Abstract
Accurate inference of the relationship between non-coding RNAs (ncRNAs) and drug resistance is essential for understanding the complicated mechanisms of drug actions and clinical treatment. Traditional biological experiments are time-consuming, laborious, and minor in scale. Although several databases provide relevant resources, computational method for predicting this type of association has not yet been developed. In this paper, we leverage the verified association data of ncRNA and drug resistance to construct a bipartite graph and then develop a linear residual graph convolution approach for predicting associations between non-coding RNA and drug resistance (LRGCPND) without introducing or defining additional data. LRGCPND first aggregates the potential features of neighboring nodes per graph convolutional layer. Next, we transform the information between layers through a linear function. Eventually, LRGCPND unites the embedding representations of each layer to complete the prediction. Results of comparison experiments demonstrate that LRGCPND has more reliable performance than seven other state-of-the-art approaches with an average AUC value of 0.8987. Case studies illustrate that LRGCPND is an effective tool for inferring the associations between ncRNA and drug resistance.
1. Introduction
Non-coding RNAs (ncRNAs) play special roles in the development, differentiation, and aging of cells. Numerous studies have shown that ncRNAs are widely involved in human pathological activities. They act as biomarkers to provide new targets for the treatment of diseases such as cancer [1]. Non-coding RNAs such as microRNAs (miRNAs), circular RNAs (circRNAs), and long ncRNAs (lncRNAs) have aroused great interest of researchers. miRNAs are short regulatory biomolecules that are involved in the post-transcriptional regulation of gene expression [2]. Compared with linear miRNAs, circRNAs [3] are more stable and may function as transporters or scaffolds [4]. They exert essential biological functions by acting as microRNA or protein inhibitors (“sponges”), regulating protein function, or being translated themselves [5]. lncRNA can play a role in regulating cooperating proteins [6]. piRNA (Piwi-Interacting RNA) has been relatively poorly studied compared to those three. piRNA can form a piRNA/PIWI complex with PIWI proteins to affect gene expression and mainly function to suppress the activity of transposons [7,8]. There are synergies among RNAs. For example, lncRNA can act as a molecular sponge of miRNA to regulate the expression of its target gene [9,10,11,12].
According to a statistical cancer report released by the American Cancer Society [13], it is estimated that there will be approximately 4950 new cancer cases and 1600 deaths due to cancer every day in the United States. Unfortunately, the development of drug resistance greatly increases the probability of recurrence and significantly reduces the cure rate. Drug resistance has become a major obstacle to clinical treatment.
With the development of sequencing technology, it has been reported that cancer resistance to treatment is related to mutations of the cell’s genome [14,15]. The instability of the genome may change the phenotype of the tumor and lead to drug resistance. Studies have shown that some ncRNAs, such as miRNAs, can act as rheostats to regulate protein output [16]. The abnormal expression of ncRNAs is not only associated with several diseases but also may promote drug resistance of cancer cells [17,18]. circRNA acts as a miRNA sponge and enhances the response of HCC (hepatocellular carcinoma) cells to chemotherapy with cisplatin [19]. lncRNA enhanced drug resistance in AML (acute myeloid leukemia) cells by inhibiting miR-186 [20]. Overexpression of miRNA-194 can make HCC cells more sensitive to sorafenib [21]. Increasing evidence suggests that drug resistance is affected by ncRNA. Exploring their interaction will provide new insights for improving the therapeutic effect.
The relationship between ncRNAs and drug resistance has been gradually discovered, and some databases already provide relevant data. The ncDR database [22] provides 135 compounds and 1050 ncRNAs. Additional information on compounds and ncRNAs, such as ncRNA genomic contexts, had also been added. NoncoRNA [23] covers the basic calculation of ncRNA, drugs, diseases, etc., and includes experimental detection techniques, drug response, and other information. However, existing knowledge is minimal compared to the unknown associations. Discovering possible relationships between ncRNAs and drugs is beneficial for understanding related drug resistance mechanisms and accelerating drug development. To some extent, the traditional biological experiments are difficult to be carried out due to the factors such as difficult control and high time cost. Computational methods are useful accelerators of this process, but very little work has been done in this area.
In recent years, association prediction methods have been greatly developed in Bioinformatics. GCMDR [24] established a three-layer latent factor model to predict miRNA-disease associations introducing features such as miRNA expression profile and drug PubChem substructure fingerprints into the model. Zhu et al. [25] utilized the matrix completion method. SDLDA [26] introduced singular value decomposition and ILNCRNADIS-FB [27] calculated the three-dimensional feature blocks to capture characteristics. In a different way, SAEMDA [28] extracts features through semantic similarity. In terms of the prediction of circRNA-disease associations, AE-RF algorithm [29] also integrates many information sources to obtain the depth features. DMFCDA [30] constructed a circRNA-disease matrix with explicit and implicit feedback to capture the non-linear features. Deng et al. [31] constructed a heterogeneous information network (HIN) containing multiple subnetworks. A great deal of research has focused on the microbe-disease association prediction. The KATZHMDA [32] introduced the Gaussian kernel to perform a complete and easy reconstruction of the microbe-disease relationship. The ABHMDA [33] is a strong classifier based on the existing model to achieve better self-adaptability. Liu et al. [34] used matrix decomposition based on neural networks to obtain nonlinear latent features to infer disease-related microbes. The NTSHMDA [35] successfully reduced the prediction error by assigning random walks according to different weights.
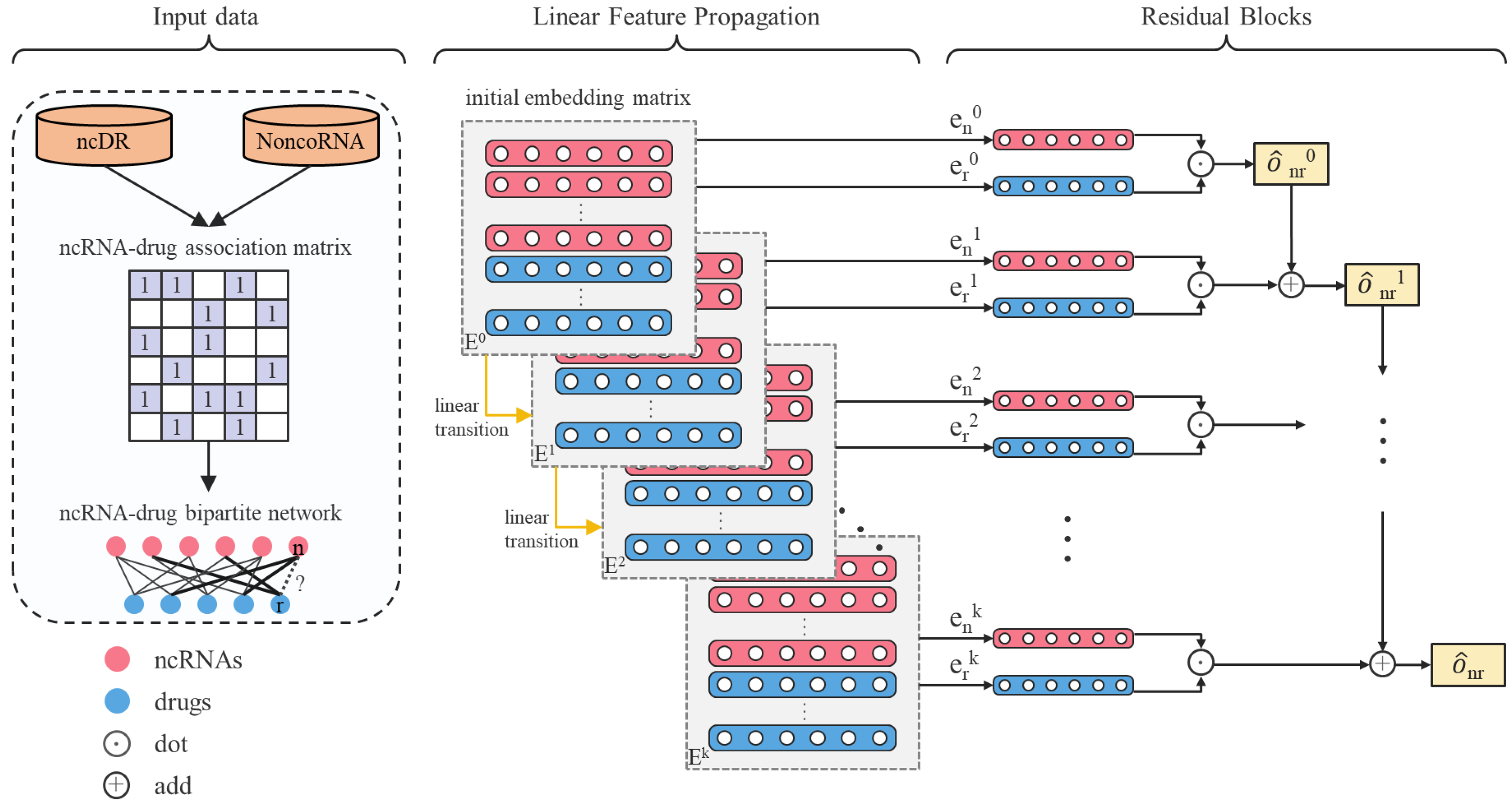
Although the above methods have achieved good results, some problems and shortcomings still hinder more comprehensive potential feature mining. The lack of relevant biological data and information leads to noise in the calculated features, which reduces prediction accuracy. Existing association predictions are more dependent on the existing similarities in the database. When the number of ncRNAs and diseases increases, the existing calculation models are difficult to draw conclusions efficiently, so they are not suitable for large-scale data sets. Therefore, these methods are not applicable when predicting the relationship between multiple ncRNAs and drug resistance. Although more and more ncRNA-drug resistance associations have been determined and existing databases provide relevant data, the existing knowledge is still very limited compared with the unknown potential associations. Here we propose an efficient approach based on a linear residual graph convolutional network, LRGCPND, which only employs ncRNA and drug resistance validated interactions. Initially, LRGCPND constructs a bipartite graph through the association network of ncRNA and drug resistance, where the edges represent the hidden interaction factors between the two types of nodes. The unconnected edges may have associations that are not obvious to identify. LRGCPND then fleetly aggregates the intrinsic characteristics of neighbor nodes in the former layer and performs the linear transition. After the specified number of iterations, it fuses the embeddings of previous convolutional layers through residual learning to favorably explore the interactions between ncRNA and drug resistance. LRGCPND achieves the best performance compared with the other advanced computational methods. Case studies of two anti-cancer drugs demonstrate the practical capability of LRGCPND. The flow chart of LRGCPND is shown in Figure 1.
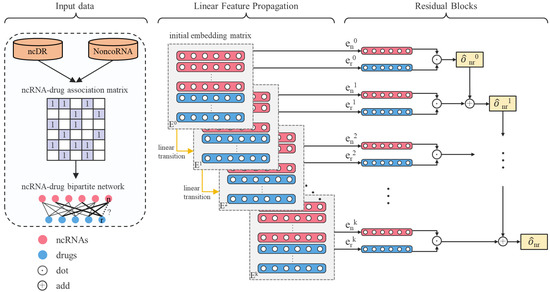
Figure 1.
Flow chart of LRGCPND. denotes the embedding matrix in layer 0. and denote the embedding of ncRNA n and drug d in layer 0, respectively. LRGCPND contains three steps: aggregation, linear transition, and residual learning. In the feature aggregation step, we use the spectral rule to aggregate the features of neighboring nodes. After that, the linear transformation is adopted to speed up the forward propagation. Finally, we add a residual block to fuse the characteristics of low-layer nodes directly, attaining higher-layer potential features.
2. Results and Discussion
2.1. Experimental Setup
To objectively and systematically evaluate the ability of LRGCPND and expedite comparison with other methods, we perform -fold cross-validation (-fold CV) on the collected dataset. All verified associations are randomly divided into parts. Each part is picked as positive samples with an equal quantity of unlabeled samples as negative samples to form the testing set. Meanwhile, the equivalent operation is performed on the remaining parts to obtain the training set. This process ends after iterations.
Even if there may be latent associations in the selected negative samples, since they account for a tiny proportion in the entire unverified sample set, the influence is negligible.
2.2. Evaluation Criteria
To observe intuitively and comprehensively, we measure the performance of models by widely adopted metrics, including AUC, AUPR, (Acc.), (P.), (R.), and scores, which are defined by the following formula:
and represent the number of correct and incorrect classifications in the related ncRNA-drug resistance pairs. In contrast, and represent the number of correct and incorrect classifications in the unrelated pairs. By adjusting the threshold, we can plot the receiver operating characteristic (ROC) curve and precision-recall (PR) curve and then calculate the area under the curves to get AUC and AUPR, respectively.
2.3. Performance Evaluation for LRGCPND
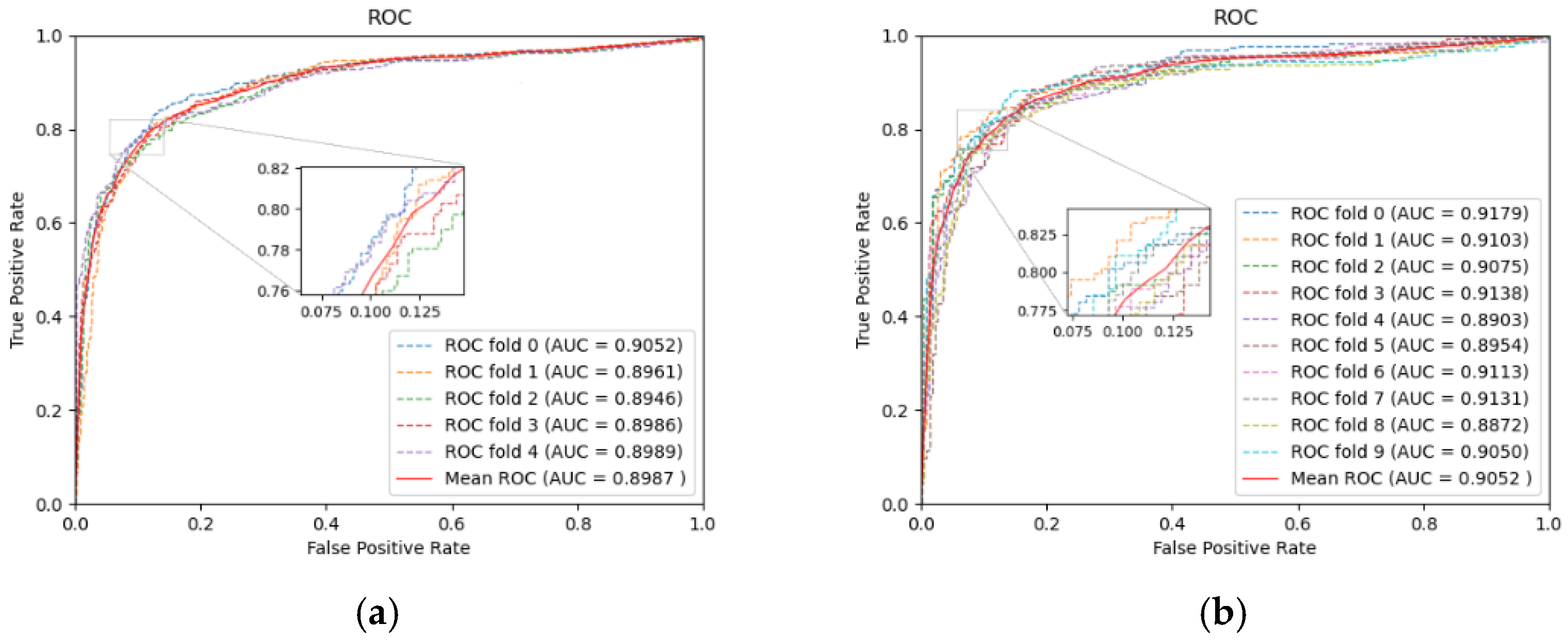
To evaluate the identification ability of our model, we performed five-fold and ten-fold CV on the dataset specified above. Table 1 lists the specific results in five-fold CV, and Figure 2 displays the ROC curves. In five-fold CV, the average values of AUC, AUPR, and Accuracy reach 0.8987, 0.9094, 0.8342, respectively. With the increasing size of the training set, training of the model will achieve a more thorough level. So, in ten-fold CV, the AUC increased to 0.9052. As seen from the above experimental results, LRGCPND can accurately and effectively identify potential ncRNAs related to drug resistance.

Table 1.
Prediction results of LRGCPND in five-fold CV.
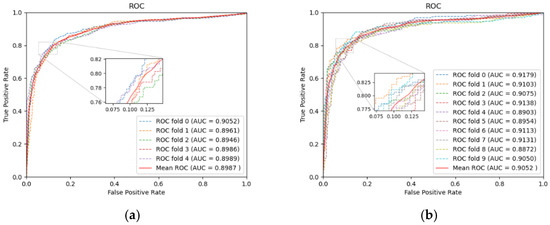
Figure 2.
Performance of LRGCPND in five-fold CV and ten-fold CV, respectively. (a) ROC curves yielded by LRGCPND in five-fold CV. (b) ROC curves yielded by LRGCPND in ten-fold CV.
2.4. Effects of Parameters
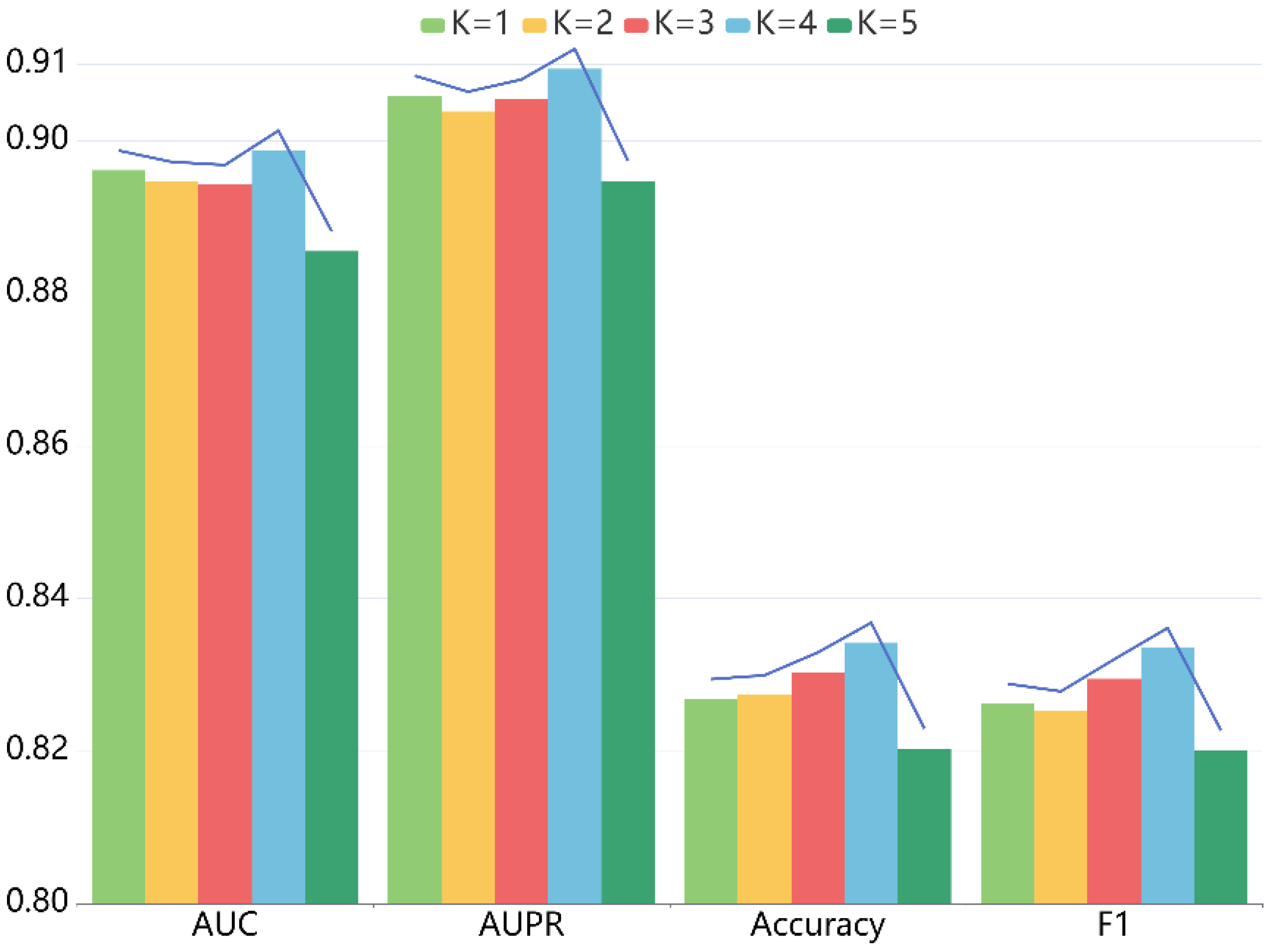
For LRGCPND, there are two crucial parameters: the depth of propagation and the dimension of embedding, which influence the prediction capability. For one thing, we explored the impact of layer depth , following the settings of other parameters constant. When ranges from 1 to 5, we performed five-fold CV. Table 2 lists the detailed data, and Figure 3 is the trend chart of different indicators. Our model achieves the best performance when is equal to 4.
Table 2.
Prediction results of LRGCPND with different depth K.
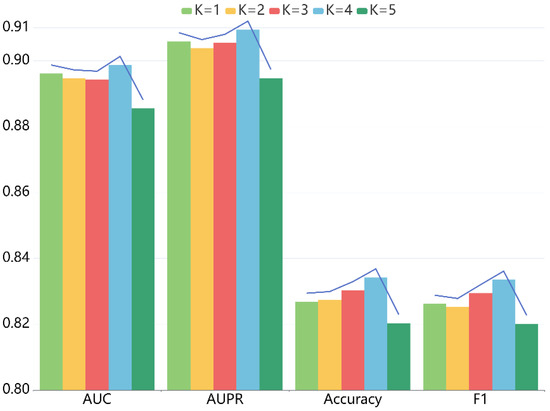
Figure 3.
Effects of depth K on the performance of LRGCPND.
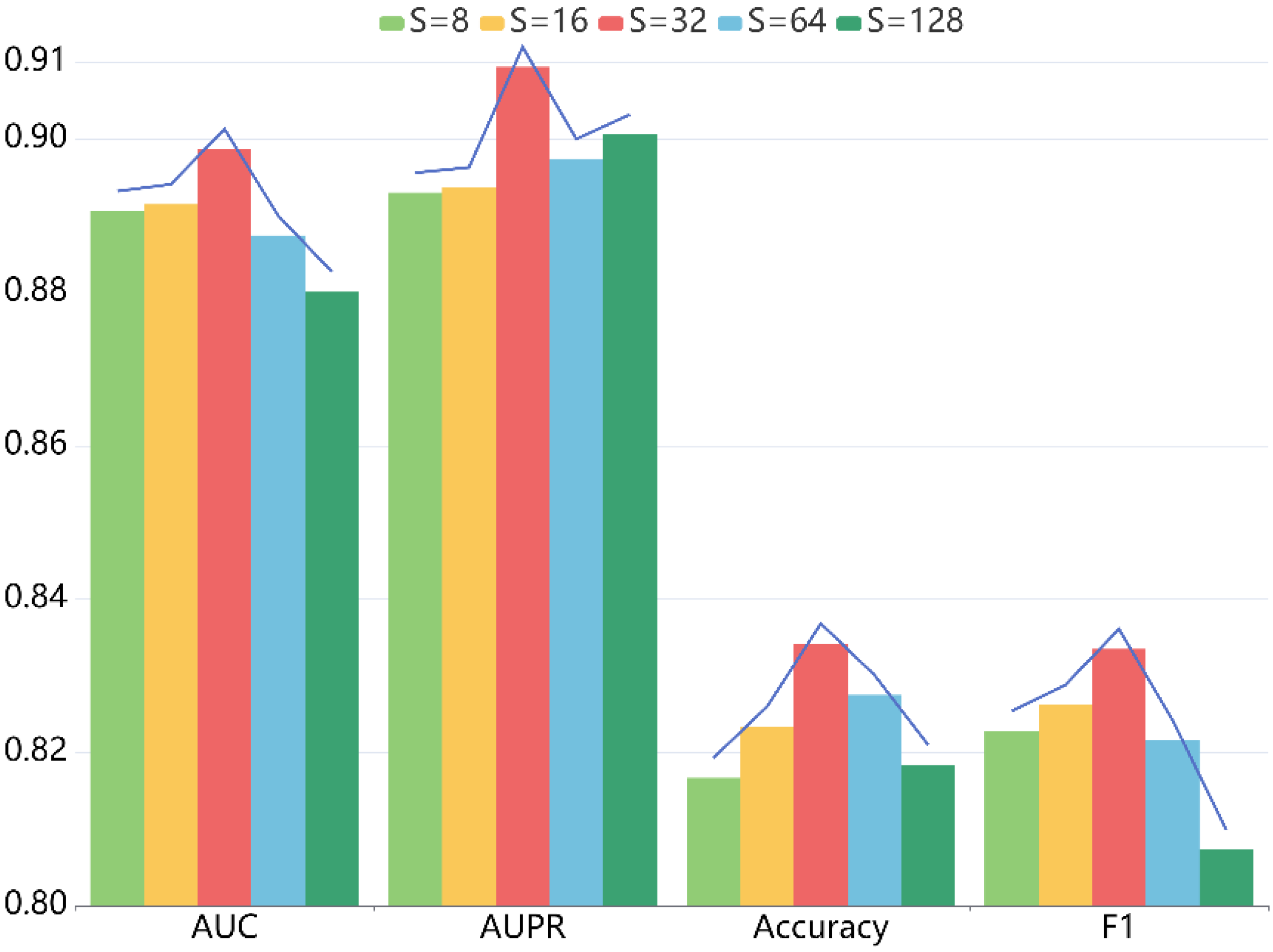
For another thing, the embedding dimension also has a critical role. When setting the value of to 8, 16, 32, 64, 128 sequentially, we conducted five-fold CV to measure the impact on the prediction ability of our model. Table 3 shows the detailed statistics, and Figure 4 indicates the trend of diverse metrics. From the results, we can conclude that when varies from 8 to 128, the performance first monotonically improves. That is because the larger embedding dimension enhances the expressivity of LRGCPND to a certain extent. When is 32, it reaches the optimum. Then as increases, it starts to produce adverse effects on the performance.
Table 3.
Prediction results of LRGCPND with different embedding size S.
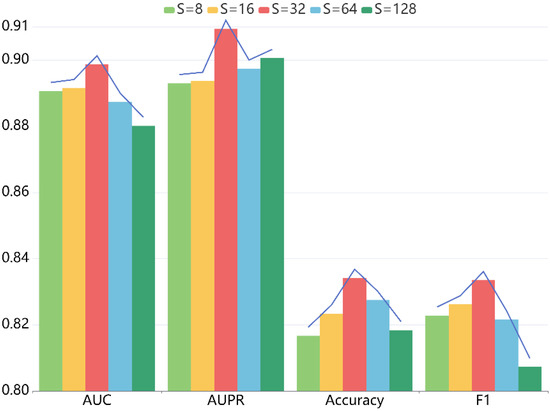
Figure 4.
Effects of embedding size S on the performance of LRGCPND.
In other experiments, we employ the optimal values obtained above as the default of model parameters.
2.5. Comparison with Other Approaches
Since inferring ncRNA-drug resistance interactions is a relatively new area, no researchers have proposed relevant solutions already. Nonetheless, reviewing other association prediction methods in bioinformatics still provides significant references for the performance of our model. To further assess the effectiveness of LRGCPND, we compared it with seven advanced approaches in directions of lncRNA-disease, circRNA-disease, and microbe-disease.
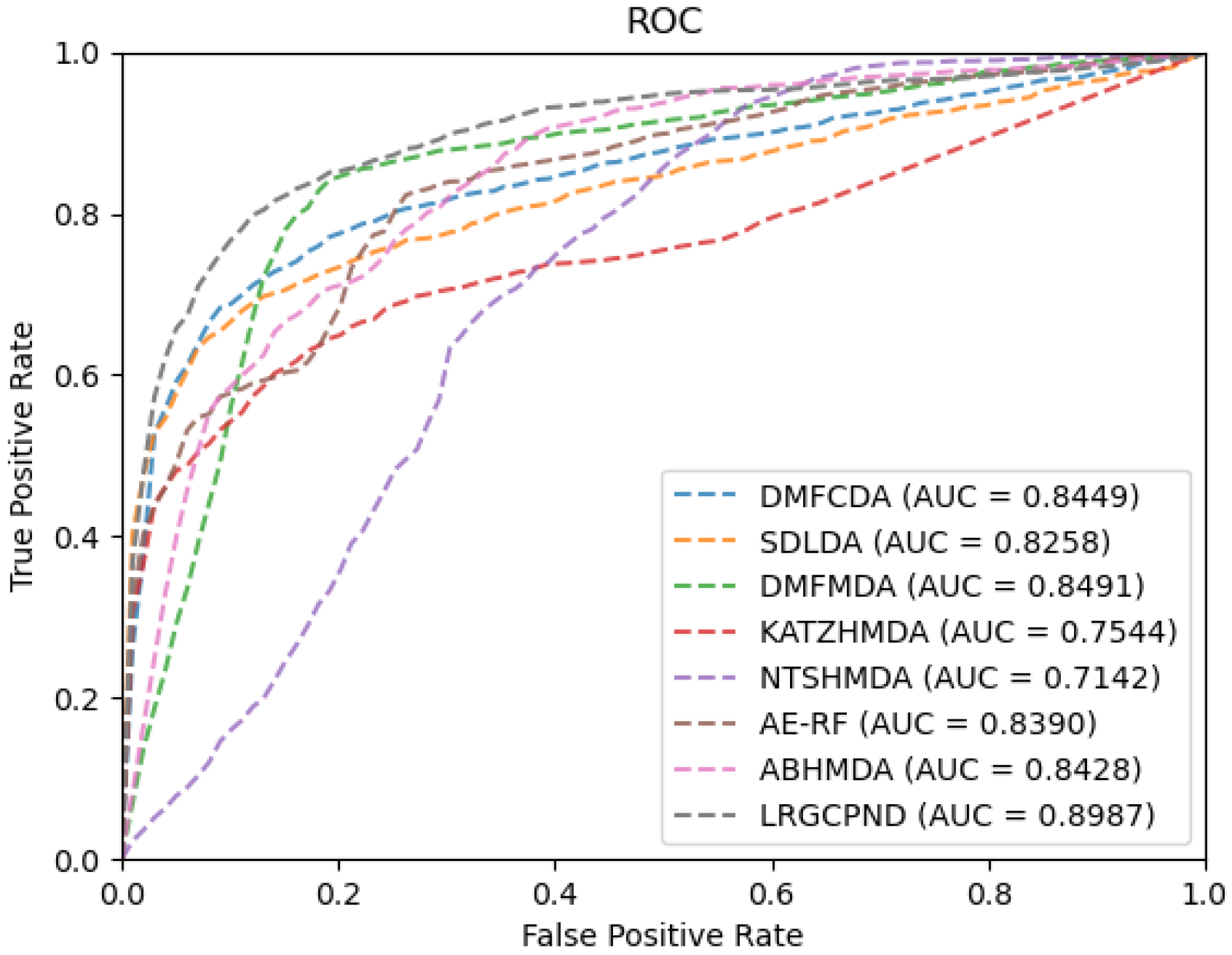
For the sake of rigor, we need to point out that since AE-RF [29] and ABHMDA [33] employ other similarity-based features besides the Gaussian interaction profile (GIP) kernel similarity. Considering the scarcity of relevant biological resources and convenience, we only calculated the GIP similarity for them in the experiments. Furthermore, the adjacency matrix allocated at the beginning of training is different, so the topology information of the interaction network needs to be re-extracted. We re-calculated the GIP similarity matrices during each cross-validation process for similarity-based methods, AE-RF, KATZHMDA [32], NTSHMDA [35], and ABHMDA. As plotted in Figure 5, it is evident that LRGCPND leads others with the average AUC value of 0.8987, which is 5.84% higher than the second-best method DMFMDA [34].
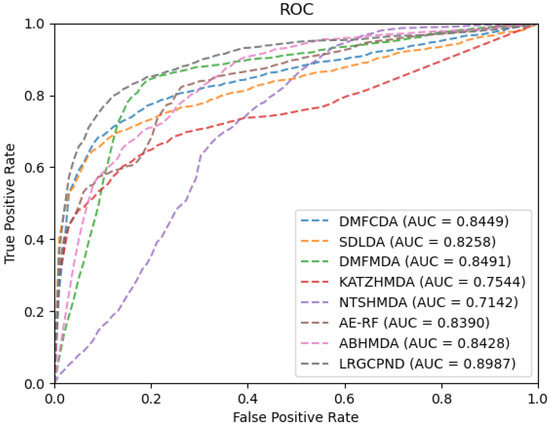
Figure 5.
ROC curves of different methods on our dataset.
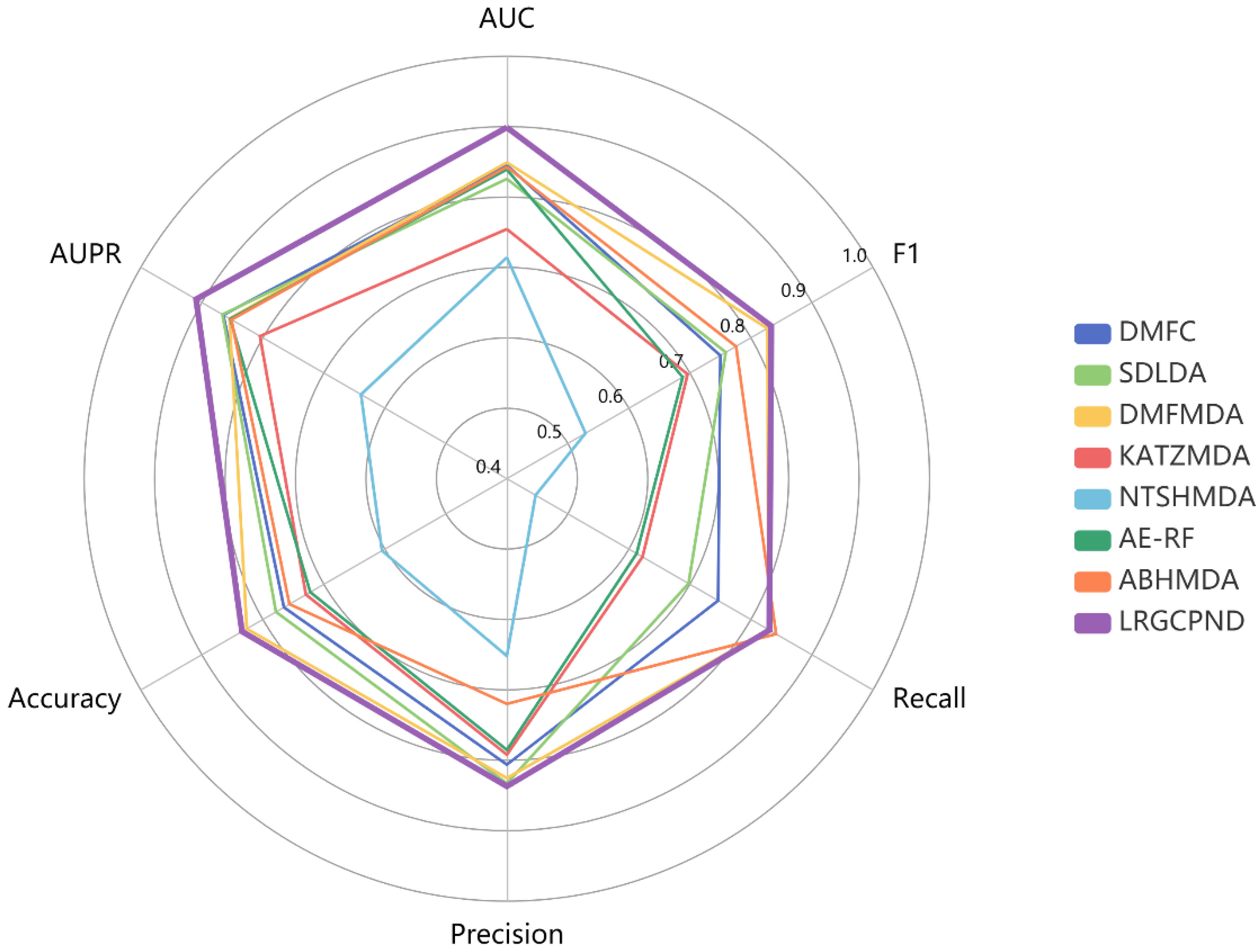
From statistics of various metrics listed in Table 4, except that the Recall value is slightly lower than ABHMDA, our model yields the optimal identification ability. Its AUPR, Accuracy, and F1 values achieve 0.9094, 0.8342, 0.8335, respectively. We also drew a radar chart to intuitively and comprehensively measure the capabilities of diverse models through various metrics, as shown in Figure 6. All six evaluation metrics range from 0.4 to 1.0. The farther the point from the center of the circle, the higher the value. It is also apparent to conclude that LRGCPND advantages over other methods.
Table 4.
Prediction results of different methods in five-fold CV.
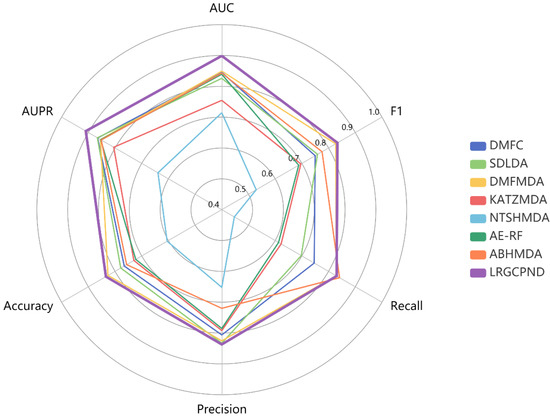
Figure 6.
Performance comparison using multiple metrics in five-fold CV.
These experimental results sufficiently demonstrate that our model is reliable and promising in inferring candidate ncRNA-drug resistance pairs.
2.6. Case Studies
The discovery of unknown associations between ncRNA and drug resistance matters tremendously for practical application. Thus, we selected two drugs, Cisplatin and Paclitaxel, and conducted case studies. Precisely, for a particular drug, to start with, we removed the known associated ncRNAs. Then, the remaining ncRNAs were sorted in descending order following the values predicted by LRGCPND. Lastly, we screened the top 15 ncRNAs and searched for supporting evidence in published literature.
Cisplatin is a common chemotherapeutic drug used to treat numerous cancers, including lung cancer, head and neck cancer, and ovarian cancer. Resistance frequently causes reduced efficacy of Cisplatin in chemotherapy [36]. Paclitaxel is another widely applied taxane medication. Chemoresistance to Paclitaxel makes its clinical application problematic [37]. Table 5 and Table 6 summarize the top 15 candidate ncRNAs of Cisplatin and Paclitaxel, respectively. We can see that 10 and 7 of the former and the latter are confirmed by existing evidence, indicating that our method has an excellent capability for predicting novel associated ncRNAs for drugs in terms of resistance. It is noteworthy that other unproven associations are likely to exist and deserve further relevant experiments.
Table 5.
The top 15 miRNAs related to Cisplatin resistance predicted by LRGCPND.
Table 6.
The top 15 miRNAs related to Paclitaxel resistance predicted by LRGCPND.
3. Materials and Methods
3.1. Datasets
NoncoRNA: NoncoRNA [23] contains 5568 ncRNAs and 154 drugs in 134 cancers. This is the first database that provides diverse ncRNAs and associations between ncRNAs and drug resistance in cancers. We use the Feb 2020 version of the NoncoRNA database, which is publicly released at http://www.ncdtcdb.cn:8080/NoncoRNA (accessed on 10 March 2021).
ncDR: Hitherto, one of the most frequently used databases is ncDR [22] in the field of drug resistance-related non-coding RNA. Here, we adopt the data downloaded from the June 2016 version of the ncDR database. The dataset contains 5864 associations between ncRNAs and drug resistance, including 877 miRNAs and 162 lncRNAs from nearly 900 pieces of published literature. It now can be available on the website http://www.jianglab.cn/ncDR (accessed on 10 March 2021).
We manually integrated a set of 2693 associations between ncRNAs and drug resistance from NoncoRNA and ncDR datasets, including 625 ncRNAs and 121 drugs. Here we choose the experimental data. Besides, we clean the dataset by removing the redundant ones and associations in which a ncRNA only contains one drug resistance binding. The dataset can be expressed as:
where represents the positive dataset, which contains 2693 ncRNA-drug resistance associations verified with wet experiment. represents the negative dataset, which contains a total of 72,932 ncRNA-drug resistance associations without verified experimentally. Earlier in Section 2.1, we have introduced the detail of sampling. Our dataset can be downloaded on the website https://github.com/TroyePlus/LRGCPND (accessed on 30 July 2021).
3.2. Problem Description
In order to predict the relationship between ncRNA and drug resistance, for a given set of m ncRNAs and n drugs, we use and respectively represent the collection of ncRNAs and drugs, and is the correlation matrix. If ncRNA is related to drug resistance , then the entry , otherwise . However, does not mean that ncRNA has no association with the drug . It may be that the relationship has not been found yet. In addition, we use to represent the linked set of ncRNA found, and to represent the non-linked set. is defined as the set of all linked ncRNA and drug resistance pairs.
3.3. Graph Construction
We use a bipartite graph to express the associations between different ncRNAs and drug resistance, where , are the previously defined ncRNA set and drug set. Every edge belonging to represents a verified association between ncRNA and drug resistance .
3.4. Graph Embedding
Matrix factorization is a common method of graph embedding. Matrix factorization only uses the linear relationship between entities and can be applied to data that only contains associations. However, the matrix factorization method cannot make full use of data information, and its ability to extract high-order features is weak. In recent years, graph-based models have become popular in the field of semi-supervised classification. The network built by graphs combined with deep learning methods can be applied to graph embedding to obtain vector representations of graphs or graph nodes [38]. Graph convolutional neural network is often used in the field of association prediction in biological information. The design of graph convolutional neural network is inspired by convolutional neural network, which is widely used in the field of computer vision. Its advantage is that it can extract the structural features of node neighborhoods and then learn higher-order relationships. But obvious disadvantages are the over-smoothing problem and time-consuming calculation. In this work, the task of ncRNA-drug resistance association is similar to the recommendation problem, where ncRNA corresponds to the user, and the drug resistance is equivalent to the project. The verified association is equal to the user’s viewing/shopping history. Therefore, the graph convolutional neural network method, which is very popular in the recommendation task, can be applied to our problem. Here, we solve the above problems with linear propagation and residual block based on GCN. We first construct the adjacency matrix of the bipartite graph as follows:
Then use to represent the embedding matrix of ncRNA and drug resistance. We generate initial values from the normal distribution given standard deviation = 0.1 to fill the initial embedding matrix with . Every epoch in training, LRGCPND treats the embedding matrix as input:
where is calculated in each iteration and will be updated.
3.5. Feature Aggregation
There is no intra-domain edge in the bipartite graph, so the message passing and node feature aggregation are only performed through the inter-domain edge for the convolution of the bipartite graph. We use the spectral rule to aggregate feature of graph:
where , is the identity matrix. is the degree matrix of . As is adopted widely in GCN, spectral rule considers not only the degree of th node, but also the degree of the th node when calculating the aggregation of the th node.
3.6. Linear Transition
We remove the nonlinear transformation functions at the end of each layer. Despite the linear propagation of LRGCPND, the “receptive field” of our model is the same as a -layer GCN. The step embedding could be calculated as:
where represents the linear transformation, is the step embedding.
Due to the linear transformation, we can get the matrix form to model each ncRNA ’s and drug-resistance ’s embedding:
where is the diagonal degree of ncRNA (drug-resistance ) in . () represents the neighbors of node () in .
3.7. Residual Block in LRGCPND
In a graph convolution network, there is an over-smoothing problem caused by network stacking. The role of GCN is equivalent to low-pass filtering, making the input signal smoother, which is an inherent advantage of the GCN model. However, after multiple executions of GCN operations, the signals will tend to be the same, so the diversity of node characteristics is lost, which is a fatal disadvantage for tasks related to node classification. From the perspective of the spectral domain, analyzing the frequency response function of GCN points out that if the smoothing operation is continuously performed on a graph signal, the graph signal will eventually become equal everywhere, ultimately losing the discrimination information between nodes. Here we adopt the residual block [39] proposed by Kaiming He to establish identity mapping. The output of our model can be described as:
3.8. Model Optimization
BPR [40] is a sorting algorithm based on matrix decomposition. It is not a global scoring optimization but a sorting optimization for each ncRNA’s related drug-resistance preferences. It is a pairwise sorting algorithm. For each triple , the model hopes to make the ncRNA ’s difference between drug-resistance and more obvious.
where , , is updated after the model backward propagation. controls the strength of regularization. denotes the positive subset for of drug set . represents the pairs containing positive sample and negative sample .
4. Conclusions
Drug resistance response has caused vital challenges to clinical treatment. Numerous studies have indicated that ncRNA plays a pivotal role in the mechanisms of drug resistance. Accurately identifying the ncRNA-drug resistance association pairs is conducive to drug development and promotes clinical treatment. In this work, we propose LRGCPND, a graph convolutional network computational framework for mining the latent associations between ncRNA and drug resistance through linear transition and residual prediction. To our best knowledge, this is the first computational prediction approach in this field. We represent the relationship between ncRNA and drug resistance in a bipartite graph and exploit limited information to learn complex latent factors for edge prediction. LRGCPND first captures the neighborhood representations by aggregation. Then, it performs feature transformation through linear operations. Finally, the embedding vectors of convolutional layers are concatenated through residual blocks to achieve prediction.
Experimental results and case studies corroborate the effectiveness of our model, to which several aspects may contribute. We utilize graph convolution to perform relatively more adequate representation learning on the original association data with inadequate information. Residual blocks enable the model to attain higher-layer potential characteristics, and linear feature propagation keeps the model lightweight and flexible to extend to datasets on a large scale. In conclusion, our model is promising and facilitates further research in predicting novel associated ncRNAs for drug resistance. Our study helps build a systematic map of ncRNA and drug resistance, provides more insights into drug resistance, and aids in identifying effective therapeutic combinations.
As with many computational prediction methods, LRGCPND also has its limitations. First, LRGCNPND only utilizes ncRNA-drug resistance association data. The quality and coverage of the association data would affect the performance. Second, LRGCPND makes predictions with ncRNAs containing subtypes. Despite this provides insights from a broader perspective, differences between subtypes would cause bias. In the future, we will combine the attention mechanism and integrate multiple heterogeneous data to improve the performance further.
Author Contributions
Conceptualization, Y.L. and L.D.; methodology, Y.L., R.W., S.Z., H.X. and L.D.; software, Y.L., R.W., S.Z. and H.X.; validation, Y.L. and L.D.; investigation, Y.L. and L.D.; resources, Y.L.; data curation, Y.L.; writing—original draft preparation, Y.L., R.W., S.Z. and H.X.; writing—review and editing, Y.L. and L.D.; supervision, L.D.; project administration, L.D.; funding acquisition, L.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Natural Science Foundation of China under grant number 61972422. The APC was funded by National Natural Science Foundation of China under grant number 61972422.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The code and data of this study are available at https://github/TroyePlus/LRGCPND (accessed on 30 July 2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wang, L.; Xuan, Z.; Zhou, S.; Kuang, L.; Pei, T. A Novel Model for Predicting LncRNA-disease Associations based on the LncRNA-MiRNA-Disease Interactive Network. Curr. Bioinform. 2019, 14, 269–278. [Google Scholar] [CrossRef]
- Treiber, T.; Treiber, N.; Meister, G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 2019, 20, 5–20. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sorrentino, J.A.; Wang, K.; Slevin, M.K.; Burd, C.E.; Liu, J.; Marzluff, W.F.; Sharpless, N.E. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA 2013, 19, 141–157. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, L.S.; Hansen, T.B.; Veno, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [Green Version]
- Dong, P.; Xiong, Y.; Konno, Y.; Ihira, K.; Xu, D.; Kobayashi, N.; Yue, J.; Watari, H. Critical Roles of PIWIL1 in Human Tumors: Expression, Functions, Mechanisms, and Potential Clinical Implications. Front. Cell Dev. Biol. 2021, 9, 656993. [Google Scholar] [CrossRef]
- Liu, Y.; Dou, M.; Song, X.; Dong, Y.; Liu, S.; Liu, H.; Tao, J.; Li, W.; Yin, X.; Xu, W. The emerging role of the piRNA/piwi complex in cancer. Mol. Cancer 2019, 18, 123. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.L. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef]
- Yang, Q.; Wu, J.; Zhao, J.; Xu, T.; Han, P.; Song, X. The Expression Profiles of lncRNAs and Their Regulatory Network During Smek1/2 Knockout Mouse Neural Stem Cells Differentiation. Curr. Bioinform. 2020, 15, 77–88. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, C.; Li, M.; Yu, X.; Liu, H.; Chen, Q.; Wang, J.; Shen, S.; Jiang, J. Integrative Analysis of miRNA-mediated Competing Endogenous RNA Network Reveals the lncRNAs-mRNAs Interaction in Glioblastoma Stem Cell Differentiation. Curr. Bioinform. 2021, 15, 1187–1196. [Google Scholar] [CrossRef]
- Tian, L.; Wang, S.-L. Exploring miRNA Sponge Networks of Breast Cancer by Combining miRNA-disease-lncRNA and miRNA-target Networks. Curr. Bioinform. 2021, 16, 385–394. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Brresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Baek, D.; Villen, J.; Shin, C.; Camargo, F.D.; Gygi, S.P.; Bartel, D.P. The impact of microRNAs on protein output. Nature 2008, 455, 64–71. [Google Scholar] [CrossRef] [Green Version]
- Farazi, T.A.; Spitzer, J.I.; Morozov, P.; Tuschl, T. miRNAs in human cancer. J. Pathol. 2011, 223, 102–115. [Google Scholar] [CrossRef]
- Qu, Y.; Tan, H.Y.; Chan, Y.T.; Jiang, H.; Wang, N.; Wang, D. The functional role of long noncoding RNA in resistance to anticancer treatment. Ther. Adv. Med. Oncol. 2020, 12, 1758835920927850. [Google Scholar] [CrossRef]
- Wang, M.; Yu, F.; Chen, X.; Li, P.; Wang, K. The Underlying Mechanisms of Noncoding RNAs in the Chemoresistance of Hepatocellular Carcinoma. Mol. Ther. Nucleic. Acids 2020, 21, 13–27. [Google Scholar] [CrossRef]
- Lin, P.; Wen, D.Y.; Li, Q.; He, Y.; Yang, H.; Chen, G. Genome-Wide Analysis of Prognostic lncRNAs, miRNAs, and mRNAs Forming a Competing Endogenous RNA Network in Hepatocellular Carcinoma. Cell Physiol. Biochem. 2018, 48, 1953–1967. [Google Scholar] [CrossRef]
- Ran, R.Z.; Chen, J.; Cui, L.J.; Lin, X.L.; Fan, M.M.; Cong, Z.Z.; Zhang, H.; Tan, W.F.; Zhang, G.Q.; Zhang, Y.J. miR-194 inhibits liver cancer stem cell expansion by regulating RAC1 pathway. Exp. Cell Res. 2019, 378, 66–75. [Google Scholar] [CrossRef]
- Dai, E.; Yang, F.; Wang, J.; Zhou, X.; Song, Q.; An, W.; Wang, L.; Jiang, W. ncDR: A comprehensive resource of non-coding RNAs involved in drug resistance. Bioinformatics 2017, 33, 4010–4011. [Google Scholar] [CrossRef]
- Li, L.; Wu, P.; Wang, Z.; Meng, X.; Zha, C.; Li, Z.; Qi, T.; Zhang, Y.; Han, B.; Li, S.; et al. NoncoRNA: A database of experimentally supported non-coding RNAs and drug targets in cancer. J. Hematol. Oncol. 2020, 13, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.-A.; Hu, P.; Chan, K.C.C.; You, Z.-H. Graph convolution for predicting associations between miRNA and drug resistance. Bioinformatics 2019, 36, 851–858. [Google Scholar] [CrossRef]
- Zhu, R.; Ji, C.; Wang, Y.; Cai, Y.; Wu, H. Heterogeneous Graph Convolutional Networks and Matrix Completion for miRNA-Disease Association Prediction. Front. Bioeng. Biotechnol. 2020, 8, 901. [Google Scholar] [CrossRef]
- Zeng, M.; Lu, C.; Zhang, F.; Li, Y.; Wu, F.X.; Li, Y.; Li, M. SDLDA: LncRNA-disease association prediction based on singular value decomposition and deep learning. Methods 2020, 179, 73–80. [Google Scholar] [CrossRef]
- Wei, H.; Liao, Q.; Liu, B. iLncRNAdis-FB: Identify lncRNA-disease associations by fusing biological feature blocks through deep neural network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020. [Google Scholar] [CrossRef]
- Ji, C.; Wang, Y.T.; Gao, Z.; Li, L.; Ni, J.C.; Zheng, C.H. A Semi-Supervised Learning Method for MiRNA-Disease Association Prediction Based on Variational Autoencoder. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021. [Google Scholar] [CrossRef]
- Deepthi, K.; Jereesh, A.S. Inferring Potential CircRNA-Disease Associations via Deep Autoencoder-Based Classification. Mol. Diagn. Ther. 2021, 25, 87–97. [Google Scholar] [CrossRef]
- Lu, C.; Zeng, M.; Zhang, F.; Wu, F.X.; Li, M.; Wang, J. Deep Matrix Factorization Improves Prediction of Human CircRNA-Disease Associations. IEEE J. Biomed. Health Inform. 2021, 25, 891–899. [Google Scholar] [CrossRef]
- Deng, L.; Yang, J.; Liu, H. Predicting circRNA-disease associations using meta path-based representation learning on heterogenous network. In Proceedings of the 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Seoul, Korea, 19 December 2020. [Google Scholar]
- Chen, X.; Huang, Y.-A.; You, Z.-H.; Yan, G.-Y.; Wang, X.-S. A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics 2016, 33, 733–739. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.H.; Yin, J.; Zhou, L.; Liu, M.X.; Zhao, Y. Human Microbe-Disease Association Prediction Based on Adaptive Boosting. Front. Microbiol. 2018, 9, 2440. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Zhang, J.; Zhang, W.; Zhou, S.; Li, W. DMFMDA: Prediction of microbe-disease associations based on deep matrix factorization using Bayesian Personalized Ranking. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020. [Google Scholar] [CrossRef]
- Luo, J.; Long, Y. NTSHMDA: Prediction of Human Microbe-Disease Association Based on Random Walk by Integrating Network Topological Similarity. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 1341–1351. [Google Scholar] [CrossRef]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef] [Green Version]
- Sangrajrang, S.; Fellous, A. Taxol resistance. Chemotherapy 2000, 46, 327–334. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, J.; Chen, S.; Gong, M.; Gao, D.; Zhu, M.; Gan, W. Review of the Applications of Deep Learning in Bioinformatics. Curr. Bioinform. 2021, 15, 898–911. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 30 June 2016. [Google Scholar]
- Rendle, S.; Freudenthaler, C.; Gantner, Z.; Schmidt-Thieme, L. BPR: Bayesian personalized ranking from implicit feedback. arXiv 2012, arXiv:1205.2618. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).