
Highly Efficient Modular Construction of Functional Drug Delivery Platform Based on Amphiphilic Biodegradable Polymers via Click Chemistry


Abstract
:1. Introduction
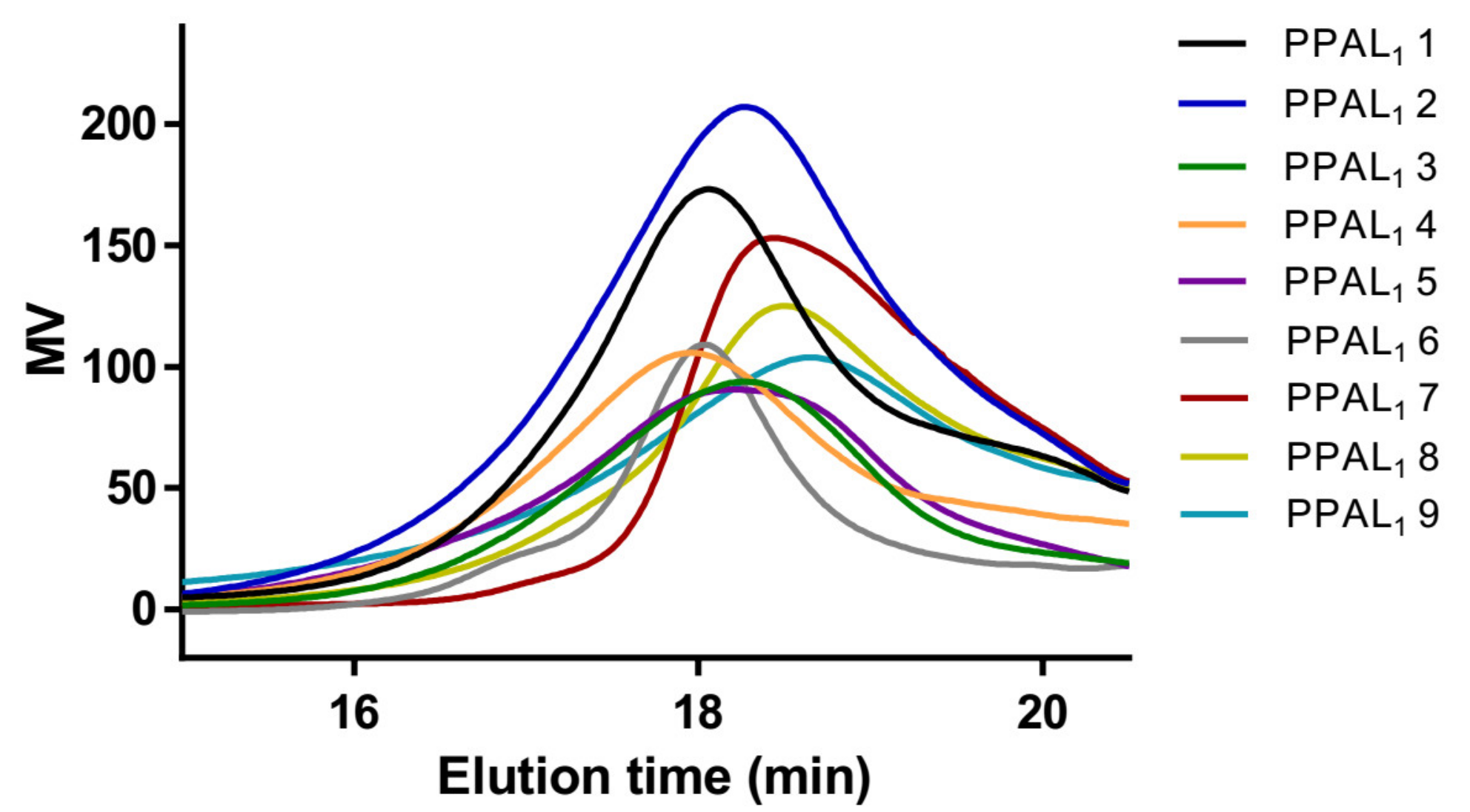
2. Results and Discussion
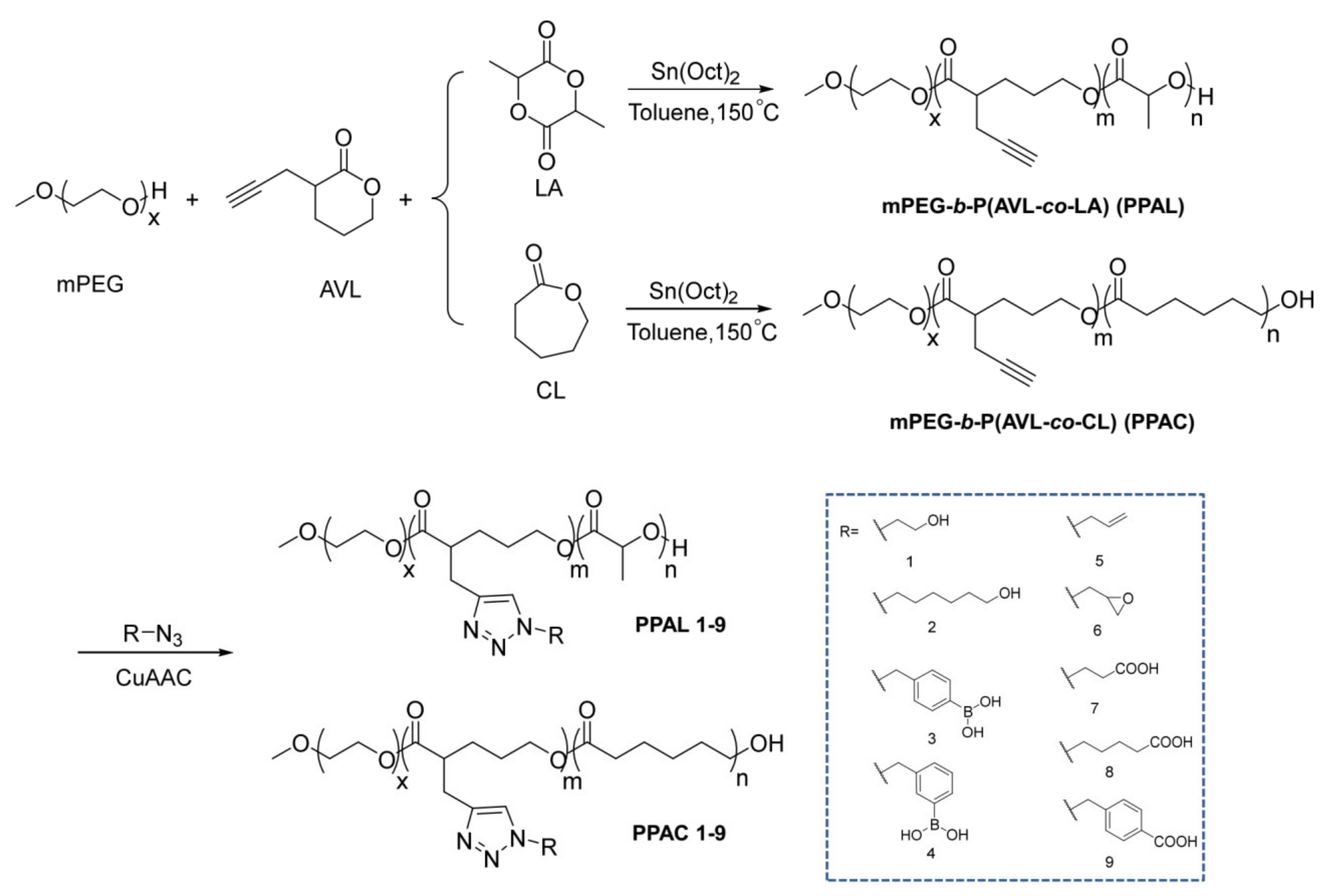
2.1. Syntheses of mPEG-b-P(AVL-co-LA) (PPAL) and mPEG-b-P(AVL-co-CL) (PPAC) Copolymers with Pendant Alkynyl Groups
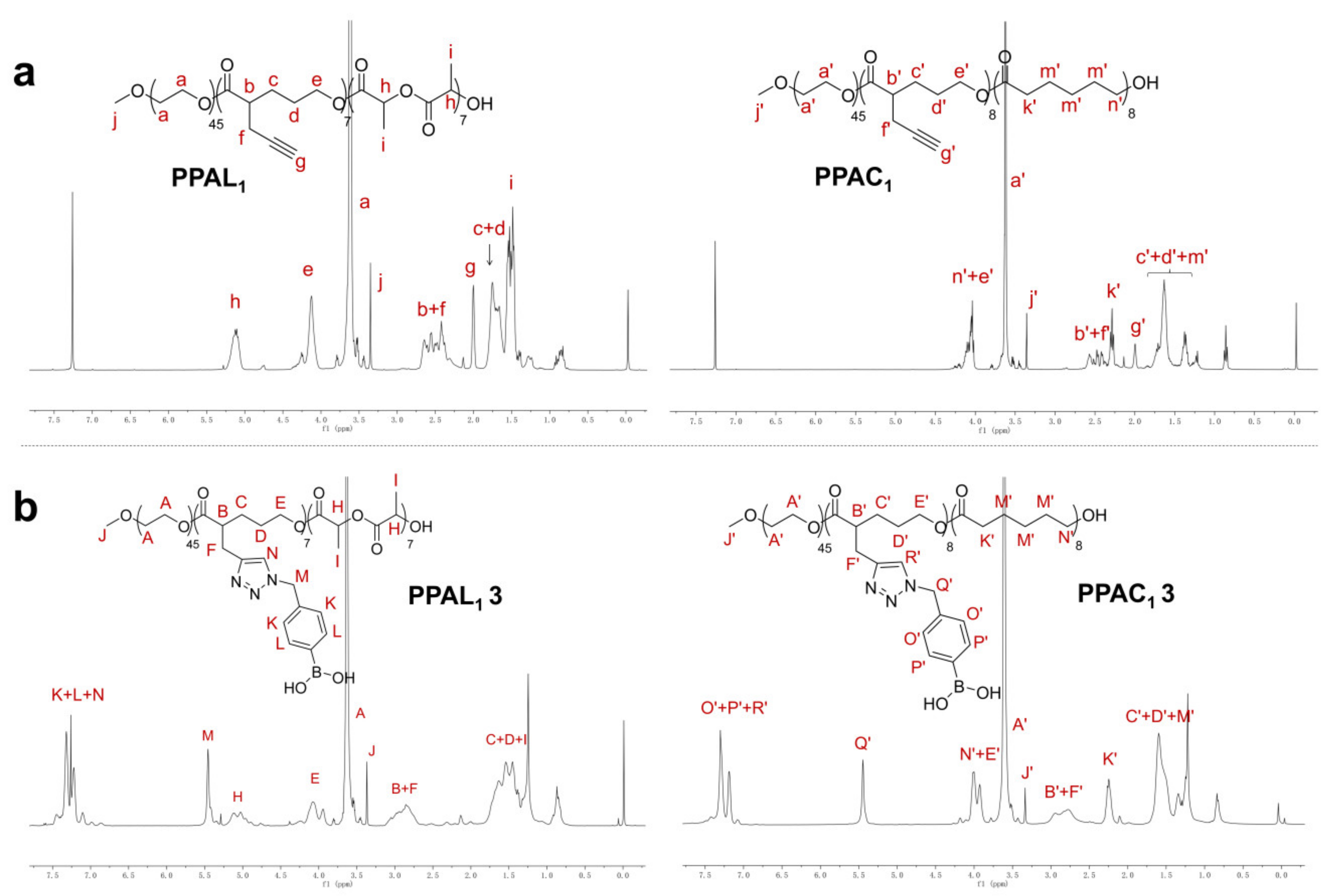
2.2. Functionalization of PPAL and PPAC Copolymers by Click Chemistry
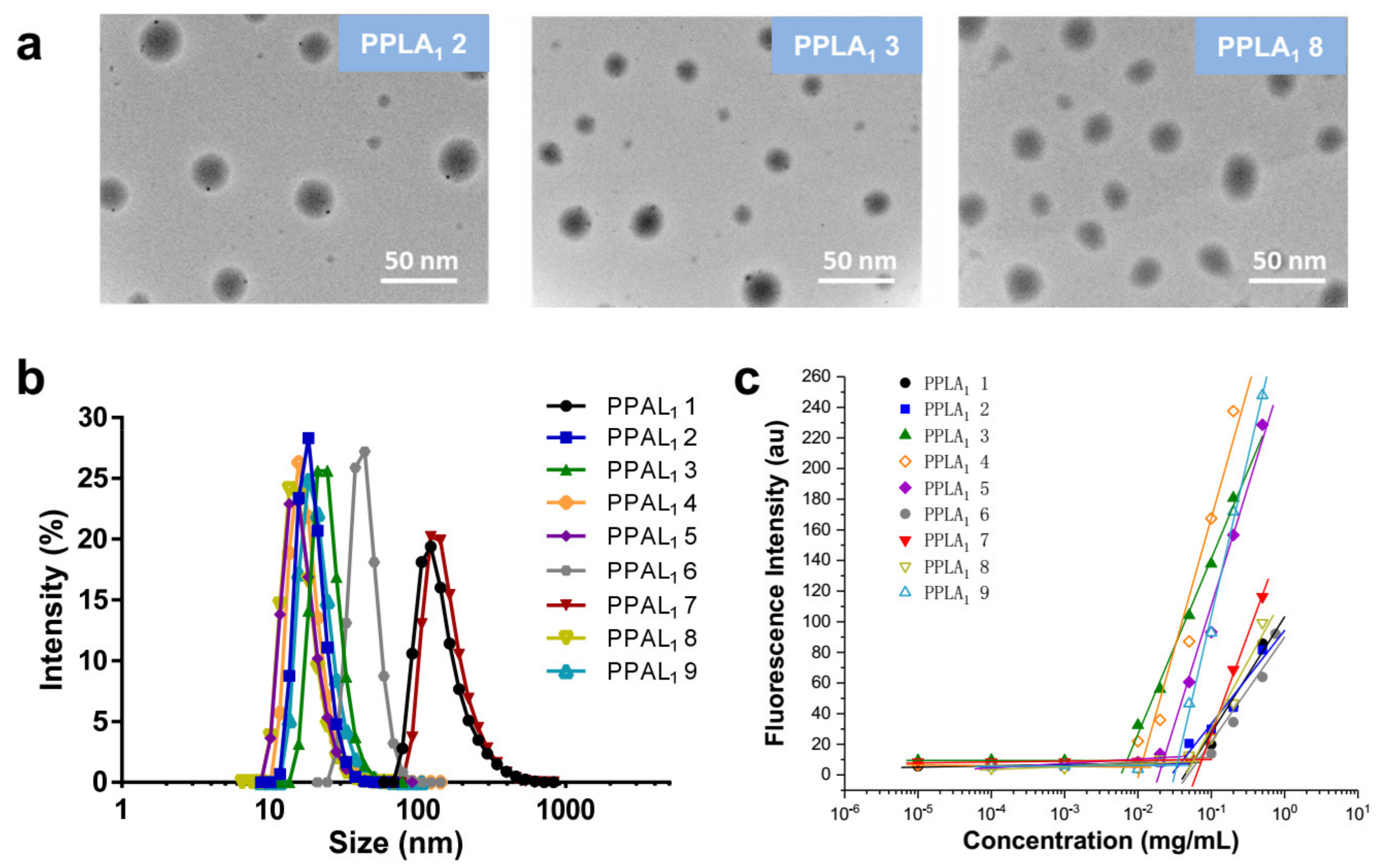
2.3. Self-Assembly of Functional Polymeric Nanoparticles
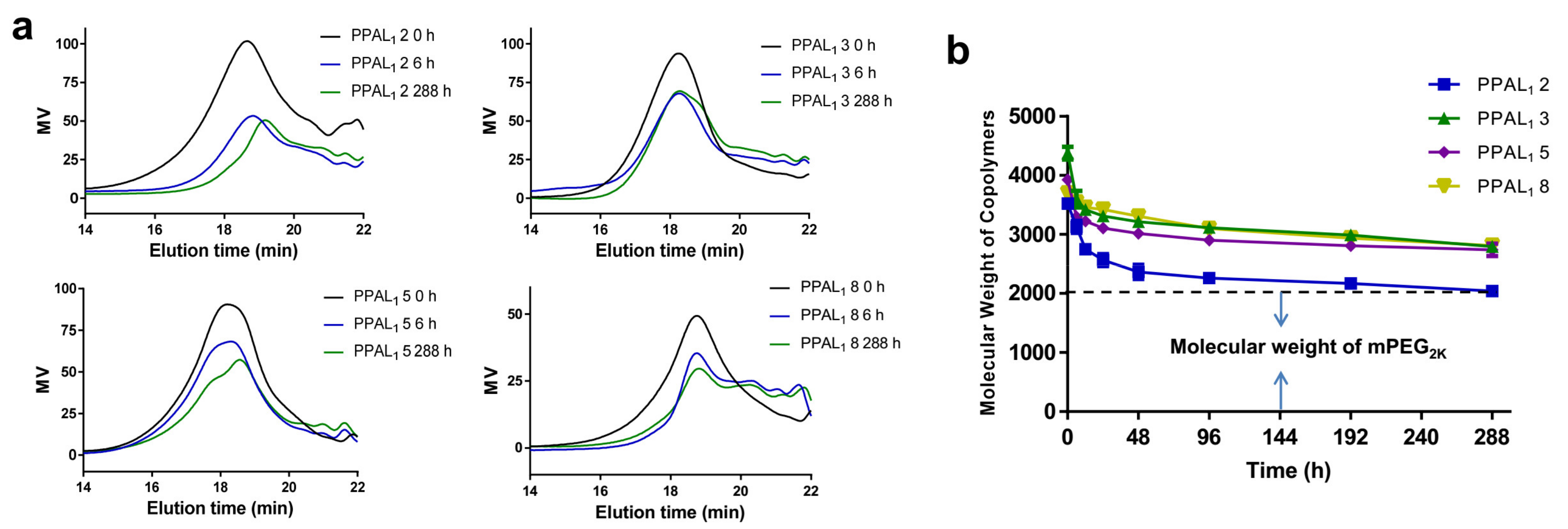
2.4. Degradation Study
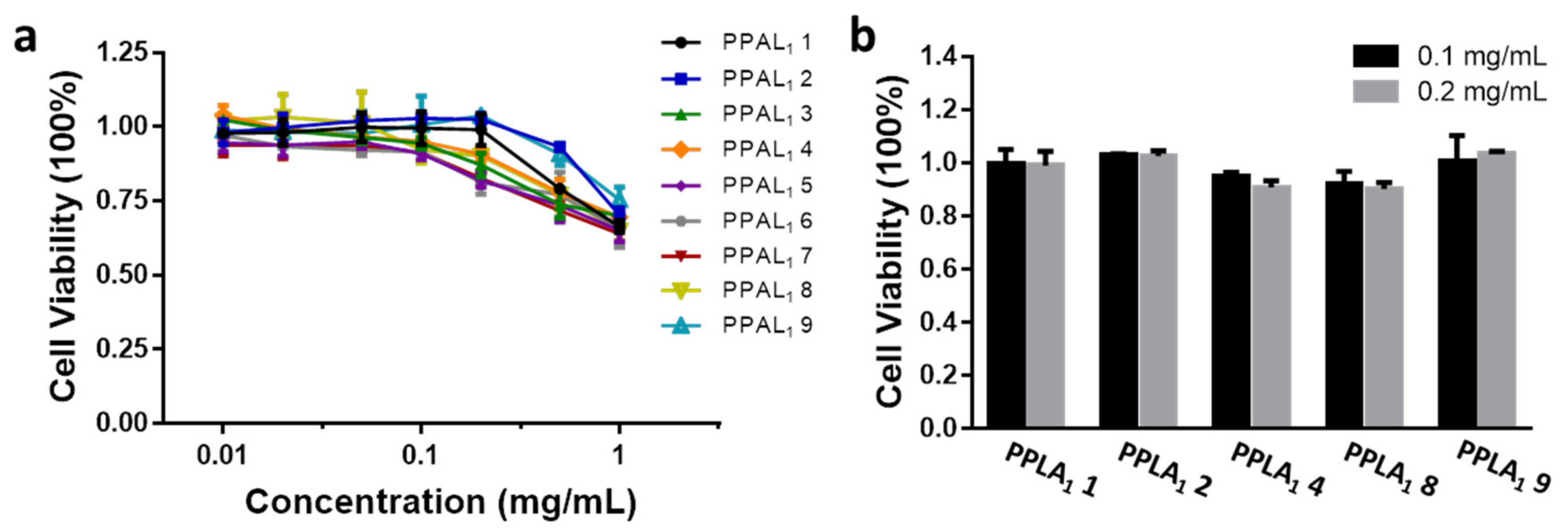
2.5. Cytotoxicity Assay
3. Materials and Methods
3.1. Materials
3.2. Instruments
3.3. Synthesis of α-Propargyl-δ-valerolactone (AVL)
3.4. General Procedure for Synthesis of mPEG-b-P(AVL-co-LA) (PPAL) and mPEG-b-P(AVL-co-CL) (PPAC) Copolymers
3.5. General Procedure for Modification of mPEG-b-P(AVL-co-LA) (PPAL) and mPEG-b-P(AVL-co-CL) (PPAC) Copolymers by CuAAC Click Chemistry
3.6. Preparation of Polymeric Nanoparticles
3.7. Characterization of Polymeric Nanoparticles
3.8. Determination of Critical Micelle Concentration (CMC)
3.9. In Vitro Degradation Study
3.10. Cell Culture
3.11. Cytotoxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kutikov, A.B.; Song, J. Biodegradable PEG-Based Amphiphilic Block Copolymers for Tissue Engineering Applications. ACS Biomater. Sci. Eng. 2015, 1, 463–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, E.; Shepherd, L.M.; Saunders, L.; Frey, M.W. Surface Functional Poly(lactic Acid) Electrospun Nanofibers for Biosensor Applications. Materials 2016, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Wang, X.; Tang, J.; Guo, Z.; Shen, Y.; Tian, H.; Zhu, W.H. Dual-channel NIR activatable theranostic prodrug for in vivo spatiotemporal tracking thiol-triggered chemotherapy. Chem. Sci. 2016, 7, 4958–4965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Sun, X.; Guo, Z.; Tang, J.; Shen, Y.; James, T.D.; Tian, H.; Zhu, W. In vivo and in situ tracking cancer chemotherapy by highly photostable NIR fluorescent theranostic prodrug. J. Am. Chem. Soc. 2014, 136, 3579–3588. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Zhu, H.; Qin, J.; Dong, H.; Du, J. Theranostic vesicles based on bovine serum albumin and poly(ethylene glycol)-block-poly(L-lactic-co-glycolic acid) for magnetic resonance imaging and anticancer drug delivery. Biomacromolecules 2014, 15, 1586–1592. [Google Scholar] [CrossRef]
- Liu, X.-M.; Pramoda, K.P.; Yang, Y.-Y.; Chow, S.Y.; He, C.B. Cholesteryl-grafted functional amphiphilic poly(N-isopropylacrylamide-co-N-hydroxylmethylacrylamide): Synthesis, temperaturesensitivity, self-assembly and encapsulation of a hydrophobic agent. Biomaterials 2004, 25, 2619–2628. [Google Scholar] [CrossRef]
- Mikhail, A.S.; Allen, C. Block copolymer micelles for delivery of cancer therapy: Transport at the whole body, tissue and cellular levels. J. Control. Release 2009, 138, 214–223. [Google Scholar] [CrossRef]
- Avgoustakis, K. Pegylated Poly(Lactide) and Poly(Lactide-Co-Glycolide) Nanoparticles: Preparation, Properties and Possible Applications in Drug Delivery Pegylated Poly(Lactide) and Poly(Lactide-Co-Glycolide) Nanoparticles: Preparation, Properties and Possible Applications in Drug Delivery. Curr. Drug Deliv. 2004, 1, 321–333. [Google Scholar]
- Grossen, P.; Witzigmann, D.; Sieber, S.; Huwyler, J. PEG-PCL-based nanomedicines: A biodegradable drug delivery system and its application. J. Control. Release 2017, 260, 46–60. [Google Scholar] [CrossRef]
- Cho, H.; Gao, J.M.; Kwon, G.S. PEG-b-PLA micelles and PLGA-b-PEG-b-PLGA sol–gels for drug delivery. J. Control. Release 2016, 240, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Danafar, H.; Schumacher, U. MPEG-PCL copolymeric nanoparticles in drug delivery systems. Cogent Med. 2016, 3, 1142411. [Google Scholar] [CrossRef]
- Xiao, R.Z.; Zeng, Z.W.; Zhou, G.L.; Wang, J.J.; Li, F.Z.; Wang, A.M. Recent advances in PEG-PLA block copolymer nanoparticles. Int. J. Nanomed. 2010, 5, 1057–1065. [Google Scholar]
- Hubbell, J.A. Bioactive biomaterials. Curr. Opin. Biotechnol. 1999, 10, 123–129. [Google Scholar] [CrossRef]
- Place, E.S.; Evans, N.D.; Stevens, M.M. Complexity in biomaterials for tissue engineering. Nat. Mater. 2009, 8, 457–470. [Google Scholar] [CrossRef]
- Kalelkar, P.P.; Geng, Z.; Finn, M.G.; Collard, D.M. Azide-Substituted Polylactide: A Biodegradable Substrate for Antimicrobial Materials via Click Chemistry Attachment of Quaternary Ammonium Groups. Biomacromolecules 2019, 20, 3366–3374. [Google Scholar] [CrossRef] [PubMed]
- Kalelkar, P.P.; Collard, D.M. Thiol-substituted copolylactide: Synthesis, characterization and post-polymerization modification using thiol–ene chemistry. Polym. Chem. 2018, 9, 1022–1031. [Google Scholar] [CrossRef]
- Yang, C.; Hu, R.; Anderson, T.; Wang, Y.; Lin, G.; Law, W.C.; Lin, W.-J.; Nguyen, Q.T.; Toh, H.T.; Yoon, H.S.; et al. Biodegradable nanoparticle-mediated K-ras down regulation for pancreatic cancer gene therapy. J. Mater. Chem. B 2015, 3, 2163–2172. [Google Scholar] [CrossRef]
- Yang, S.; Meel, R.V.D.; Theek, B.; Blenke, E.O.; Pieters, E.H.E.; Fens, M.H.A.M.; Ehling, J.; Schiffelers, R.M.; Storm, G.; Nostrum, C.F.V.; et al. Complete Regression of Xenograft Tumors upon Targeted Delivery of Paclitaxel via Π-Π Stacking Stabilized Polymeric Micelles. ACS Nano 2015, 9, 3740–3752. [Google Scholar]
- Lv, S.; Wu, Y.; Cai, K.; He, H.; Li, Y.; Lan, M.; Chen, X.; Cheng, J.; Yin, L. High Drug Loading and Sub-Quantitative Loading Efficiency of Polymeric Micelles Driven by Donor–Receptor Coordination Interactions. J. Am. Chem. Soc. 2018, 140, 1235–1238. [Google Scholar] [CrossRef]
- Li, Y.; Ding, J.; Zhu, J.; Tian, H.; Chen, X. Photothermal Effect-Triggered Drug Release from Hydrogen Bonding-Enhanced Polymeric Micelles. Biomacromolecules 2018, 19, 1950–1958. [Google Scholar] [CrossRef]
- Dong, Y.; Du, P.; Pei, M.; Liu, P. Design, postpolymerization conjugation and self-assembly of a di-block copolymer-based prodrug for tumor intracellular acid-triggered DOX release. J. Mater. Chem. B 2019, 7, 5640. [Google Scholar] [CrossRef]
- Gerhardt, W.W.; Noga, D.E.; Hardcastle, K.I.; Garcia, A.J.; Collard, D.M.; Weck, M. Functional lactide monomers: Methodology and polymerization. Biomacromolecules 2006, 7, 1735–1742. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.L.; Liu, S.; Chen, X.S.; Mo, G.J.; Xie, Z.G.; Jing, X.B. Biodegradable amphiphilic block copolymers bearing protected hydroxyl groups: Synthesis and characterization. Biomacromolecules 2008, 9, 553–560. [Google Scholar] [CrossRef]
- Zhang, X.J.; Mei, H.J.; Hu, C.; Zhong, Z.L.; Zhuo, R.X. Amphiphilic Triblock Copolycarbonates with Poly(glycerol carbonate) as Hydrophilic Blocks. Macromolecules 2009, 42, 1010–1016. [Google Scholar] [CrossRef]
- Wang, D.; Feng, X.-D. Synthesis of Poly(glycolic acid-alt-l-aspartic acid) from a Morpholine-2,5-dione Derivative. Macromolecules 1997, 30, 5688–5692. [Google Scholar] [CrossRef]
- Noga, D.E.; Petrie, T.A.; Kumar, A.; Weck, M.; Garcia, A.J.; Collard, D.M. Synthesis and modification of functional poly(lactide) copolymers: Toward biofunctional materials. Biomacromolecules 2008, 9, 2056–2062. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, A.; Xiong, X.-B.; Lavasanifar, A. Self-Associating Poly(ethylene oxide)-block-poly(-caprolactone) Block Copolymers with Functional Side Groups on the Polyester Block for Drug Delivery. Macromolecules 2006, 39, 9419–9428. [Google Scholar] [CrossRef]
- Trollsås, M.; Lee, V.Y.; Mecerreyes, D.; Löwenhielm, P.; Möller, M.; Miller, R.D.; Hedrick, J.L. Hydrophilic Aliphatic Polyesters: Design, Synthesis, and Ring-Opening Polymerization of Functional Cyclic Esters. Macromolecules 2000, 33, 4619–4627. [Google Scholar] [CrossRef]
- Chen, W.; Yang, H.; Wang, R.; Cheng, R.; Meng, F.; Wei, W.; Zhong, Z. Versatile Synthesis of Functional Biodegradable Polymers by Combining Ring-Opening Polymerization and Postpolymerization Modification via Michael-Type Addition Reaction. Macromolecules 2010, 43, 201–207. [Google Scholar] [CrossRef]
- Gauthier, M.A.; Gibson, M.I.; Klok, H.-A. Synthesis of Functional Polymers by Post-Polymerization Modification. Angew. Chem. Int. Ed. 2009, 48, 48–58. [Google Scholar] [CrossRef]
- Fishman, J.M.; Zwick, D.B.; Kruger, A.G.; Kiessling, L.L. Chemoselective, Postpolymerization Modification of Bioactive, Degradable Polymers. Biomacromolecules 2019, 20, 1018–1027. [Google Scholar] [CrossRef]
- Tempelaar, S.; Mespouille, L.; Dubois, P.; Dove, A.P. Organocatalytic Synthesis and Postpolymerization Functionalization of Allyl-Functional Poly(carbonate)s. Macromolecules 2011, 44, 2084–2091. [Google Scholar] [CrossRef]
- Thomas, A.W.; Dove, A.P. Postpolymerization Modifications of Alkene-Functional Polycarbonates for the Development of Advanced Materials Biomaterials. Macromol. Biosci. 2016, 16, 1762–1775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, W.H.; Sachsenhofer, R. ‘Click’ Chemistry in Polymer and Materials Science. Macromol. Rapid Commun. 2007, 28, 15–54. [Google Scholar] [CrossRef]
- Narayanan, A.; Maiti, B.; De, P. Exploring the post-polymerization modification of side-chain amino acid containing polymers via Michael addition reactions. React. Funct. Polym. 2015, 91–92, 35–42. [Google Scholar] [CrossRef]
- Riva, R.; Lussis, P.; Lenoir, S.; Jérôme, C.; Jérôme, R.; Lecomte, P. Contribution of ‘click chemistry’ to the synthesis of antimicrobial aliphatic copolyester. Polymer 2008, 49, 2023–2028. [Google Scholar] [CrossRef]
- Mantovani, G.; Ladmiral, V.; Tao, L.; Haddleton, D.M. One-pot tandem living radical polymerisation–Huisgens cycloaddition process (“click”) catalysed by N-alkyl-2-pyridylmethanimine/Cu(I)Br complexes. Chem. Commun. 2005, 2089–2091. [Google Scholar] [CrossRef] [PubMed]
- Döhler, D.; Michael, P.; Binder, W.H. CuAAC-Based Click Chemistry in Self-Healing Polymers. Acc. Chem. Res. 2017, 50, 2610–2620. [Google Scholar] [CrossRef]
- Cao, X.; Shi, Y.; Gan, W.; Naguib, H.; Wang, X.; Graff, R.W.; Gao, H. Effect of Monomer Structure on the CuAAC Polymerization To Produce Hyperbranched Polymers. Macromolecules 2016, 49, 5342–5349. [Google Scholar] [CrossRef]
- Zeng, M.; Cao, X.; Xu, H.; Gan, W.; Smith, B.D.; Gao, H.; Yuan, J. Synthesis and direct assembly of linear–dendritic copolymers via CuAAC click polymerization induced self-assembly (CPISA). Polym. Chem. 2020, 11, 936–943. [Google Scholar] [CrossRef]
- Riva, R.; Schmeits, S.; Jérôme, C.; Jérôme, R.; Lecomte, P. Combination of ringopening polymerization and ‘click chemistry’: Toward functionalization and grafting of poly(e-caprolactone). Macromolecules 2007, 40, 796–803. [Google Scholar] [CrossRef]
- Suksiriworapong, J.; Sripha, K.; Junyaprasert, V.B. Synthesis and characterization of bioactive molecules grafted on poly(e-caprolactone) by click chemistry. Polymer 2010, 51, 2286–2295. [Google Scholar] [CrossRef]
- Chen, D.; Chang, C.-C.; Cooper, B.; Silvers, A.; Emrick, T.; Hayward, R.C. Photopatternable Biodegradable Aliphatic Polyester with Pendent Benzophenone Groups. Biomacromolecules 2015, 16, 3329–3335. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, Y.; Li, Z.; Yang, G.; Kochovski, Z.; Chen, G.; Jiang, M. “Sweet” Architecture-Dependent Uptake of Glycocalyx-Mimicking Nanoparticles Based on Biodegradable Aliphatic Polyesters by Macrophages. J. Am. Chem. Soc. 2017, 139, 14684–14692. [Google Scholar] [CrossRef]
- Yu, Y.; Chen, C.-K.; Law, W.-C.; Sun, H.; Prasadb, P.N.; Cheng, C. A degradable brush polymer–drug conjugate for pH-responsive release of doxorubicin. Polym. Chem. 2015, 6, 953–961. [Google Scholar] [CrossRef]
- Ganivada, M.N.; Rao, N.V.; Dinda, H.; Kumar, P.; Sarma, J.D.; Shunmugam, R. Biodegradable Magnetic Nanocarrier for Stimuli Responsive Drug Release. Macromolecules 2014, 47, 2703–2711. [Google Scholar] [CrossRef]
- Şanal, T.; Koçak, İ.; Hazer, B. Synthesis of comb-type amphiphilic graft copolymers derived from chlorinated poly(ε-caprolactone) via click reaction. Polym. Bull. 2017, 74, 977–995. [Google Scholar] [CrossRef]
- Meduri, A.; Fuoco, T.; Lamberti, M.; Pellecchia, C.; Pappalardo, D. Versatile Copolymerization of Glycolide and rac-Lactide by Dimethyl(salicylaldiminato)aluminum Compounds. Macromolecules 2014, 47, 534–543. [Google Scholar] [CrossRef]
- Cao, J.; Gao, X.; Cheng, M.; Niu, X.; Li, X.; Zhang, Y.; Liu, Y.; Wang, W.; Yuan, Z. Reversible Shielding between Dual Ligands for Enhanced Tumor Accumulation of ZnPc-Loaded Micelles. Nano Lett. 2019, 19, 1665–1674. [Google Scholar] [CrossRef]
- Adams, M.L.; Lavasanifar, A.; Kwon, G.S. Amphiphilic block copolymers for drug delivery. J. Pharm. Sci. 2003, 92, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Shuai, Q.; Zhao, G.; Lian, X.; Wan, J.; Cen, B.; Zhang, W.; Liu, J.; Su, W.; Wang, H. Self-assembling poly(ethylene glycol)-block-polylactide-cabazitaxel conjugate nanoparticles for anticancer therapy with high efficacy and low in vivo toxicity. Int. J. Pharm. 2020, 574, 118879. [Google Scholar] [CrossRef]
- Torchilin, V.P. Structure and design of polymeric surfactant-based drug delivery systems. J. Control. Release 2001, 73, 137–172. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, X.; Graff, R.W.; Phillip, W.A.; Gao, H. Synthesis of degradable molecular brushes via a combination of ring-opening polymerization and click chemistry. J. Am. Chem. Soc. 2005, 127, 7404–7410. [Google Scholar] [CrossRef]
- Arote, R.; Kim, T.-H.; Kim, Y.-K.; Hwang, S.-K.; Jiang, H.-L.; Song, H.-H.; Nah, J.-W.; Cho, M.-H.; Cho, C.-S. A biodegradable poly(ester amine) based on polycaprolactone and polyethylenimine as a gene carrier. Biomaterials 2007, 28, 735–744. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Copolymer | [LA] or [CL]/[AVL] Molar Ratio in the Feed | Mn,NMR(a) (g moL−1) | Mn,GPC (b) (g moL−1) | PDI (c) | Number of Alkynyl (d) |
|---|---|---|---|---|---|
| mPEG2K-b-P(AVL-co-LA)2K (AVL:LA = 1:1) (PPAL1) | 1/1.9 | 4100 | 3600 | 1.33 | 7 |
| mPEG2K-b-P(AVL-co-LA)2K (AVL:LA = 1:3) (PPAL2) | 3/1.9 | 3900 | 4200 | 1.31 | 4 |
| mPEG4K-b-P(AVL-co-LA)4K (AVL:LA = 1:1) (PPAL3) | 1/1.9 | 7800 | 6900 | 1.25 | 14 |
| mPEG4K-b-P(AVL-co-LA)4K (AVL:LA = 1:3) (PPAL4) | 3/1.9 | 7600 | 8000 | 1.30 | 7 |
| mPEG2K-b-P(AVL-co-CL)2K (AVL:CL = 1:1) (PPAC1) | 1/1.9 | 3900 | 4200 | 1.27 | 7 |
| mPEG2K-b-P(AVL-co-CL)2K (AVL:CL = 1:3) (PPAC2) | 3/1.9 | 3900 | 4300 | 1.25 | 4 |
| mPEG4K-b-P(AVL-co-CL)4K (AVL:CL = 1:1) (PPAC3) | 1/1.9 | 7500 | 7000 | 1.23 | 12 |
| mPEG4K-b-P(AVL-co-CL)4K (AVL:CL = 1:3) (PPAC4) | 3/1.9 | 7800 | 8100 | 1.31 | 7 |
| mPEG2K-b-P(AVL-co-LA)2K (AVL:LA = 1:1) (PPAL1) | |||||
|---|---|---|---|---|---|
| Azido Molecule | Grafting Efficiency (a) (%) | Yield (%) | Mn,NMR (b) (g mol−1) | Mn,GPC(c) (g mol−1) | PDI (d) |
| 1 | 98.6 | 65.5 | 4100 | 5800 | 1.25 |
| 2 | 96.7 | 69.5 | 5100 | 5600 | 1.28 |
| 3 | 100.0 | 82.5 | 5300 | 5300 | 1.28 |
| 4 | 93.9 | 75.0 | 5100 | 4400 | 1.32 |
| 5 | 100.0 | 74.0 | 5100 | 5900 | 1.24 |
| 6 | 98.3 | 85.6 | 5100 | 4800 | 1.14 |
| 7 | 86.3 | 70.0 | 5100 | 4900 | 1.20 |
| 8 | 83.6 | 91.5 | 4600 | 5100 | 1.22 |
| 9 | 98.4 | 70.2 | 4700 | 4500 | 1.22 |
| Functional Copolymer | Particle Size (nm) | PDI | CMC (mg mL−1) |
|---|---|---|---|
| PPAL1 1 | 159.65 ± 5.6 | 0.153 ± 0.02 | 0.059 |
| PPAL1 2 | 18.83 ± 1.2 | 0.312 ± 0.04 | 0.037 |
| PPAL1 3 | 27.27 ± 3.2 | 0.140 ± 0.02 | 0.007 |
| PPAL1 4 | 18.14 ± 3.0 | 0.259 ± 0.01 | 0.010 |
| PPAL1 5 | 16.75 ± 0.7 | 0.301 ± 0.04 | 0.023 |
| PPAL1 6 | 35.13 ± 2.9 | 0.248 ± 0.05 | 0.071 |
| PPAL1 7 | 139.7 ± 5.4 | 0.141 ± 0.03 | 0.075 |
| PPAL1 8 | 16.69 ± 3.3 | 0.218 ± 0.01 | 0.058 |
| PPAL1 9 | 22.09 ± 4.2 | 0.261 ± 0.02 | 0.034 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, G.; Ge, T.; Yan, Y.; Shuai, Q.; Su, W.-K. Highly Efficient Modular Construction of Functional Drug Delivery Platform Based on Amphiphilic Biodegradable Polymers via Click Chemistry. Int. J. Mol. Sci. 2021, 22, 10407. https://doi.org/10.3390/ijms221910407
Zhao G, Ge T, Yan Y, Shuai Q, Su W-K. Highly Efficient Modular Construction of Functional Drug Delivery Platform Based on Amphiphilic Biodegradable Polymers via Click Chemistry. International Journal of Molecular Sciences. 2021; 22(19):10407. https://doi.org/10.3390/ijms221910407
Chicago/Turabian StyleZhao, Guangkuo, Tongtong Ge, Yunfeng Yan, Qi Shuai, and Wei-Ke Su. 2021. "Highly Efficient Modular Construction of Functional Drug Delivery Platform Based on Amphiphilic Biodegradable Polymers via Click Chemistry" International Journal of Molecular Sciences 22, no. 19: 10407. https://doi.org/10.3390/ijms221910407