
Macrophages, Metabolites, and Nucleosomes: Chromatin at the Intersection between Aging and Inflammation


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
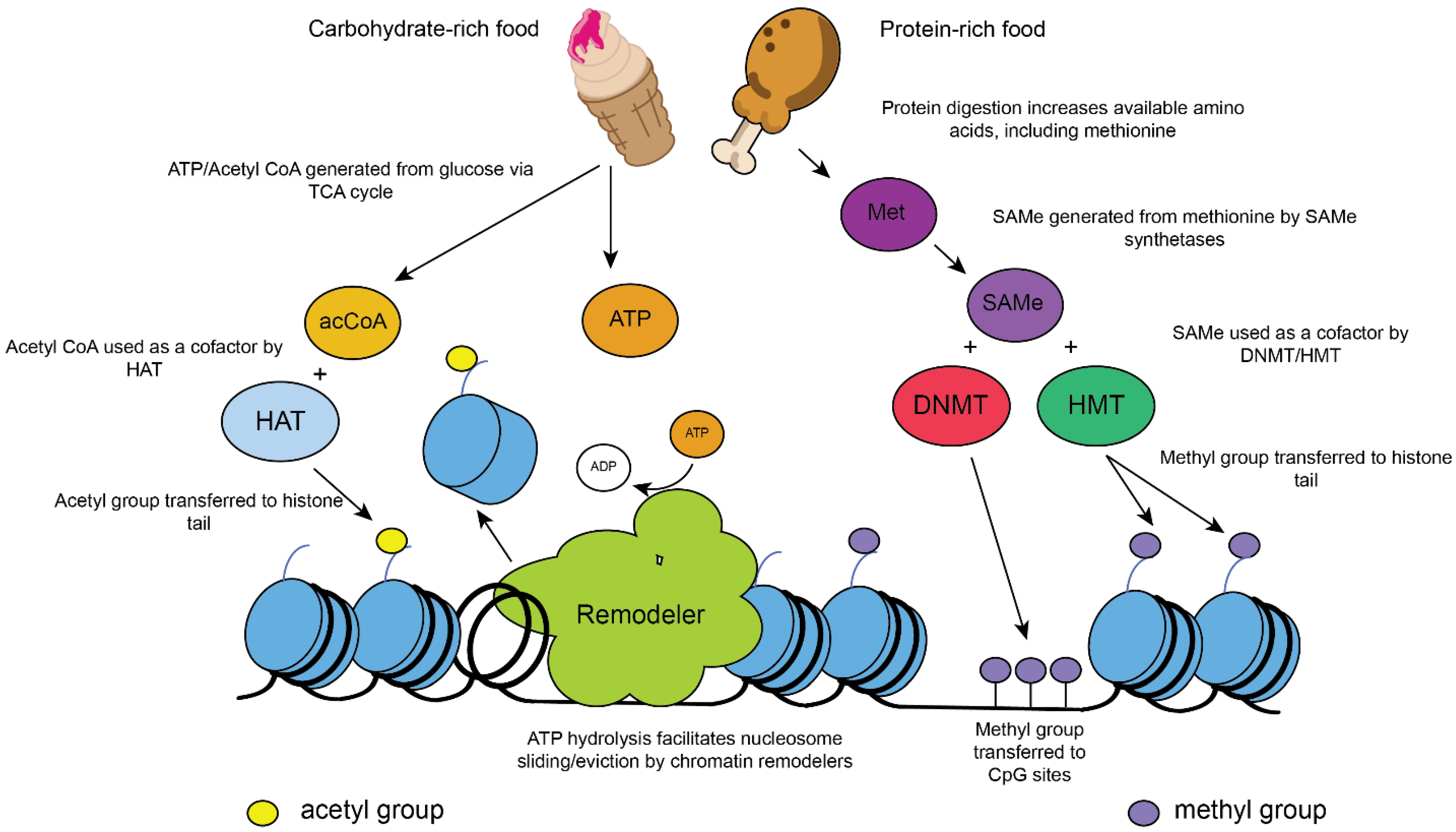
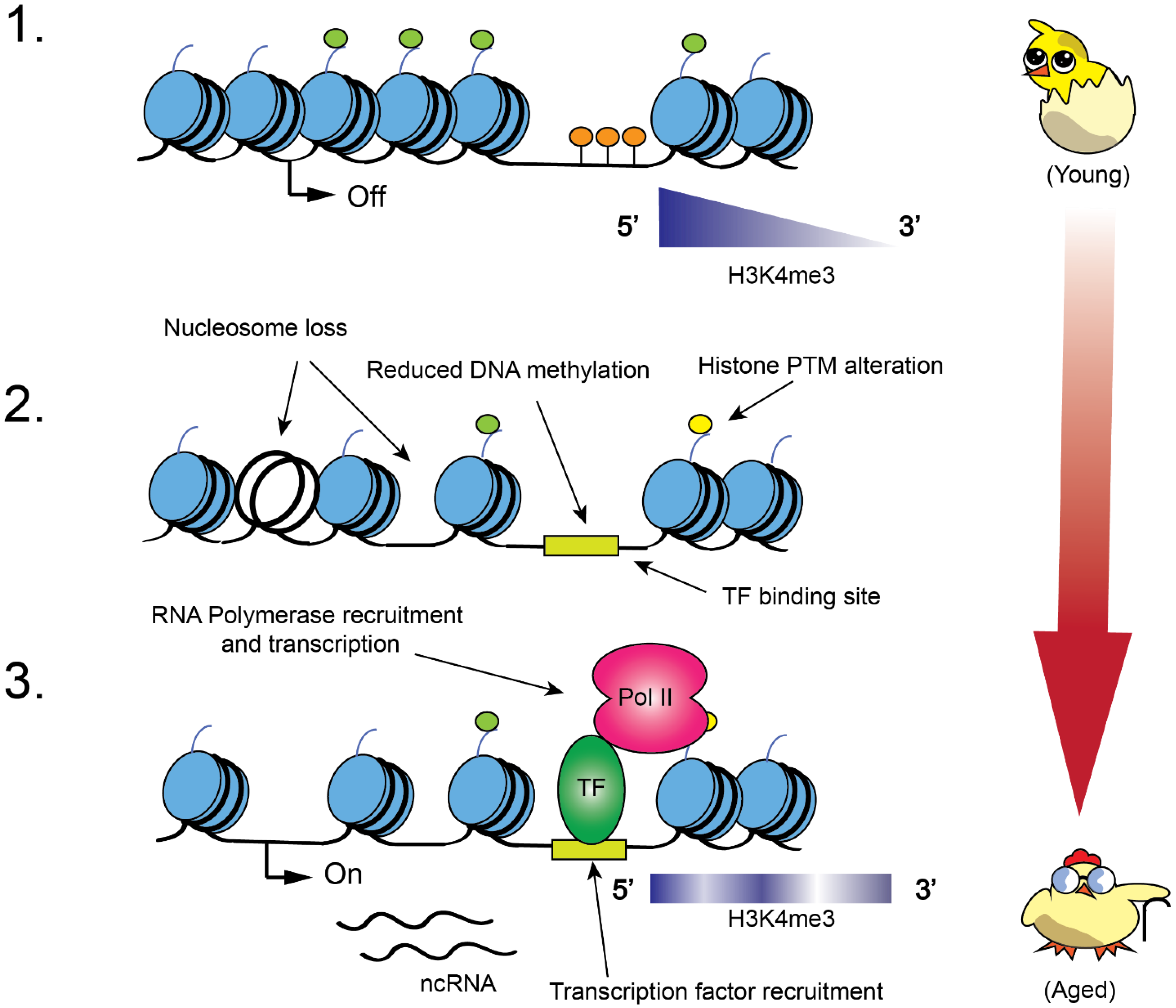
2. Role of Chromatin Modifications in Metabolism and Aging
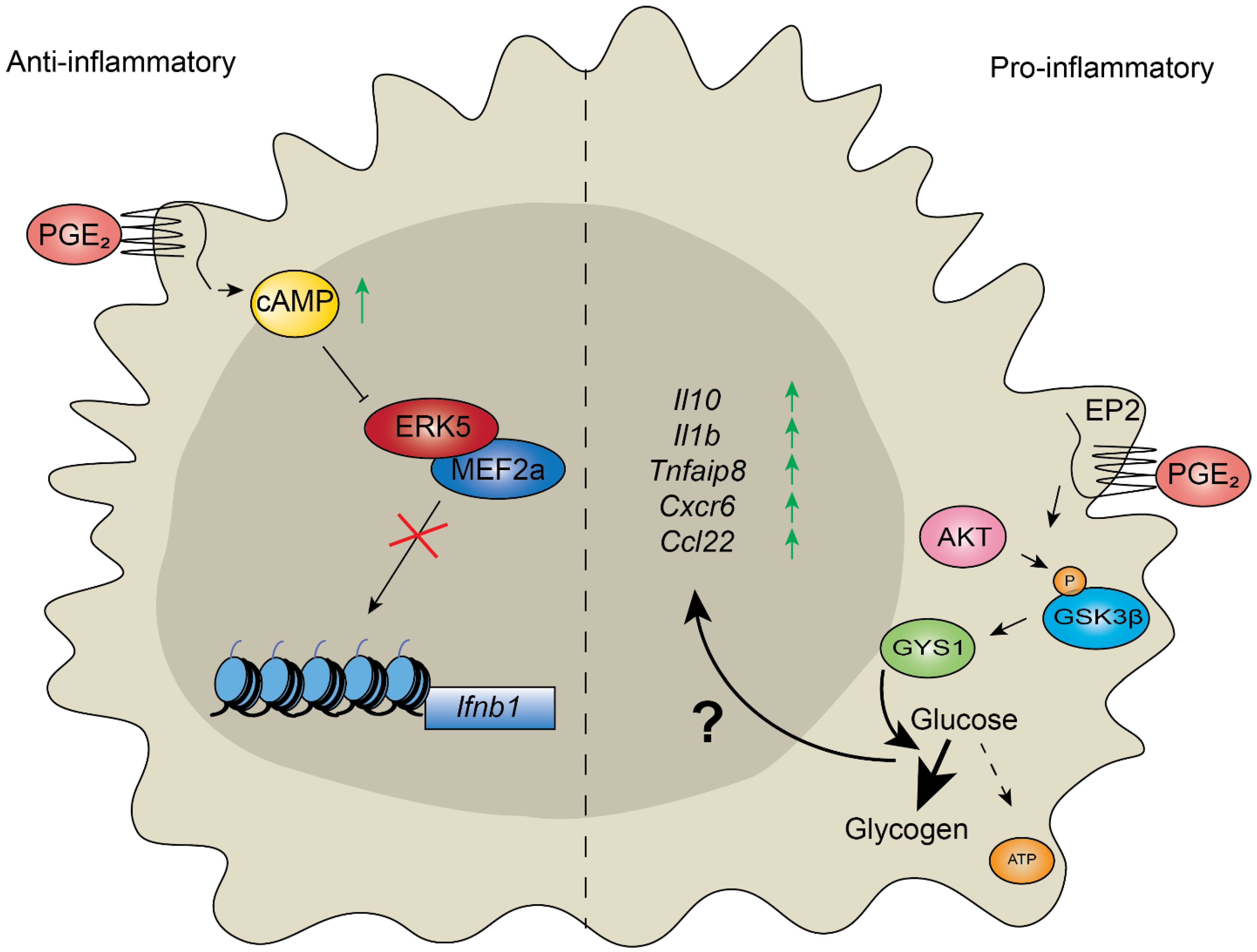
3. Control of Inflammation Depends on Metabolic Reprogramming of Chromatin in Response to Stimuli
4. Chromatin Modifiers Regulate Aging and Chronic Inflammatory Response
5. Chromatin Modifiers Associated with RNA Synthesis and Metabolic Sinks May Contribute to Inflammaging
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar]
- Medzhitov, R. Inflammation 2010: New Adventures of an Old Flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef] [Green Version]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8, 1960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Wang, C.; Mao, X.; Hao, Y. B Cell Dysfunction Associated With Aging and Autoimmune Diseases. Front. Immunol. 2019, 10, 318. [Google Scholar] [CrossRef] [Green Version]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Tsuchida, T.; Ishiyama, A.; Taniguchi, T.; Suzuki, S.; Horiuchi, Y.; Matsuo, Y.; Yoshizawa, N.; Suganuma, T.; Omae, M.; et al. Endoscopic mucosal resection and endoscopic submucosal dissection for en bloc resection of superficial pharyngeal carcinomas. Endoscopy 2012, 44, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Fredman, G.; Tabas, I. Boosting Inflammation Resolution in Atherosclerosis: The Next Frontier for Therapy. Am. J. Pathol. 2017, 187, 1211–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nathan, C.; Ding, A. Nonresolving inflammation. Cell 2010, 140, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Yang, J.; Wei, Y.; Wei, X. Epigenetic regulation of macrophages: From homeostasis maintenance to host defense. Cell. Mol. Immunol. 2020, 17, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Ding, N.; Zhang, H.; Liu, K.; Xiong, J.; Ma, S.; Yang, A.; Zhang, H.; Jiang, Y. SNF5 promotes IL-1β expression via H3K4me1 in atherosclerosis induced by homocysteine. Int. J. Biochem. Cell Biol. 2021, 135, 105974. [Google Scholar] [CrossRef] [PubMed]
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Investig. 2017, 127, 3240–3249. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Salvioli, S.; Monti, D.; Lanzarini, C.; Conte, M.; Pirazzini, C.; Bacalini, M.G.; Garagnani, P.; Giuliani, C.; Fontanesi, E.; Ostan, R.; et al. Immune system, cell senescence, aging and longevity—Inflamm-aging reappraised. Curr. Pharm. Des. 2013, 19, 1675–1679. [Google Scholar]
- Lettieri-Barbato, D.; Giovannetti, E.; Aquilano, K. Effects of dietary restriction on adipose mass and biomarkers of healthy aging in human. Aging 2016, 8, 3341–3355. [Google Scholar] [CrossRef] [Green Version]
- Davis, F.M.; Tsoi, L.C.; Wasikowski, R.; Dendekker, A.; Joshi, A.; Wilke, C.; Deng, H.; Wolf, S.; Obi, A.; Huang, S.; et al. Epigenetic regulation of the PGE2 pathway modulates macrophage phenotype in normal and pathologic wound repair. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Carey, M.; Workman, J.L. The Role of Chromatin during Transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Luger, K.; Rechsteiner, T.J.; Flaus, A.; Waye, M.M.Y.; Richmond, T.J. Characterization of nucleosome core particles containing histone proteins made in bacteria. J. Mol. Biol. 1997, 272, 301–311. [Google Scholar] [CrossRef]
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Zhao, Y.; Garcia, B.A. Comprehensive Catalog of Currently Documented Histone Modifications. Cold Spring Harb. Perspect. Biol. 2015, 7, a025064. [Google Scholar] [CrossRef] [Green Version]
- Sabari, B.R.; Zhang, D.; Allis, C.D.; Zhao, Y. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017, 18, 90–101. [Google Scholar] [CrossRef] [Green Version]
- Ye, C.; Sutter, B.M.; Wang, Y.; Kuang, Z.; Zhao, X.; Yu, Y.; Tu, B.P. Demethylation of the Protein Phosphatase PP2A Promotes Demethylation of Histones to Enable Their Function as a Methyl Group Sink. Mol. Cell 2019, 73, 1115–1126.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarsland, D.; Creese, B.; Politis, M.; Chaudhuri, K.R.; Ffytche, D.H.; Weintraub, D.; Ballard, C. Cognitive decline in Parkinson disease. Nat. Rev. Neurol. 2017, 13, 217–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauterbach, M.A.; Hanke, J.E.; Serefidou, M.; Mangan, M.S.; Kolbe, C.-C.; Hess, T.; Rothe, M.; Kaiser, R.; Hoss, F.; Gehlen, J.; et al. Toll-like Receptor Signaling Rewires Macrophage Metabolism and Promotes Histone Acetylation via ATP-Citrate Lyase. Immunity 2019, 51, 997–1011.e7. [Google Scholar] [CrossRef] [PubMed]
- Suganuma, T.; Workman, J.L. Signals and Combinatorial Functions of Histone Modifications. Annu. Rev. Biochem. 2011, 80, 473–499. [Google Scholar] [CrossRef]
- Dutta, A.; Gogol, M.; Kim, J.-H.; Smolle, M.; Venkatesh, S.; Gilmore, J.; Florens, L.; Washburn, M.; Workman, J.L. Swi/Snf dynamics on stress-responsive genes is governed by competitive bromodomain interactions. Genes Dev. 2014, 28, 2314–2330. [Google Scholar] [CrossRef] [Green Version]
- Sharma, T.; Robinson, D.C.L.; Witwicka, H.; Dilworth, F.J.; Imbalzano, A.N. The Bromodomains of the mammalian SWI/SNF (mSWI/SNF) ATPases Brahma (BRM) and Brahma Related Gene 1 (BRG1) promote chromatin interaction and are critical for skeletal muscle differentiation. Nucleic Acids Res. 2021, 49, 8060–8077. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lee, R.S.; Alver, B.; Haswell, J.R.; Wang, S.; Mieczkowski, J.; Drier, Y.; Gillespie, S.; Archer, T.C.; Wu, J.N.; et al. SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 2017, 49, 289–295. [Google Scholar] [CrossRef]
- Ogiwara, H.; Takahashi, K.; Sasaki, M.; Kuroda, T.; Yoshida, H.; Watanabe, R.; Maruyama, A.; Makinoshima, H.; Chiwaki, F.; Sasaki, H.; et al. Targeting the Vulnerability of Glutathione Metabolism in ARID1A-Deficient Cancers. Cancer Cell 2019, 35, 177–190.e8. [Google Scholar] [CrossRef] [Green Version]
- Neigeborn, L.; Carlson, M. Genes affecting the regulation of SUC2 gene expression by glucose repression in Saccharomyces cerevisiae. Genetics 1984, 108, 845–858. [Google Scholar] [CrossRef]
- Gavin, I.M.; Simpson, R.T. Interplay of yeast global transcriptional regulators Ssn6p-Tup1p and Swi-Snf and their effect on chromatin structure. EMBO J. 1997, 16, 6263–6271. [Google Scholar] [CrossRef]
- Suganuma, T.; Workman, J.L. Chromatin and Metabolism. Annu. Rev. Biochem. 2018, 87, 27–49. [Google Scholar] [CrossRef] [PubMed]
- Yi, S.-J.; Kim, K. New Insights into the Role of Histone Changes in Aging. Int. J. Mol. Sci. 2020, 21, 8241. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, R.J.; Kubicek, S.; Schreiber, S.L.; Karlseder, J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat. Struct. Mol. Biol. 2010, 17, 1218–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, W.; Steffen, K.K.; Perry, R.; Dorsey, J.A.; Johnson, F.B.; Shilatifard, A.; Kaeberlein, M.; Kennedy, B.; Berger, S.L. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009, 459, 802–807. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Dang, W.; Donahue, G.; Dai, J.; Dorsey, J.; Cao, X.; Liu, W.; Cao, K.; Perry, R.; Lee, J.Y.; et al. H3K36 methylation promotes longevity by enhancing transcriptional fidelity. Genes Dev. 2015, 29, 1362–1376. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.D.; Tsuchiya, M.; Fox, L.A.; Dang, N.; Hu, D.; Kerr, E.O.; Johnston, E.D.; Tchao, B.N.; Pak, D.N.; Welton, K.L.; et al. Quantitative evidence for conserved longevity pathways between divergent eukaryotic species. Genome Res. 2008, 18, 564–570. [Google Scholar] [CrossRef] [Green Version]
- Cruz, C.; Della Rosa, M.; Krueger, C.; Gao, Q.; Horkai, D.; King, M.; Field, L.; Houseley, J. Tri-methylation of histone H3 lysine 4 facilitates gene expression in ageing cells. Elife 2018, 7, 7. [Google Scholar] [CrossRef]
- Greer, E.; Maures, T.J.; Hauswirth, A.G.; Green, E.; Leeman, D.S.; Maro, G.S.; Han, S.; Banko, M.R.; Gozani, O.; Brunet, A. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature 2010, 466, 383–387. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.G.; Mayerbarber, K.D.; Sung, H.; Beura, L.K.; James, B.R.; Taylor, J.J.; Qunaj, L.; Griffith, T.S.; Vezys, V.; Barber, D.L.; et al. Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 2014, 9, 209–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adelman, E.R.; Huang, H.-T.; Roisman, A.; Olsson, A.; Colaprico, A.; Qin, T.; Lindsley, R.C.; Bejar, R.; Salomonis, N.; Grimes, H.L.; et al. Aging Human Hematopoietic Stem Cells Manifest Profound Epigenetic Reprogramming of Enhancers That May Predispose to Leukemia. Cancer Discov. 2019, 9, 1080–1101. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Pastor, R.; Burchfiel, E.T.; Neef, D.W.; Jaeger, A.M.; Cabiscol, E.; McKinstry, S.U.; Doss, A.; Aballay, A.; Lo, D.C.; Akimov, S.S.; et al. Abnormal degradation of the neuronal stress-protective transcription factor HSF1 in Huntington’s disease. Nat. Commun. 2017, 8, 14405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, M.; Wang, M.; Wang, W.; Velayudhan, S.S.; Lee, S.S. Unique patterns of trimethylation of histone H3 lysine 4 are prone to changes during aging in Caenorhabditis elegans somatic cells. PLoS Genet. 2018, 14, e1007466. [Google Scholar] [CrossRef] [Green Version]
- Booth, L.N.; Brunet, A. The Aging Epigenome. Mol. Cell 2016, 62, 728–744. [Google Scholar] [CrossRef] [Green Version]
- Salameh, Y.; Bejaoui, Y.; El Hajj, N. DNA Methylation Biomarkers in Aging and Age-Related Diseases. Front. Genet. 2020, 11, 171. [Google Scholar] [CrossRef]
- Bogdanović, O.; Lister, R. DNA methylation and the preservation of cell identity. Curr. Opin. Genet. Dev. 2017, 46, 9–14. [Google Scholar] [CrossRef]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef]
- Aggarwal, V.K. Winning in Asia, U.S. Style: Market and Nonmarket Strategies for Success; Palgrave Macmillan: New York, NY, USA, 2003; Volume xiii, p. 279. [Google Scholar]
- Varela, M.L.; Mogildea, M.; Moreno, I.; Lopes, A. Acute Inflammation and Metabolism. Inflammation 2018, 41, 1115–1127. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Funes, S.C.; Rios, M.; Escobar-Vera, J.; Kalergis, A.M. Implications of macrophage polarization in autoimmunity. Immunology 2018, 154, 186–195. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zhang, S.; Wu, H.; Rong, X.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [Green Version]
- Foster, S.L.; Hargreaves, D.C.; Medzhitov, R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature 2007, 447, 972–978. [Google Scholar] [CrossRef]
- Cilenti, F.; Barbiera, G.; Caronni, N.; Iodice, D.; Montaldo, E.; Barresi, S.; Lusito, E.; Cuzzola, V.; Vittoria, F.M.; Mezzanzanica, L.; et al. A PGE 2-MEF2A axis enables context-dependent control of inflammatory gene expression. Immunity 2021, 54, 1665–1682.e14. [Google Scholar] [CrossRef]
- Luo, L.; Feng, S.; Wu, Y.; Su, Y.; Jing, F.; Yi, Q. Serum Levels of Syndecan-1 in Patients with Kawasaki Disease. Pediatr. Infect. Dis. J. 2019, 38, 89–94. [Google Scholar] [CrossRef]
- Ankrum, J.A.; Miranda, O.R.; Ng, K.S.; Sarkar, D.; Xu, C.; Karp, J.M. Engineering cells with intracellular agent–loaded microparticles to control cell phenotype. Nat. Protoc. 2014, 9, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Ansari, K.M.; Sung, Y.M.; He, G.; Fischer, S.M. Prostaglandin receptor EP2 is responsible for cyclooxygenase-2 induction by prostaglandin E2 in mouse skin. Carcinogenesis 2007, 28, 2063–2068. [Google Scholar] [CrossRef] [Green Version]
- Iyer, N.G.; Özdag, H.; Caldas, C. p300/CBP and cancer. Oncogene 2004, 23, 4225–4231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minhas, P.S.; Latif-Hernandez, A.; McReynolds, M.R.; Durairaj, A.S.; Wang, Q.; Rubin, A.; Joshi, A.U.; He, J.Q.; Gauba, E.; Liu, L.; et al. Restoring metabolism of myeloid cells reverses cognitive decline in ageing. Nature 2021, 590, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Revisiting atherosclerosis and dementia. Nat. Neurosci. 2020, 23, 691–692. [Google Scholar] [CrossRef] [PubMed]
- Bouchareychas, L.; Raffai, R.L. Apolipoprotein E and Atherosclerosis: From Lipoprotein Metabolism to MicroRNA Control of Inflammation. J. Cardiovasc. Dev. Dis. 2018, 5, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallner, S.; Schröder, C.; Leitão, E.; Berulava, T.; Haak, C.; Beißer, D.; Rahmann, S.; Richter, A.S.; Manke, T.; Bönisch, U.; et al. Epigenetic dynamics of monocyte-to-macrophage differentiation. Epigenet. Chromatin 2016, 9, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrzak, J.; Płoszaj, T.; Pułaski, Ł.; Robaszkiewicz, A. EP300-HDAC1-SWI/SNF functional unit defines transcription of some DNA repair enzymes during differentiation of human macrophages. Biochim. Biophys. Acta Gene Regul. Mech. 2019, 1862, 198–208. [Google Scholar] [CrossRef]
- Hine, C.; Harputlugil, E.; Zhang, Y.; Ruckenstuhl, C.; Lee, B.C.; Brace, L.; Longchamp, A.; Treviño-Villarreal, J.H.; Mejia, P.; Ozaki, C.K.; et al. Endogenous Hydrogen Sulfide Production Is Essential for Dietary Restriction Benefits. Cell 2015, 160, 132–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganuma, T.; Swanson, S.K.; Gogol, M.; Garrett, T.J.; Conkright-Fincham, J.; Florens, L.; Washburn, M.P.; Workman, J.L. MPTAC Determines APP Fragmentation via Sensing Sulfur Amino Acid Catabolism. Cell Rep. 2018, 24, 1585–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casolini, P.; Catalani, A.; Zuena, A.R.; Angelucci, L. Inhibition of COX-2 reduces the age-dependent increase of hippocampal inflammatory markers, corticosterone secretion, and behavioral impairments in the rat. J. Neurosci. Res. 2002, 68, 337–343. [Google Scholar] [CrossRef]
- Wu, D.; Meydani, S.N. Mechanism of age-associated up-regulation in macrophage PGE2 synthesis. Brain Behav. Immun. 2004, 18, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Nagai, S.; Davis, R.E.; Mattei, P.J.; Eagen, K.; Kornberg, R.D. Chromatin potentiates transcription. Proc. Natl. Acad. Sci. USA 2017, 114, 1536–1541. [Google Scholar] [CrossRef] [Green Version]
- Day, F.; Ruth, K.S.; Thompson, D.J.; Lunetta, K.L.; Pervjakova, N.; Chasman, D.I.; Stolk, L.; Finucane, H.K.; Sulem, P.; Bulik-Sullivan, B.; et al. Large-scale genomic analyses link reproductive aging to hypothalamic signaling, breast cancer susceptibility and BRCA1-mediated DNA repair. Nat. Genet. 2015, 47, 1294–1303. [Google Scholar] [CrossRef]
- Loo, T.M.; Kamachi, F.; Watanabe, Y.; Yoshimoto, S.; Kanda, H.; Arai, Y.; Nakajima-Takagi, Y.; Iwama, A.; Koga, T.; Sugimoto, Y.; et al. Gut Microbiota Promotes Obesity-Associated Liver Cancer through PGE2-Mediated Suppression of Antitumor Immunity. Cancer Discov. 2017, 7, 522–538. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; LaFleur, M.; Nguyen, T.; Chen, S.; Chakravarthy, A.; Conway, J.; Li, Y.; Chen, H.; Yang, H.; Hsu, P.-H.; et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018, 174, 549–563.e19. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Huen, S.; Luan, H.H.; Yu, S.; Zhang, C.; Gallezot, J.-D.; Booth, C.J.; Medzhitov, R. Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell 2016, 166, 1512–1525.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Asouli, Y.; Banai, Y.; Pel-Or, Y.; Shir, A.; Kaempfer, R. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell 2002, 108, 221–232. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Furuhashi, M.; Li, P.; Cao, H.; Tuncman, G.; Sonenberg, N.; Gorgun, C.Z.; Hotamisligil, G.S. Double-Stranded RNA-Dependent Protein Kinase Links Pathogen Sensing with Stress and Metabolic Homeostasis. Cell 2010, 140, 338–348. [Google Scholar] [CrossRef] [Green Version]
- Suganuma, T.; Swanson, S.K.; Florens, L.; Washburn, M.; Workman, J.L. Moco biosynthesis and the ATAC acetyltransferase engage translation initiation by inhibiting latent PKR activity. J. Mol. Cell Biol. 2015, 8, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Wang, Z.; Zhang, K.; Chi, Z.; Xu, T.; Jiang, D.; Chen, S.; Li, W.; Yang, X.; Zhang, X.; et al. One-Carbon Metabolism Supports S-Adenosylmethionine and Histone Methylation to Drive Inflammatory Macrophages. Mol. Cell 2019, 75, 1147–1160.e5. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Church, M.C.; Workman, J.L.; Suganuma, T. Macrophages, Metabolites, and Nucleosomes: Chromatin at the Intersection between Aging and Inflammation. Int. J. Mol. Sci. 2021, 22, 10274. https://doi.org/10.3390/ijms221910274
Church MC, Workman JL, Suganuma T. Macrophages, Metabolites, and Nucleosomes: Chromatin at the Intersection between Aging and Inflammation. International Journal of Molecular Sciences. 2021; 22(19):10274. https://doi.org/10.3390/ijms221910274
Chicago/Turabian StyleChurch, Michael C., Jerry L. Workman, and Tamaki Suganuma. 2021. "Macrophages, Metabolites, and Nucleosomes: Chromatin at the Intersection between Aging and Inflammation" International Journal of Molecular Sciences 22, no. 19: 10274. https://doi.org/10.3390/ijms221910274
APA StyleChurch, M. C., Workman, J. L., & Suganuma, T. (2021). Macrophages, Metabolites, and Nucleosomes: Chromatin at the Intersection between Aging and Inflammation. International Journal of Molecular Sciences, 22(19), 10274. https://doi.org/10.3390/ijms221910274