method
3. Discussion
4. Material and Methods
4.1. Cheminformatics Modeling—Computer Aided Drug Design (CADD)
4.1.1. Enalos+ Similarity KNIME Node
4.1.2. Molecular Modeling
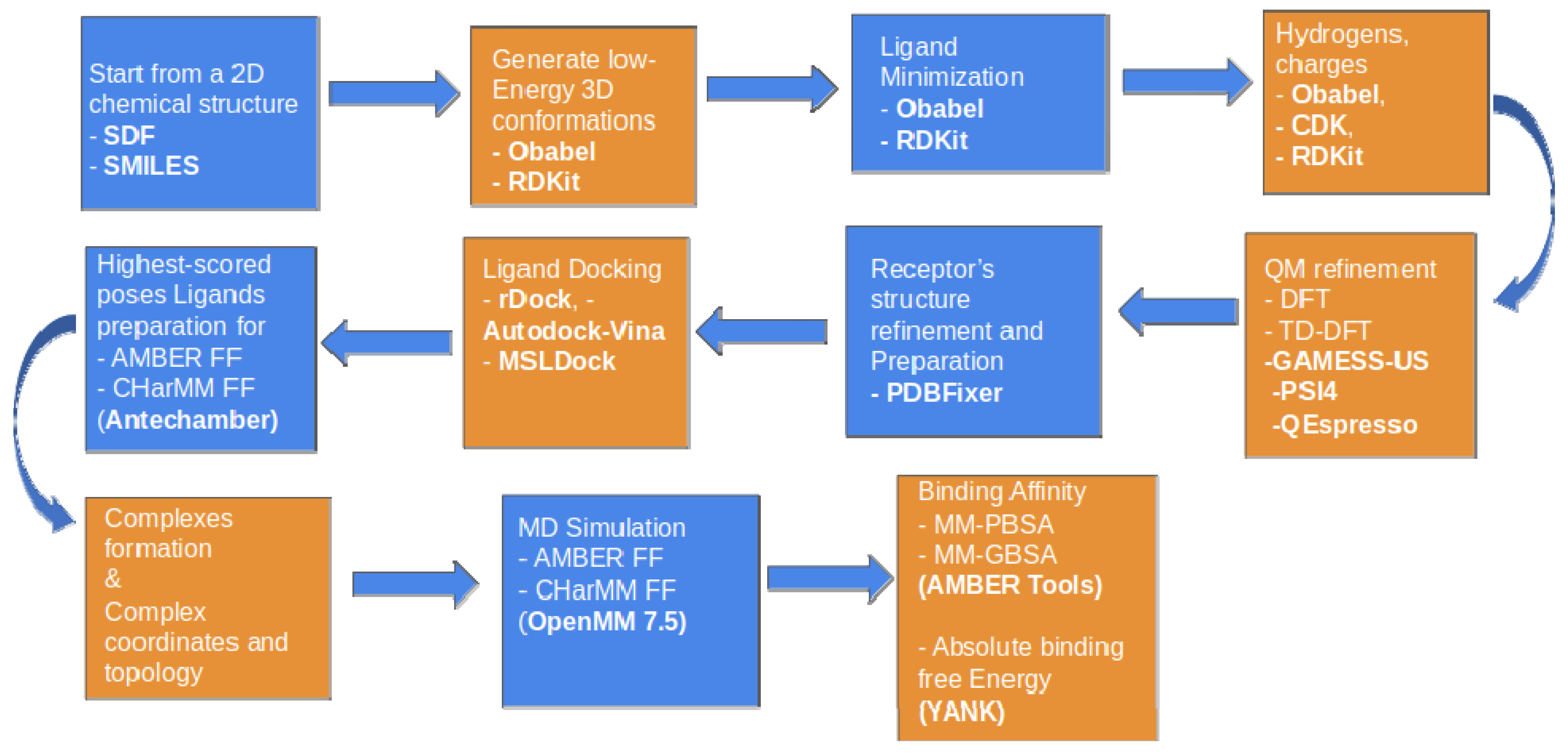
4.1.3. Enalos Asclepios KNIME Workflow
4.1.4. Molecular Docking with Enalos Asclepios KNIME Workflow
4.1.5. Molecular Dynamics
4.1.6. MM-GBSA Method
4.1.7. Absolute Binding Free Energies Calculation
4.2. Pharmacological Testing
4.2.1. Cell Lines
4.2.2. Mice
4.2.3. Chemokine Level Assay
4.2.4. Natural Products
4.2.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayden, M.S.; Ghosh, S. Regulation of NF-ΚB by TNF Family Cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolen, J.S.; Landewé, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; van Vollenhoven, R.F.; de Wit, M.; et al. EULAR Recommendations for the Management of Rheumatoid Arthritis with Synthetic and Biological Disease-Modifying Antirheumatic Drugs: 2019 Update. Ann. Rheum. Dis. 2020, 79, 685–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roskoski, R. Classification of Small Molecule Protein Kinase Inhibitors Based upon the Structures of Their Drug-Enzyme Complexes. Pharmacol. Res. 2016, 103, 26–48. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.; Milsark, I.W.; Cerami, A.C. Passive Immunization against Cachectin/Tumor Necrosis Factor Protects Mice from Lethal Effect of Endotoxin. Science 1985, 229, 869–871. [Google Scholar] [CrossRef] [PubMed]
- Eck, M.J.; Sprang, S.R. The Structure of Tumor Necrosis Factor-α at 2.6 Å Resolution. J. Biol. Chem. 1989, 264, 17595–17605. [Google Scholar] [CrossRef]
- He, M.M.; Smith, A.S.; Oslob, J.D.; Flanagan, W.M.; Braisted, A.C.; Whitty, A.; Cancilla, M.T.; Wang, J.; Lugovskoy, A.A.; Yoburn, J.C.; et al. Small-Molecule Inhibition of TNF-α. Science 2005, 310, 1022–1025. [Google Scholar] [CrossRef]
- Willrich, M.A.V.; Murray, D.L.; Snyder, M.R. Tumor Necrosis Factor Inhibitors: Clinical Utility in Autoimmune Diseases. Transl. Res. 2015, 165, 270–282. [Google Scholar] [CrossRef]
- Sfikakis, P.P. The First Decade of Biologic TNF Antagonists in Clinical Practice: Lessons Learned, Unresolved Issues and Future Directions. TNF Pathophysiol. 2010, 11, 180–210. [Google Scholar] [CrossRef]
- Melagraki, G.; Ntougkos, E.; Papadopoulou, D.; Rinotas, V.; Leonis, G.; Douni, E.; Afantitis, A.; Kollias, G. In Silico Discovery of Plant-Origin Natural Product Inhibitors of Tumor Necrosis Factor (TNF) and Receptor Activator of NF-ΚB Ligand (RANKL). Front. Pharmacol. 2018, 9, 800. [Google Scholar] [CrossRef]
- Davis, J.M.; Colangelo, J. Small-Molecule Inhibitors of the Interaction between TNF and TNFR. Future Med. Chem. 2012, 5, 69–79. [Google Scholar] [CrossRef]
- Richmond, V.; Michelini, F.M.; Bueno, C.A.; Alché, L.E.; Ramírez, J.A. Small Molecules as Anti-TNF Drugs. Curr. Med. Chem. 2015, 22, 2920–2942. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, J.D.; Longenecker, K.L.; Wilson, N.S.; Goess, C.; Panchal, S.C.; Swann, S.L.; Petros, A.M.; Hobson, A.D.; Ihle, D.; Song, D.; et al. Development of Orally Efficacious Allosteric Inhibitors of TNFα via Fragment-Based Drug Design. J. Med. Chem. 2021, 64, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Steeland, S.; Libert, C.; Vandenbroucke, R.E. A New Venue of TNF Targeting. IJMS 2018, 19, 1442. [Google Scholar] [CrossRef] [Green Version]
- Goel, N.; Stephens, S. Certolizumab Pegol. MAbs 2010, 2, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazumdar, S.; Greenwald, D. Golimumab. MAbs 2009, 1, 422–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, N.J.; Stein, C.M. New Drugs for Rheumatoid Arthritis. N. Engl. J. Med. 2004, 350, 2167–2179. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Shah, N.; Deshpande, G.; Tang, D.H.; Eisenberg, D.F. Comparing Biologic Cost Per Treated Patient Across Indications Among Adult US Managed Care Patients: A Retrospective Cohort Study. Drugs Real World Outcomes 2016, 3, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Steenholdt, C.; Svenson, M.; Bendtzen, K.; Thomsen, O.Ø.; Brynskov, J.; Ainsworth, M.A. Acute and Delayed Hypersensitivity Reactions to Infliximab and Adalimumab in a Patient with Crohn’s Disease. J. Crohns Colitis 2012, 6, 108–111. [Google Scholar] [CrossRef] [Green Version]
- Ben-Horin, S.; Kopylov, U.; Chowers, Y. Optimizing Anti-TNF Treatments in Inflammatory Bowel Disease. Autoimmun. Rev. 2014, 13, 24–30. [Google Scholar] [CrossRef]
- Murdaca, G.; Spanò, F.; Contatore, M.; Guastalla, A.; Penza, E.; Magnani, O.; Puppo, F. Immunogenicity of Infliximab and Adalimumab: What Is Its Role in Hypersensitivity and Modulation of Therapeutic Efficacy and Safety? Expert Opin. Drug Saf. 2016, 15, 43–52. [Google Scholar] [CrossRef]
- Melagraki, G.; Leonis, G.; Ntougkos, E.; Rinotas, V.; Papaneophytou, C.; Mavromoustakos, T.; Kontopidis, G.; Douni, E.; Kollias, G.; Afantitis, A. Current Status and Future Prospects of Small–Molecule Protein–Protein Interaction (PPI) Inhibitors of Tumor Necrosis Factor (TNF) and Receptor Activator of NF-ΚB Ligand (RANKL). Curr. Top. Med. Chem. 2018, 18, 661–673. [Google Scholar] [CrossRef]
- Supply Chain Management in the Drug Industry: Delivering Patient Value for Pharmaceuticals and Biologics|Wiley. Available online: https://www.wiley.com/en-bm/Supply+Chain+Management+in+the+Drug+Industry%3A+Delivering+Patient+Value+for+Pharmaceuticals+and+Biologics-p-9780470555170 (accessed on 27 May 2021).
- Hawking, F. Suramin: With Special Reference to Onchocerciasis. In Advances in Pharmacology; Garattini, S., Goldin, A., Hawking, F., Kopin, I.J., Schnitzer, I.J., Eds.; Academic Press: Cambridge, MA, USA, 1978; Volume 15, pp. 289–322. [Google Scholar] [CrossRef]
- Grazioli, L.; Alzani, R.; Ciomei, M.; Mariani, M.; Restivo, A.; Cozzi, E.; Marcucci, F. Inhibitory Effect of Suramin on Receptor Binding and Cytotoxic Activity of Tumor Necrosis Factor α. Int. J. Immunopharmacol. 1992, 14, 637–642. [Google Scholar] [CrossRef]
- Mancini, F.; Toro, C.M.; Mabilia, M.; Giannangeli, M.; Pinza, M.; Milanese, C. Inhibition of Tumor Necrosis Factor-α (TNF-α)/ TNF-α Receptor Binding by Structural Analogues of Suramin§§Abbreviations: TNF-α, Tumor Necrosis Factor-α; and MC/EM, MonteCarlo/Energy Minimization. Biochem. Pharmacol. 1999, 58, 851–859. [Google Scholar] [CrossRef]
- Shah, B.A.; Chib, R.; Gupta, P.; Sethi, V.K.; Koul, S.; Andotra, S.S.; Nargotra, A.; Sharma, S.; Pandey, A.; Bani, S.; et al. Saponins as Novel TNF-α Inhibitors: Isolation of Saponins and a nor-Pseudoguaianolide from Parthenium Hysterophorus. Org. Biomol. Chem. 2009, 7, 3230. [Google Scholar] [CrossRef]
- O’Connell, J.; Porter, J.; Kroeplien, B.; Norman, T.; Rapecki, S.; Davis, R.; McMillan, D.; Arakaki, T.; Burgin, A.; Fox, D., III; et al. Small Molecules That Inhibit TNF Signalling by Stabilising an Asymmetric Form of the Trimer. Nat. Commun. 2019, 10, 5795. [Google Scholar] [CrossRef] [PubMed]
- Mouhsine, H.; Guillemain, H.; Moreau, G.; Fourati, N.; Zerrouki, C.; Baron, B.; Desallais, L.; Gizzi, P.; Ben Nasr, N.; Perrier, J.; et al. Identification of an in Vivo Orally Active Dual-Binding Protein-Protein Interaction Inhibitor Targeting TNFα through Combined in Silico/in Vitro/in Vivo Screening. Sci. Rep. 2017, 7, 3424. [Google Scholar] [CrossRef]
- Blevitt, J.M.; Hack, M.D.; Herman, K.L.; Jackson, P.F.; Krawczuk, P.J.; Lebsack, A.D.; Liu, A.X.; Mirzadegan, T.; Nelen, M.I.; Patrick, A.N.; et al. Structural Basis of Small-Molecule Aggregate Induced Inhibition of a Protein–Protein Interaction. J. Med. Chem. 2017, 60, 3511–3517. [Google Scholar] [CrossRef]
- Xiao, H.-Y.; Li, N.; Duan, J.J.-W.; Jiang, B.; Lu, Z.; Ngu, K.; Tino, J.; Kopcho, L.M.; Lu, H.; Chen, J.; et al. Biologic-like In Vivo Efficacy with Small Molecule Inhibitors of TNFα Identified Using Scaffold Hopping and Structure-Based Drug Design Approaches. J. Med. Chem. 2020, 63, 15050–15071. [Google Scholar] [CrossRef]
- Rodrigues, T.; Reker, D.; Schneider, P.; Schneider, G. Counting on Natural Products for Drug Design. Nat. Chem. 2016, 8, 531–541. [Google Scholar] [CrossRef]
- Mohammad, H.B.; Khurshid, A.; Sudeep, R.; Jalaluddin, M.A.; Mohd, A.; Mohammad, H.S.; Saif, K.; Mohammad, A.K.; Ivo, P.; Inho, C. Computer Aided Drug Design: Success and Limitations. Curr. Pharm. Des. 2016, 22, 572–581. [Google Scholar]
- Kollias, G.; Papadaki, P.; Apparailly, F.; Vervoordeldonk, M.J.; Holmdahl, R.; Baumans, V.; Desaintes, C.; Di Santo, J.; Distler, J.; Garside, P.; et al. Animal Models for Arthritis: Innovative Tools for Prevention and Treatment. Ann. Rheum. Dis. 2011, 70, 1357–1362. [Google Scholar] [CrossRef]
- Choi, H.; Lee, Y.; Park, H.; Oh, D.-S. Discovery of the Inhibitors of Tumor Necrosis Factor Alpha with Structure-Based Virtual Screening. Bioorganic Med. Chem. Lett. 2010, 20, 6195–6198. [Google Scholar] [CrossRef] [PubMed]
- Saddala, M.S.; Huang, H. Identification of Novel Inhibitors for TNFα, TNFR1 and TNFα-TNFR1 Complex Using Pharmacophore-Based Approaches. J. Transl. Med. 2019, 17, 215. [Google Scholar] [CrossRef] [PubMed]
- Melagraki, G.; Ntougkos, E.; Rinotas, V.; Papaneophytou, C.; Leonis, G.; Mavromoustakos, T.; Kontopidis, G.; Douni, E.; Afantitis, A.; Kollias, G. Cheminformatics-Aided Discovery of Small-Molecule Protein-Protein Interaction (PPI) Dual Inhibitors of Tumor Necrosis Factor (TNF) and Receptor Activator of NF-ΚB Ligand (RANKL). PLoS Comput. Biol. 2017, 13, e1005372. [Google Scholar] [CrossRef] [Green Version]
- Afantitis, A.; Tsoumanis, A.; Melagraki, G. Enalos Suite of Tools: Enhancing Cheminformatics and Nanoinformatics through KNIME. Curr. Med. Chem. 2020, 27, 6523–6535. [Google Scholar] [CrossRef]
- Wells, J.A.; McClendon, C.L. Reaching for High-Hanging Fruit in Drug Discovery at Protein–Protein Interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef]
- Clackson, T.; Wells, J.A. A Hot Spot of Binding Energy in a Hormone-Receptor Interface. Science 1995, 267, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Berg, T. Modulation of Protein–Protein Interactions with Small Organic Molecules. Angew. Chem. Int. Ed. 2003, 42, 2462–2481. [Google Scholar] [CrossRef] [PubMed]
- Buchwald, P. Small-Molecule Protein–Protein Interaction Inhibitors: Therapeutic Potential in Light of Molecular Size, Chemical Space, and Ligand Binding Efficiency Considerations. IUBMB Life 2010, 62, 724–731. [Google Scholar] [CrossRef]
- Jones, S.; Thornton, J.M. Principles of Protein-Protein Interactions. Proc. Natl. Acad. Sci. USA 1996, 93, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, C.; Dong, G.; Miao, Z.; Zhang, W.; Wang, W. State-of-the-Art Strategies for Targeting Protein–Protein Interactions by Small-Molecule Inhibitors. Chem. Soc. Rev. 2015, 44, 8238–8259. [Google Scholar] [CrossRef]
- Magkrioti, C.; Kaffe, E.; Stylianaki, E.-A.; Sidahmet, C.; Melagraki, G.; Afantitis, A.; Matralis, A.N.; Aidinis, V. Structure-Based Discovery of Novel Chemical Classes of Autotaxin Inhibitors. Int. J. Mol. Sci. 2020, 21, 7002. [Google Scholar] [CrossRef]
- Mouchlis, V.D.; Afantitis, A.; Serra, A.; Fratello, M.; Papadiamantis, A.G.; Aidinis, V.; Lynch, I.; Greco, D.; Melagraki, G. Advances in De Novo Drug Design: From Conventional to Machine Learning Methods. Int. J. Mol. Sci. 2021, 22, 1676. [Google Scholar] [CrossRef] [PubMed]
- Mouchlis, V.D.; Melagraki, G.; Zacharia, L.C.; Afantitis, A. Computer-Aided Drug Design of β-Secretase, γ-Secretase and Anti-Tau Inhibitors for the Discovery of Novel Alzheimer’s Therapeutics. Int. J. Mol. Sci. 2020, 21, 703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varsou, D.-D.; Nikolakopoulos, S.; Tsoumanis, A.; Melagraki, G.; Afantitis, A. Enalos Suite: New Cheminformatics Platform for Drug Discovery and Computational Toxicology. Methods Mol. Biol. 2018, 1800, 287–311. [Google Scholar] [CrossRef]
- Varsou, D.-D.; Nikolakopoulos, S.; Tsoumanis, A.; Melagraki, G.; Afantitis, A. Enalos+ KNIME Nodes: New Cheminformatics Tools for Drug Discovery. Methods Mol. Biol. 2018, 1824, 113–138. [Google Scholar] [CrossRef]
- Lagarias, P.; Papadiamantis, A.G.; Tsoumanis, A.; Melagraki, G.; Afantitis, A. Enalos+ KNIME Nodes: User-Friendly Cheminformatics and Nanoinformatics Tools for Drug Discovery and in Silico Workflows. In Abstract Book—Proceedings of the 18th Hellenic Symposium on Medicinal Chemistry, Athens, Greece, 25–27 February 2021; Hellenic Society of Medicinal Chemistry: Athens, Greece, 2021; p. 328. [Google Scholar]
- Johnson, M.A.; Maggiora, G.M. (Eds.) Concepts and Applications of Molecular Similarity; Wiley: Hoboken, NJ, USA, 1990. [Google Scholar]
- Willett, P.; Barnard, J.M.; Downs, G.M. Chemical Similarity Searching. J. Chem. Inf. Comput. Sci. 1998, 38, 983–996. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. RDock: A Fast, Versatile and Open Source Program for Docking Ligands to Proteins and Nucleic Acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [Green Version]
- Armaka, M.; Gkretsi, V.; Kontoyiannis, D.; Kollias, G. A Standardized Protocol for the Isolation and Culture of Normal and Arthritogenic Murine Synovial Fibroblasts. Protoc. Exch. 2009. [Google Scholar] [CrossRef]
- Keffer, J.; Probert, L.; Cazlaris, H.; Georgopoulos, S.; Kaslaris, E.; Kioussis, D.; Kollias, G. Transgenic Mice Expressing Human Tumour Necrosis Factor: A Predictive Genetic Model of Arthritis. EMBO J. 1991, 10, 4025–4031. [Google Scholar] [CrossRef]
- Karagianni, N.; Kranidioti, K.; Fikas, N.; Tsochatzidou, M.; Chouvardas, P.; Denis, M.C.; Kollias, G.; Nikolaou, C. An Integrative Transcriptome Analysis Framework for Drug Efficacy and Similarity Reveals Drug-Specific Signatures of Anti-TNF Treatment in a Mouse Model of Inflammatory Polyarthritis. PLoS Comput. Biol. 2019, 15, e1006933. [Google Scholar] [CrossRef] [Green Version]
- Armaka, M.; Apostolaki, M.; Jacques, P.; Kontoyiannis, D.L.; Elewaut, D.; Kollias, G. Mesenchymal Cell Targeting by TNF as a Common Pathogenic Principle in Chronic Inflammatory Joint and Intestinal Diseases. J. Exp. Med. 2008, 205, 331–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J.; Zhou, Q.; Li, P.; Wang, Z.; Liu, S.; He, C.; Zhang, C.; Xiao, P. Update on Phytochemistry and Pharmacology of Naturally Occurring Resveratrol Oligomers. Molecules 2017, 22, 2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dávid, C.Z.; Hohmann, J.; Vasas, A. Chemistry and Pharmacology of Cyperaceae Stilbenoids: A Review. Molecules 2021, 26, 2794. [Google Scholar] [CrossRef] [PubMed]
- Niesen, D.B.; Hessler, C.; Seeram, N.P. Beyond Resveratrol: A Review of Natural Stilbenoids Identified from 2009–2013. J. Berry Res. 2013, 3, 181–196. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Li, B.; Chen, Y. Flexuosol A, a New Tetrastilbene from Vitis Flexuosa. J. Nat. Prod. 1998, 61, 646–647. [Google Scholar] [CrossRef]
- Yamada, M.; Hayashi, K.; Hayashi, H.; Ikeda, S.; Hoshino, T.; Tsutsui, K.; Tsutsui, K.; Iinuma, M.; Nozaki, H. Stilbenoids of Kobresia Nepalensis (Cyperaceae) Exhibiting DNA Topoisomerase II Inhibition. Phytochemistry 2006, 67, 307–313. [Google Scholar] [CrossRef]
- Silva, A.A.; Haraguchi, S.K.; Cellet, T.S.P.; Schuquel, I.T.A.; Sarragiotto, M.H.; Vidotti, G.J.; de Melo, J.O.; Bersani-Amado, C.A.; Zanoli, K.; Nakamura, C.V. Resveratrol-Derived Stilbenoids and Biological Activity Evaluation of Seed Extracts of Cenchrus echinatus L. Nat. Prod. Res. 2012, 26, 865–868. [Google Scholar] [CrossRef]
- Keylor, M.H.; Matsuura, B.S.; Griesser, M.; Chauvin, J.-P.R.; Harding, R.A.; Kirillova, M.S.; Zhu, X.; Fischer, O.J.; Pratt, D.A.; Stephenson, C.R.J. Synthesis of Resveratrol Tetramers via a Stereoconvergent Radical Equilibrium. Science 2016, 354, 1260–1265. [Google Scholar] [CrossRef] [Green Version]
- Mattivi, F.; Vrhovsek, U.; Malacarne, G.; Masuero, D.; Zulini, L.; Stefanini, M.; Moser, C.; Velasco, R.; Guella, G. Profiling of Resveratrol Oligomers, Important Stress Metabolites, Accumulating in the Leaves of Hybrid Vitis Vinifera (Merzling × Teroldego) Genotypes Infected with Plasmopara Viticola. J. Agric. Food Chem. 2011, 59, 5364–5375. [Google Scholar] [CrossRef]
- Pawlus, A.D.; Sahli, R.; Bisson, J.; Rivière, C.; Delaunay, J.-C.; Richard, T.; Gomès, E.; Bordenave, L.; Waffo-Téguo, P.; Mérillon, J.-M. Stilbenoid Profiles of Canes from Vitis and Muscadinia Species. J. Agric. Food Chem. 2013, 61, 501–511. [Google Scholar] [CrossRef]
- Meng, Y.; Bourne, P.C.; Whiting, P.; Šik, V.; Dinan, L. Identification and Ecdysteroid Antagonist Activity of Three Oligostilbenes from the Seeds of Carex Pendula (Cyperaceae). Phytochemistry 2001, 57, 393–400. [Google Scholar] [CrossRef]
- Kulanthaivel, P.; Janzen, W.P.; Ballas, L.M.; Jiang, J.B.; Hu, C.-Q.; Darges, J.W.; Seldin, J.C.; Cofield, D.J.; Adams, L.M. Naturally Occurring Protein Kinase C Inhibitors; II1. Isolation of Oligomeric Stilbenes from Caragana Sinica2. Planta Med. 1995, 61, 41–44. [Google Scholar] [CrossRef] [PubMed]
- Sáez, V.; Pastene, E.; Vergara, C.; Mardones, C.; Hermosín-Gutiérrez, I.; Gómez-Alonso, S.; Gómez, M.V.; Theoduloz, C.; Riquelme, S.; von Baer, D. Oligostilbenoids in Vitis vinifera L. Pinot Noir Grape Cane Extract: Isolation, Characterization, in Vitro Antioxidant Capacity and Anti-Proliferative Effect on Cancer Cells. Food Chem. 2018, 265, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Syah, Y.M.; Aminah, N.S.; Hakim, E.H.; Aimi, N.; Kitajima, M.; Takayama, H.; Achmad, S.A. Two Oligostilbenes, Cis- and Trans-Diptoindonesin B, from Dryobalanops Oblongifolia. Phytochemistry 2003, 63, 913–917. [Google Scholar] [CrossRef]
- Abdjan, M.I.; Aminah, N.S.; Siswanto, I.; Kristanti, A.N.; Takaya, Y.; Choudhary, M.I. Exploration of Stilbenoid Trimers as Potential Inhibitors of Sirtuin1 Enzyme Using a Molecular Docking and Molecular Dynamics Simulation Approach. RSC Adv. 2021, 11, 19323–19332. [Google Scholar] [CrossRef]
- Cho, H.; Park, J.-H.; Ahn, E.-K.; Oh, J.S. Kobophenol A Isolated from Roots of Caragana Sinica (Buc’hoz) Rehder Exhibits Anti-Inflammatory Activity by Regulating NF-ΚB Nuclear Translocation in J774A.1 Cells. Toxicol. Rep. 2018, 5, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Henry, G.E.; Seeram, N.P. Identification and Bioactivities of Resveratrol Oligomers and Flavonoids from Carex Folliculata Seeds. J. Agric. Food Chem. 2009, 57, 7282–7287. [Google Scholar] [CrossRef]
- Lee, S.R.; Kwak, J.H.; Noh, S.J.; Pronto, J.R.; Ko, K.S.; Rhee, B.D.; Xu, Z.; Kim, N.; Han, J. Kobophenol A Inhibits Sodium Nitroprusside-Induced Cardiac H9c2 Cell Death through Suppressing Activation of JNK and Preserving Mitochondrial Anti-Apoptotic Bcl-2 and Mcl-1. Chem. Pharm. Bull. 2014, 62, 713–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.-R.; Kwak, J.-H.; Park, D.-S.; Pyo, S. Protective Effect of Kobophenol A on Nitric Oxide-Induced Cell Apoptosis in Human Osteoblast-like MG-63 Cells: Involvement of JNK, NF-ΚB and AP-1 Pathways. Int. Immunopharmacol. 2011, 11, 1251–1259. [Google Scholar] [CrossRef]
- Kwak, J.-H.; Lee, S.-R.; Park, H.-J.; Byun, H.-E.; Sohn, E.-H.; Kim, B.-O.; Rhee, D.-K.; Pyo, S. Kobophenol A Enhances Proliferation of Human Osteoblast-like Cells with Activation of the P38 Pathway. Int. Immunopharmacol. 2013, 17, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, J.; Ichikawa, S.; Kurihara, H.; Mizutani, J. Kobophenol A, a Unique Tetrastilbene from Carex Kobomugi Ohwi (Cyperaceae). Tetrahedron Lett. 1989, 30, 3785–3788. [Google Scholar] [CrossRef]
- Gangadevi, S.; Badavath, V.N.; Thakur, A.; Yin, N.; De Jonghe, S.; Acevedo, O.; Jochmans, D.; Leyssen, P.; Wang, K.; Neyts, J.; et al. Kobophenol A Inhibits Binding of Host ACE2 Receptor with Spike RBD Domain of SARS-CoV-2, a Lead Compound for Blocking COVID-19. J. Phys. Chem. Lett. 2021, 12, 1793–1802. [Google Scholar] [CrossRef]
- Kawabata, J.; Mishima, M.; Kurihara, H.; Mizutani, J. Stereochemistry of Two Tetrastilbenes from Carex Species. Phytochemistry 1995, 40, 1507–1510. [Google Scholar] [CrossRef]
- Ku, K.-T.; Huang, Y.-L.; Huang, Y.-J.; Chiou, W.-F. Miyabenol A Inhibits LPS-Induced NO Production via IKK/IκB Inactivation in RAW 264.7 Macrophages: Possible Involvement of the P38 and PI3K Pathways. J. Agric. Food Chem. 2008, 56, 8911–8918. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-L.; Tsai, W.-J.; Shen, C.-C.; Chen, C.-C. Resveratrol Derivatives from the Roots of Vitis Thunbergii. J. Nat. Prod. 2005, 68, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Bakker, J.; Timberlake, C.F. Isolation, Identification, and Characterization of New Color-Stable Anthocyanins Occurring in Some Red Wines. J. Agric. Food Chem. 1997, 45, 35–43. [Google Scholar] [CrossRef]
- Fulcrand, H.; Benabdeljalil, C.; Rigaud, J.; Cheynier, V.; Moutounet, M. A New Class of Wine Pigments Generated by Reaction between Pyruvic Acid and Grape Anthocyanins. Phytochemistry 1998, 47, 1401–1407. [Google Scholar] [CrossRef]
- Oliveira, J.; de Freitas, V.; Mateus, N. A Novel Synthetic Pathway to Vitisin B Compounds. Tetrahedron Lett. 2009, 50, 3933–3935. [Google Scholar] [CrossRef]
- Azevedo, J.; Oliveira, J.; Cruz, L.; Teixeira, N.; Brás, N.F.; De Freitas, V.; Mateus, N. Antioxidant Features of Red Wine Pyranoanthocyanins: Experimental and Theoretical Approaches. J. Agric. Food Chem. 2014, 62, 7002–7009. [Google Scholar] [CrossRef]
- Hehner, S.P.; Hofmann, T.G.; Ratter, F.; Dumont, A.; Dröge, W.; Schmitz, M.L. Tumor Necrosis Factor-α-Induced Cell Killing and Activation of Transcription Factor NF-ΚB Are Uncoupled in L929 Cells *. J. Biol. Chem. 1998, 273, 18117–18121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanlangenakker, N.; Bertrand, M.J.M.; Bogaert, P.; Vandenabeele, P.; Vanden Berghe, T. TNF-Induced Necroptosis in L929 Cells Is Tightly Regulated by Multiple TNFR1 Complex I and II Members. Cell Death Dis. 2011, 2, e230. [Google Scholar] [CrossRef]
- Alexiou, P.; Papakyriakou, A.; Ntougkos, E.; Papaneophytou, C.P.; Liepouri, F.; Mettou, A.; Katsoulis, I.; Maranti, A.; Tsiliouka, K.; Strongilos, A.; et al. Rationally Designed Less Toxic SPD-304 Analogs and Preliminary Evaluation of Their TNF Inhibitory Effects. Arch. Pharm. 2014, 347, 798–805. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.S.; Jenney, A.P.; Swantek, J.L.; Burke, J.M.; Lauffenburger, D.A.; Sorger, P.K. Profiling Drugs for Rheumatoid Arthritis That Inhibit Synovial Fibroblast Activation. Nat. Chem. Biol. 2017, 13, 38–45. [Google Scholar] [CrossRef]
- Ntari, L.; Nikolaou, C.; Kranidioti, K.; Papadopoulou, D.; Christodoulou-Vafeiadou, E.; Chouvardas, P.; Meier, F.; Geka, C.; Denis, M.C.; Karagianni, N.; et al. Combination of Subtherapeutic Anti-TNF Dose with Dasatinib Restores Clinical and Molecular Arthritogenic Profiles Better than Standard Anti-TNF Treatment. J. Transl. Med. 2021, 19, 165. [Google Scholar] [CrossRef]
- Kollias, G.; Kontoyiannis, D. Role of TNF/TNFR in Autoimmunity: Specific TNF Receptor Blockade May Be Advantageous to Anti-TNF Treatments. Cytokine Growth Factor Rev. 2002, 13, 315–321. [Google Scholar] [CrossRef]
- Amber Advanced Tutorials-Tutorial 3-MM-PBSA—Introduction. Available online: http://ambermd.org/tutorials/advanced/tutorial3/ (accessed on 9 September 2021).
- Heinzelmann, G.; Gilson, M.K. Automation of Absolute Protein-Ligand Binding Free Energy Calculations for Docking Refinement and Compound Evaluation. Sci. Rep. 2021, 11, 1116. [Google Scholar] [CrossRef]
- Forouzesh, N.; Mishra, N. An Effective MM/GBSA Protocol for Absolute Binding Free Energy Calculations: A Case Study on SARS-CoV-2 Spike Protein and the Human ACE2 Receptor. Molecules 2021, 26, 2383. [Google Scholar] [CrossRef] [PubMed]
- Shirts, M.R.; Chodera, J.D. Statistically Optimal Analysis of Samples from Multiple Equilibrium States. J. Chem. Phys. 2008, 129, 124105. [Google Scholar] [CrossRef] [Green Version]
- Rizzi, A.; Grinaway, P.; Parton, D.; Shirts, M.R.; Wang, K.; Eastman, P.; Friedrichs, M.; Pande, V.S.; Branson, K.; Mobley, D.; et al. YANK: A GPU-Accelerated Platform for Alchemical Free Energy Calculations. In preparation.
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Eastman, P.; Friedrichs, M.S.; Chodera, J.D.; Radmer, R.J.; Bruns, C.M.; Ku, J.P.; Beauchamp, K.A.; Lane, T.J.; Wang, L.-P.; Shukla, D.; et al. OpenMM 4: A Reusable, Extensible, Hardware Independent Library for High Performance Molecular Simulation. J. Chem. Theory Comput. 2013, 9, 461–469. [Google Scholar] [CrossRef] [PubMed]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barca, G.M.J.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent Developments in the General Atomic and Molecular Electronic Structure System. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef] [Green Version]
- Alexeev, Y.; Mazanetz, P.M.; Ichihara, O.; Fedorov, G.D. GAMESS As a Free Quantum-Mechanical Platform for Drug Research. Curr. Top. Med. Chem. 2012, 12, 2013–2033. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.; Duke, R.E.; Ghoreishi, D.; Giambasu, G.; et al. AMBER; University of California: San Francisco, CA, USA, 2019. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Automatic Atom Type and Bond Type Perception in Molecular Mechanical Calculations. J. Mol. Graph. Model. 2006, 25, 247–260. [Google Scholar] [CrossRef]
- Eastman, P.; Swails, J.; Chodera, J.D.; McGibbon, R.T.; Zhao, Y.; Beauchamp, K.A.; Wang, L.-P.; Simmonett, A.C.; Harrigan, M.P.; Stern, C.D.; et al. OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS Comput. Biol. 2017, 13, e1005659. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin Stabilization of Molecular Dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Tian, C.; Kasavajhala, K.; Belfon, K.A.A.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. Ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Xu, L.; Sun, H.; Li, Y.; Wang, J.; Hou, T. Assessing the Performance of MM/PBSA and MM/GBSA Methods. 3. The Impact of Force Fields and Ligand Charge Models. J. Phys. Chem. B 2013, 117, 8408–8421. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.Py: An Efficient Program for End-State Free Energy Calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring Protein Native States and Large-Scale Conformational Changes with a Modified Generalized Born Model. Proteins Struct. Funct. Bioinform. 2004, 55, 383–394. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Buckner, J.K.; Boudon, S.; Tirado-Rives, J. Efficient Computation of Absolute Free Energies of Binding by Computer Simulations. Application to the Methane Dimer in Water. J. Chem. Phys. 1988, 89, 3742–3746. [Google Scholar] [CrossRef]
- Wang, K.; Chodera, J.D.; Yang, Y.; Shirts, M.R. Identifying Ligand Binding Sites and Poses Using GPU-Accelerated Hamiltonian Replica Exchange Molecular Dynamics. J. Comput. Aided Mol. Des. 2013, 27, 989–1007. [Google Scholar] [CrossRef] [Green Version]
- Fujitani, H.; Tanida, Y.; Matsuura, A. Massively Parallel Computation of Absolute Binding Free Energy with Well-Equilibrated States. Phys. Rev. E 2009, 79, 021914. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Average kcal/mol | Std.Dev | Average kcal/mol | Std.Dev | Average kcal/mol | Std.Dev | Average kcal/mol | Std.Dev | Average kcal/mol | Std.Dev | Average kcal/mol | Std.Dev | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Miyabenol A | Nepalensinol B | Flexuosol A | Kobophenol A | Ampelopsin H | SPD304 | |||||||
| EvDW | −44.514 | 6.56 | −62.4408 | 3.1846 | −31.391 | 6.848 | −46.87 | 3.6866 | −45.9798 | 3.461 | −43.9821 | 2.5799 |
| EEL | −15.963 | 6.307 | −22.2184 | 5.5841 | −19.287 | 10.104 | −18.6796 | 6.4461 | −13.0541 | 4.7604 | −111.752 | 5.2156 |
| EGB | 45.629 | 6.965 | 56.5623 | 4.9438 | 39.683 | 9.983 | 50.9639 | 6.0277 | 45.0963 | 6.6032 | 144.1032 | 5.9402 |
| ΔGGAS | −60.484 | 9.413 | −84.712 | 6.0403 | −50.661 | 11.583 | −65.5466 | 7.5489 | −59.0364 | 6.6642 | −155.734 | 6.5496 |
| ΔGSOLV | 39.698 | 6.587 | 49.2238 | 4.8589 | 35.445 | 9.584 | 44.7208 | 5.8006 | 39.3336 | 6.47 | 139.36 | 5.8403 |
| ΔGTOTAL | −20.786 | 4.184 | −35.4882 | 3.3524 | −15.216 | 7.09 | −20.8258 | 3.7012 | −19.7027 | 2.6903 | −16.3744 | 2.8002 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papadopoulou, D.; Drakopoulos, A.; Lagarias, P.; Melagraki, G.; Kollias, G.; Afantitis, A. In Silico Identification and Evaluation of Natural Products as Potential Tumor Necrosis Factor Function Inhibitors Using Advanced Enalos Asclepios KNIME Nodes. Int. J. Mol. Sci. 2021, 22, 10220. https://doi.org/10.3390/ijms221910220
Papadopoulou D, Drakopoulos A, Lagarias P, Melagraki G, Kollias G, Afantitis A. In Silico Identification and Evaluation of Natural Products as Potential Tumor Necrosis Factor Function Inhibitors Using Advanced Enalos Asclepios KNIME Nodes. International Journal of Molecular Sciences. 2021; 22(19):10220. https://doi.org/10.3390/ijms221910220
Chicago/Turabian StylePapadopoulou, Dimitra, Antonios Drakopoulos, Panagiotis Lagarias, Georgia Melagraki, George Kollias, and Antreas Afantitis. 2021. "In Silico Identification and Evaluation of Natural Products as Potential Tumor Necrosis Factor Function Inhibitors Using Advanced Enalos Asclepios KNIME Nodes" International Journal of Molecular Sciences 22, no. 19: 10220. https://doi.org/10.3390/ijms221910220
APA StylePapadopoulou, D., Drakopoulos, A., Lagarias, P., Melagraki, G., Kollias, G., & Afantitis, A. (2021). In Silico Identification and Evaluation of Natural Products as Potential Tumor Necrosis Factor Function Inhibitors Using Advanced Enalos Asclepios KNIME Nodes. International Journal of Molecular Sciences, 22(19), 10220. https://doi.org/10.3390/ijms221910220