Lynch Syndrome: Its Impact on Urothelial Carcinoma

 ,
,  , ,
, , 
Abstract
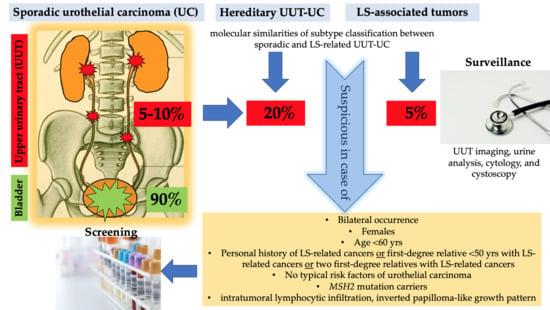
:
1. Introduction
1.1. Materials and Methods
1.2. Lynch Syndrome and Genetics
1.3. Lynch Syndrome and Cancer Susceptibility
1.4. Diagnosis of Lynch Syndrome
1.5. Lynch Syndrome and UC of the UUT and Bladder
1.6. Recommendations of Screening in LS
1.7. Recommendations of Urological Surveillance in LS
1.8. Lynch Syndrome and Immunotherapy
2. Case Presentation
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACII | Amsterdam Criteria II |
| AUA | American Urological Association |
| CT | computed tomography |
| CRC | colorectal cancer |
| CTLA-4 | cytotoxic T lymphocyte antigen 4 |
| DNA | deoxyribonucleic acid |
| EC | endometrial cancer |
| FAP | familial adenomatous polyposis |
| FGFR3 | fibroblast growth factor receptor 3 |
| HPF | high-power field |
| ICI | immune checkpoint inhibitors |
| IHC | immunohistochemistry |
| LS | Lynch syndrome |
| MLH1 | mutL homolog 1 |
| MMR | mismatch repair |
| MSH2 | mutS homolog 2 |
| MSH6 | mutS homolog 6 |
| MSI | microsatellite instability |
| MSI-H | high-frequency MSI |
| MSI-L | low-frequency MSI |
| MSS | microsatellite stable |
| ORR | objective response rate |
| PD-1 | programmed cell death 1 protein |
| PD-L1 | programmed cell death ligand 1 protein |
| PMS2 | postmeiotic segregation increased 2 |
| RBC | red blood cells |
| Th1 | Type 1 T helper cells |
| TIL | tumor infiltrating lymphocytes |
| TMB | tumor mutational burden |
| UC | urothelial cancer |
| UUT | upper urothelial tract |
References
- Fodde, R. The APC gene in colorectal cancer. Eur. J. Cancer 2002, 38, 867–871. [Google Scholar] [CrossRef]
- Kloth, M.; Ruesseler, V.; Engel, C.; Koenig, K.; Peifer, M.; Mariotti, E.; Kuenstlinger, H.; Florin, A.; Rommerscheidt-Fuss, U.; Koitzsch, U.; et al. Activating ERBB2/HER2 mutations indicate susceptibility to pan-HER inhibitors in Lynch and Lynch-like colorectal cancer. Gut 2016, 65, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Mangold, E.; Pagenstecher, C.; Friedl, W.; Mathiak, M.; Buettner, R.; Engel, C.; Loeffler, M.; Holinski-Feder, E.; Müller-Koch, Y.; Keller, G.; et al. Spectrum and frequencies of mutations in MSH2 and MLH1 identified in 1,721 German families suspected of hereditary nonpolyposis colorectal cancer. Int. J. Cancer 2005, 116, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R. Recent discoveries in the molecular genetics of Lynch syndrome. Fam. Cancer 2016, 15, 395–403. [Google Scholar] [CrossRef]
- Modrich, P. Mechanisms in eukaryotic mismatch repair. J. Biol. Chem. 2006, 281, 30305–30309. [Google Scholar] [CrossRef] [Green Version]
- Bridge, G.; Rashid, S.; Martin, S.A. DNA mismatch repair and oxidative DNA damage: Implications for cancer biology and treatment. Cancers 2014, 6, 1597–1614. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Valentin, M.; Sampson, J.R.; Seppälä, T.T.; Ten Broeke, S.W.; Plazzer, J.-P.; Nakken, S.; Engel, C.; Aretz, S.; Jenkins, M.A.; Sunde, L.; et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: Findings from the Prospective Lynch Syndrome Database. Genet. Med. 2020, 22, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Lynch, H.T.; Ens, J.A.; Lynch, J.F. The Lynch syndrome II and urological malignancies. J. Urol. 1990, 143, 24–28. [Google Scholar] [CrossRef]
- Li, X.-L.; Zhou, J.; Chen, Z.-R.; Chng, W.-J. P53 mutations in colorectal cancer—Molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84–93. [Google Scholar] [CrossRef]
- Ahadova, A.; Gallon, R.; Gebert, J.; Ballhausen, A.; Endris, V.; Kirchner, M.; Stenzinger, A.; Burn, J.; von Knebel Doeberitz, M.; Bläker, H.; et al. Three molecular pathways model colorectal carcinogenesis in Lynch syndrome. Int. J. Cancer 2018, 143, 139–150. [Google Scholar] [CrossRef] [Green Version]
- Ten Broeke, S.W.; van Bavel, T.C.; Jansen, A.M.L.; Gómez-García, E.; Hes, F.J.; van Hest, L.P.; Letteboer, T.G.W.; Olderode-Berends, M.J.W.; Ruano, D.; Spruijt, L.; et al. Molecular Background of Colorectal Tumors from Patients with Lynch Syndrome Associated with Germline Variants in PMS2. Gastroenterology 2018, 155, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.K.; Roy, H.K.; Lynch, H.T. Lynch syndrome in the 21st century: Clinical perspectives. QJM 2016, 109, 151–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, D.C.; Rustgi, A.K. DNA mismatch repair and cancer. Gastroenterology 1995, 109, 1685–1699. [Google Scholar] [CrossRef]
- Pinto, D.; Pinto, C.; Guerra, J.; Pinheiro, M.; Santos, R.; Vedeld, H.M.; Yohannes, Z.; Peixoto, A.; Santos, C.; Pinto, P.; et al. Contribution of MLH1 constitutional methylation for Lynch syndrome diagnosis in patients with tumor MLH1 downregulation. Cancer Med. 2018, 7, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Joost, P.; Therkildsen, C.; Dominguez-Valentin, M.; Jönsson, M.; Nilbert, M. Urinary Tract Cancer in Lynch Syndrome; Increased Risk in Carriers of MSH2 Mutations. Urology 2015, 86, 1212–1217. [Google Scholar] [CrossRef]
- Bonadona, V.; Bonaïti, B.; Olschwang, S.; Grandjouan, S.; Huiart, L.; Longy, M.; Guimbaud, R.; Buecher, B.; Bignon, Y.-J.; Caron, O.; et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA 2011, 305, 2304–2310. [Google Scholar] [CrossRef]
- Ten Broeke, S.W.; van der Klift, H.M.; Tops, C.M.J.; Aretz, S.; Bernstein, I.; Buchanan, D.D.; de la Chapelle, A.; Capella, G.; Clendenning, M.; Engel, C.; et al. Cancer Risks for PMS2-Associated Lynch Syndrome. J. Clin. Oncol. 2018, 36, 2961–2968. [Google Scholar] [CrossRef] [Green Version]
- de Jong, A.E.; van Puijenbroek, M.; Hendriks, Y.; Tops, C.; Wijnen, J.; Ausems, M.G.E.M.; Meijers-Heijboer, H.; Wagner, A.; van Os, T.A.M.; Bröcker-Vriends, A.H.J.T.; et al. Microsatellite instability, immunohistochemistry, and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer. Clin. Cancer Res. 2004, 10, 972–980. [Google Scholar] [CrossRef] [Green Version]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer-the stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Hoeijmakers, J.H. Genome maintenance mechanisms for preventing cancer. Nature 2001, 411, 366–374. [Google Scholar] [CrossRef]
- Peltomäki, P. Update on Lynch syndrome genomics. Fam. Cancer 2016, 15, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobocińska, J.; Kolenda, T.; Teresiak, A.; Badziąg-Leśniak, N.; Kopczyńska, M.; Guglas, K.; Przybyła, A.; Filas, V.; Bogajewska-Ryłko, E.; Lamperska, K.; et al. Diagnostics of Mutations in MMR/EPCAM Genes and Their Role in the Treatment and Care of Patients with Lynch Syndrome. Diagnostics 2020, 10, 786. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lee, L.H.; Vyas, M.; Zhang, L.; Ganesh, K.; Firat, C.; Segal, N.H.; Desai, A.; Hechtman, J.F.; Ntiamoah, P.; et al. Colorectal carcinoma with double somatic mismatch repair gene inactivation: Clinical and pathological characteristics and response to immune checkpoint blockade. Mod. Pathol. 2019, 32, 1551–1562. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, D.A.; George, T.J.; Dunne, R.F.; Frampton, G.; Sun, J.; Gowen, K.; Kennedy, M.; Greenbowe, J.; Schrock, A.B.; Hezel, A.F.; et al. Beyond microsatellite testing: Assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J. Gastrointest. Oncol. 2018, 9, 610–617. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O’Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: A single-arm, multicentre, phase 2 trial. Lancet 2016, 387, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted Next Generation Sequencing Identifies Markers of Response to PD-1 Blockade. Cancer Immunol. Res. 2016, 4, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Boussios, S.; Mikropoulos, C.; Samartzis, E.; Karihtala, P.; Moschetta, M.; Sheriff, M.; Karathanasi, A.; Sadauskaite, A.; Rassy, E.; Pavlidis, N. Wise Management of Ovarian Cancer: On the Cutting Edge. J. Pers. Med. 2020, 10, 41. [Google Scholar] [CrossRef]
- Møller, P.; Seppälä, T.T.; Bernstein, I.; Holinski-Feder, E.; Sala, P.; Gareth Evans, D.; Lindblom, A.; Macrae, F.; Blanco, I.; Sijmons, R.H.; et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: A report from the Prospective Lynch Syndrome Database. Gut 2018, 67, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Salem, M.E.; Bodor, J.N.; Puccini, A.; Xiu, J.; Goldberg, R.M.; Grothey, A.; Korn, W.M.; Shields, A.F.; Worrilow, W.M.; Kim, E.S.; et al. Relationship between MLH1, PMS2, MSH2 and MSH6 gene-specific alterations and tumor mutational burden in 1057 microsatellite instability-high solid tumors. Int. J. Cancer 2020, 147, 2948–2956. [Google Scholar] [CrossRef]
- Smyrk, T.C.; Watson, P.; Kaul, K.; Lynch, H.T. Tumor-infiltrating lymphocytes are a marker for microsatellite instability in colorectal carcinoma. Cancer 2001, 91, 2417–2422. [Google Scholar] [CrossRef]
- Kim, H.; Jen, J.; Vogelstein, B.; Hamilton, S.R. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am. J. Pathol. 1994, 145, 148–156. [Google Scholar] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Li, L.S.; Morales, J.C.; Veigl, M.; Sedwick, D.; Greer, S.; Meyers, M.; Wagner, M.; Fishel, R.; Boothman, D.A. DNA mismatch repair (MMR)-dependent 5-fluorouracil cytotoxicity and the potential for new therapeutic targets. Br. J. Pharmacol. 2009, 158, 679–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasen, H.F.; Mecklin, J.P.; Khan, P.M.; Lynch, H.T. The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer (ICG-HNPCC). Dis. Colon Rectum 1991, 34, 424–425. [Google Scholar] [CrossRef]
- Vasen, H.; Watson, P.; Mecklin, J.; Lynch, H. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC. Gastroenterology 1999, 116, 1453–1456. [Google Scholar] [CrossRef]
- Church, J.; McGannon, E. Family history of colorectal cancer: How often and how accurately is it recorded? Dis. Colon Rectum 2000, 43, 1540–1544. [Google Scholar] [CrossRef]
- Sanchez, J.A.; Vogel, J.D.; Kalady, M.F.; Bronner, M.P.; Skacel, M.; Church, J.M. Identifying Lynch syndrome: We are all responsible. Dis. Colon Rectum 2008, 51, 1750–1756. [Google Scholar] [CrossRef]
- Gaf, R.; Maestri, I.; Matteuzzi, M.; Santini, A.; Ferretti, S.; Cavazzini, L.; Lanza, G. Sporadic colorectal adenocarcinomas with high-frequency microsatellite instability. Cancer 2000, 89, 2025–2037. [Google Scholar] [CrossRef]
- Truta, B.; Chen, Y.-Y.; Blanco, A.M.; Deng, G.; Conrad, P.G.; Kim, Y.H.; Park, E.T.; Kakar, S.; Kim, Y.S.; Velayos, F.; et al. Tumor histology helps to identify Lynch syndrome among colorectal cancer patients. Fam. Cancer 2008, 7, 267–274. [Google Scholar] [CrossRef]
- Hugen, N.; van Beek, J.J.P.; de Wilt, J.H.W.; Nagtegaal, I.D. Insight into mucinous colorectal carcinoma: Clues from etiology. Ann. Surg. Oncol. 2014, 21, 2963–2970. [Google Scholar] [CrossRef]
- Ott, C.; Gerken, M.; Hirsch, D.; Fest, P.; Fichtner-Feigl, S.; Munker, S.; Schnoy, E.; Stroszczynski, C.; Vogelhuber, M.; Herr, W.; et al. Advanced Mucinous Colorectal Cancer: Epidemiology, Prognosis and Efficacy of Chemotherapeutic Treatment. Digestion 2018, 98, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Bigas, M.A.; Boland, C.R.; Hamilton, S.R.; Henson, D.E.; Jass, J.R.; Khan, P.M.; Lynch, H.; Perucho, M.; Smyrk, T.; Sobin, L.; et al. A National Cancer Institute Workshop on Hereditary Nonpolyposis Colorectal Cancer Syndrome: Meeting highlights and Bethesda guidelines. J. Natl. Cancer Inst. 1997, 89, 1758–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umar, A.; Boland, C.R.; Terdiman, J.P.; Syngal, S.; de La Chapelle, A.; Rüschoff, J.; Fishel, R.; Lindor, N.M.; Burgart, L.J.; Hamelin, R.; et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J. Natl. Cancer Inst. 2004, 96, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Kievit, W.; de Bruin, J.H.F.M.; Adang, E.M.M.; Severens, J.L.; Kleibeuker, J.H.; Sijmons, R.H.; Ruers, T.J.; Nagengast, F.M.; Vasen, H.F.A.; van Krieken, J.H.J.M.; et al. Cost effectiveness of a new strategy to identify HNPCC patients. Gut 2005, 54, 97–102. [Google Scholar] [CrossRef] [PubMed]
- van Lier, M.G.F.; Wagner, A.; van Leerdam, M.E.; Biermann, K.; Kuipers, E.J.; Steyerberg, E.W.; Dubbink, H.J.; Dinjens, W.N.M. A review on the molecular diagnostics of Lynch syndrome: A central role for the pathology laboratory. J. Cell Mol. Med. 2010, 14, 181–197. [Google Scholar] [CrossRef] [Green Version]
- Lynch, H.T.; de La Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef]
- Gryfe, R.; Kim, H.; Hsieh, E.T.; Aronson, M.D.; Holowaty, E.J.; Bull, S.B.; Redston, M.; Gallinger, S. Tumor microsatellite instability and clinical outcome in young patients with colorectal cancer. N. Engl. J. Med. 2000, 342, 69–77. [Google Scholar] [CrossRef]
- Germano, G.; Lamba, S.; Rospo, G.; Barault, L.; Magrì, A.; Maione, F.; Russo, M.; Crisafulli, G.; Bartolini, A.; Lerda, G.; et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 2017, 552, 116–120. [Google Scholar] [CrossRef]
- Kang, S.; Na, Y.; Joung, S.Y.; Lee, S.I.; Oh, S.C.; Min, B.W. The significance of microsatellite instability in colorectal cancer after controlling for clinicopathological factors. Medicine (Baltimore) 2018, 97, e0019. [Google Scholar] [CrossRef]
- Kuismanen, S.A.; Moisio, A.-L.; Schweizer, P.; Truninger, K.; Salovaara, R.; Arola, J.; Butzow, R.; Jiricny, J.; Nyström-Lahti, M.; Peltomäki, P. Endometrial and Colorectal Tumors from Patients with Hereditary Nonpolyposis Colon Cancer Display Different Patterns of Microsatellite Instability. Am. J. Pathol. 2002, 160, 1953–1958. [Google Scholar] [CrossRef] [Green Version]
- Boussios, S.; Ozturk, M.A.; Moschetta, M.; Karathanasi, A.; Zakynthinakis-Kyriakou, N.; Katsanos, K.H.; Christodoulou, D.K.; Pavlidis, N. The Developing Story of Predictive Biomarkers in Colorectal Cancer. J. Pers. Med. 2019, 9, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Nakagawa, H.; Sotamaa, K.; Prior, T.W.; Westman, J. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piñol, V.; Castells, A.; Andreu, M.; Castellví-Bel, S.; Alenda, C.; Llor, X.; Xicola, R.M.; Rodríguez-Moranta, F.; Payá, A.; Jover, R.; et al. Accuracy of revised Bethesda guidelines, microsatellite instability, and immunohistochemistry for the identification of patients with hereditary nonpolyposis colorectal cancer. JAMA 2005, 293, 1986–1994. [Google Scholar] [CrossRef] [PubMed]
- Shia, J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome. Part, I. The utility of immunohistochemistry. J. Mol. Diagn. 2008, 10, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, W.; Burgart, L.J.; Krause-Paulus, R.; Thibodeau, S.N.; Almeida, M.; Edmonston, T.B.; Boland, C.R.; Sutter, C.; Jass, J.R.; Lindblom, A.; et al. The reliability of immunohistochemistry as a prescreening method for the diagnosis of hereditary nonpolyposis colorectal cancer (HNPCC)--results of an international collaborative study. Fam. Cancer 2001, 1, 87–92. [Google Scholar] [CrossRef]
- Overbeek, L.I.H.; Ligtenberg, M.J.L.; Willems, R.W.; Hermens, R.P.M.G.; Blokx, W.A.M.; Dubois, S.V.; van der Linden, H.; Meijer, J.W.R.; Mlynek-Kersjes, M.L.; Hoogerbrugge, N.; et al. Interpretation of immunohistochemistry for mismatch repair proteins is only reliable in a specialized setting. Am. J. Surg. Pathol. 2008, 32, 1246–1251. [Google Scholar] [CrossRef]
- Dinh, T.A.; Rosner, B.I.; Atwood, J.C.; Boland, C.R.; Syngal, S.; Vasen, H.F.A.; Gruber, S.B.; Burt, R.W. Health benefits and cost-effectiveness of primary genetic screening for Lynch syndrome in the general population. Cancer Prev. Res. 2011, 4, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Strafford, J.C. Genetic testing for lynch syndrome, an inherited cancer of the bowel, endometrium, and ovary. Rev. Obstet. Gynecol. 2012, 5, 42–49. [Google Scholar]
- Xicola, R.M.; Llor, X. Cancer risk assessment in Lynch syndrome: Does the gene matter? JAMA 2011, 305, 2351–2352. [Google Scholar] [CrossRef]
- Hampel, H. Population Screening for Hereditary Colorectal Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 319–325. [Google Scholar] [CrossRef]
- Buchanan, D.D.; Rosty, C.; Clendenning, M.; Spurdle, A.B.; Win, A.K. Clinical problems of colorectal cancer and endometrial cancer cases with unknown cause of tumor mismatch repair deficiency (suspected Lynch syndrome). Appl. Clin. Genet. 2014, 7, 183–193. [Google Scholar] [PubMed] [Green Version]
- Gray, P.N.; Tsai, P.; Chen, D.; Wu, S.; Hoo, J.; Mu, W.; Li, B.; Vuong, H.; Lu, H.-M.; Batth, N.; et al. TumorNext-Lynch-MMR: A comprehensive next generation sequencing assay for the detection of germline and somatic mutations in genes associated with mismatch repair deficiency and Lynch syndrome. Oncotarget 2018, 9, 20304–20322. [Google Scholar] [CrossRef] [PubMed]
- Salvador, M.U.; Truelson, M.R.F.; Mason, C.; Souders, B.; LaDuca, H.; Dougall, B.; Black, M.H.; Fulk, K.; Profato, J.; Gutierrez, S.; et al. Comprehensive Paired Tumor/Germline Testing for Lynch Syndrome: Bringing Resolution to the Diagnostic Process. J. Clin. Oncol. 2019, 37, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Rouprêt, M.; Babjuk, M.; Compérat, E.; Zigeuner, R.; Sylvester, R.J.; Burger, M.; Cowan, N.C.; Gontero, P.; van Rhijn, B.W.G.; Mostafid, A.H.; et al. European Association of Urology Guidelines on Upper Urinary Tract Urothelial Carcinoma: 2017 Update. Eur. Urol. 2018, 73, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Soria, F.; Shariat, S.F.; Lerner, S.P.; Fritsche, H.-M.; Rink, M.; Kassouf, W.; Spiess, P.E.; Lotan, Y.; Ye, D.; Fernández, M.I.; et al. Epidemiology, diagnosis, preoperative evaluation and prognostic assessment of upper-tract urothelial carcinoma (UTUC). World J. Urol. 2017, 35, 379–387. [Google Scholar] [CrossRef]
- Margulis, V.; Shariat, S.F.; Matin, S.F.; Kamat, A.M.; Zigeuner, R.; Kikuchi, E.; Lotan, Y.; Weizer, A.; Raman, J.D.; Wood, C.G. Outcomes of radical nephroureterectomy: A series from the Upper Tract Urothelial Carcinoma Collaboration. Cancer 2009, 115, 1224–1233. [Google Scholar] [CrossRef]
- Park, S.; Hong, B.; Kim, C.-S.; Ahn, H. The impact of tumor location on prognosis of transitional cell carcinoma of the upper urinary tract. J. Urol. 2004, 171, 621–625. [Google Scholar] [CrossRef]
- Audenet, F.; Colin, P.; Yates, D.R.; Ouzzane, A.; Pignot, G.; Long, J.-A.; Soulie, M.; Phé, V.; Bensadoun, H.; Guy, L.; et al. A proportion of hereditary upper urinary tract urothelial carcinomas are misclassified as sporadic according to a multi-institutional database analysis: Proposal of patient-specific risk identification tool. BJU Int. 2012, 110, E583–E589. [Google Scholar] [CrossRef]
- Hassler, M.R.; Bray, F.; Catto, J.W.F.; Grollman, A.P.; Hartmann, A.; Margulis, V.; Matin, S.F.; Roupret, M.; Sfakianos, J.P.; Shariat, S.F.; et al. Molecular Characterization of Upper Tract Urothelial Carcinoma in the Era of Next-generation Sequencing: A Systematic Review of the Current Literature. Eur. Urol. 2020, 78, 209–220. [Google Scholar] [CrossRef]
- Watson, P.; Vasen, H.F.A.; Mecklin, J.-P.; Bernstein, I.; Aarnio, M.; Järvinen, H.J.; Myrhøj, T.; Sunde, L.; Wijnen, J.T.; Lynch, H.T. The risk of extra-colonic, extra-endometrial cancer in the Lynch syndrome. Int. J. Cancer 2008, 123, 444–449. [Google Scholar] [CrossRef] [Green Version]
- Crockett, D.G.; Wagner, D.G.; Holmäng, S.; Johansson, S.L.; Lynch, H.T. Upper urinary tract carcinoma in Lynch syndrome cases. J. Urol. 2011, 185, 1627–1630. [Google Scholar] [CrossRef] [PubMed]
- Sijmons, R.H.; Kiemeney, L.A.; Witjes, J.A.; Vasen, H.F. Urinary tract cancer and hereditary nonpolyposis colorectal cancer: Risks and screening options. J. Urol. 1998, 160, 466–470. [Google Scholar] [CrossRef]
- Therkildsen, C.; Eriksson, P.; Höglund, M.; Jönsson, M.; Sjödahl, G.; Nilbert, M.; Liedberg, F. Molecular subtype classification of urothelial carcinoma in Lynch syndrome. Mol. Oncol. 2018, 12, 1286–1295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krabbe, L.-M.; Lotan, Y.; Bagrodia, A.; Gayed, B.A.; Darwish, O.M.; Youssef, R.F.; Bolenz, C.; Sagalowsky, A.I.; Raj, G.V.; Shariat, S.F.; et al. Prospective comparison of molecular signatures in urothelial cancer of the bladder and the upper urinary tract—Is there evidence for discordant biology? J. Urol. 2014, 191, 926–931. [Google Scholar] [CrossRef]
- Sanford, T.; Porten, S.; Meng, M.V. Molecular Analysis of Upper Tract and Bladder Urothelial Carcinoma: Results from a Microarray Comparison. PLoS ONE 2015, 10, e0137141. [Google Scholar] [CrossRef]
- Sfakianos, J.P.; Cha, E.K.; Iyer, G.; Scott, S.N.; Zabor, E.C.; Shah, R.H.; Ren, Q.; Bagrodia, A.; Kim, P.H.; Hakimi, A.A.; et al. Genomic Characterization of Upper Tract Urothelial Carcinoma. Eur. Urol. 2015, 68, 970–977. [Google Scholar] [CrossRef] [Green Version]
- Mork, M.; Hubosky, S.G.; Rouprêt, M.; Margulis, V.; Raman, J.; Lotan, Y.; O’Brien, T.; You, N.; Shariat, S.F.; Matin, S.F.; et al. Lynch Syndrome: A Primer for Urologists and Panel Recommendations. J. Urol. 2015, 194, 21–29. [Google Scholar] [CrossRef]
- Dailey, L.; Ambrosetti, D.; Mansukhani, A.; Basilico, C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. 2005, 16, 233–247. [Google Scholar] [CrossRef]
- van Rhijn, B.W.G.; Montironi, R.; Zwarthoff, E.C.; Jöbsis, A.C.; van der Kwast, T.H. Frequent FGFR3 mutations in urothelial papilloma. J. Pathol. 2002, 198, 245–251. [Google Scholar] [CrossRef]
- Knowles, M.A. Role of FGFR3 in urothelial cell carcinoma: Biomarker and potential therapeutic target. World J. Urol. 2007, 25, 581–593. [Google Scholar] [CrossRef] [Green Version]
- Moss, T.J.; Qi, Y.; Xi, L.; Peng, B.; Kim, T.-B.; Ezzedine, N.E.; Mosqueda, M.E.; Guo, C.C.; Czerniak, B.A.; Ittmann, M.; et al. Comprehensive Genomic Characterization of Upper Tract Urothelial Carcinoma. Eur. Urol. 2017, 72, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Harper, H.L.; McKenney, J.K.; Heald, B.; Stephenson, A.; Campbell, S.C.; Plesec, T.; Magi-Galluzzi, C. Upper tract urothelial carcinomas: Frequency of association with mismatch repair protein loss and lynch syndrome. Mod. Pathol. 2017, 30, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Vasen, H.F.; Stormorken, A.; Menko, F.H.; Nagengast, F.M.; Kleibeuker, J.H.; Griffioen, G.; Taal, B.G.; Moller, P.; Wijnen, J.T. MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: A study of hereditary nonpolyposis colorectal cancer families. J. Clin. Oncol. 2001, 19, 4074–4080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Post, R.S.; Kiemeney, L.A.; Ligtenberg, M.J.L.; Witjes, J.A.; Hulsbergen-van de Kaa, C.A.; Bodmer, D.; Schaap, L.; Kets, C.M.; van Krieken, J.H.J.M.; Hoogerbrugge, N. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J. Med. Genet. 2010, 47, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Carlo, M.I.; Ravichandran, V.; Srinavasan, P.; Bandlamudi, C.; Kemel, Y.; Ceyhan-Birsoy, O.; Mukherjee, S.; Mandelker, D.; Chaim, J.; Knezevic, A.; et al. Cancer Susceptibility Mutations in Patients with Urothelial Malignancies. J. Clin. Oncol. 2020, 38, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Amira, N.; Rivet, J.; Soliman, H.; Cancel-Tassin, G.; Le Duc, A.; Janin, A.; Cussenot, O. Microsatellite instability in urothelial carcinoma of the upper urinary tract. J. Urol. 2003, 170, 1151–1154. [Google Scholar] [CrossRef]
- Hartmann, A.; Zanardo, L.; Bocker-Edmonston, T.; Blaszyk, H.; Dietmaier, W.; Stoehr, R.; Cheville, J.C.; Junker, K.; Wieland, W.; Knuechel, R.; et al. Frequent microsatellite instability in sporadic tumors of the upper urinary tract. Cancer Res. 2002, 62, 6796–6802. [Google Scholar]
- Hartmann, A.; Dietmaier, W.; Hofstädter, F.; Burgart, L.J.; Cheville, J.C.; Blaszyk, H. Urothelial carcinoma of the upper urinary tract: Inverted growth pattern is predictive of microsatellite instability. Hum. Pathol. 2003, 34, 222–227. [Google Scholar] [CrossRef]
- Huang, D.; Matin, S.F.; Lawrentschuk, N.; Roupret, M. Systematic Review: An Update on the Spectrum of Urological Malignancies in Lynch Syndrome. Bladder Cancer 2018, 4, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Geary, J.; Sasieni, P.; Houlston, R.; Izatt, L.; Eeles, R.; Payne, S.J.; Fisher, S.; Hodgson, S.V. Gene-related cancer spectrum in families with hereditary non-polyposis colorectal cancer (HNPCC). Fam. Cancer 2008, 7, 163–172. [Google Scholar] [CrossRef]
- Skeldon, S.C.; Semotiuk, K.; Aronson, M.; Holter, S.; Gallinger, S.; Pollett, A.; Kuk, C.; van Rhijn, B.; Bostrom, P.; Cohen, Z.; et al. Patients with Lynch syndrome mismatch repair gene mutations are at higher risk for not only upper tract urothelial cancer but also bladder cancer. Eur. Urol. 2013, 63, 379–385. [Google Scholar] [CrossRef]
- Bermejo, J.L.; Eng, C.; Hemminki, K. Cancer characteristics in Swedish families fulfilling criteria for hereditary nonpolyposis colorectal cancer. Gastroenterology 2005, 129, 1889–1899. [Google Scholar] [CrossRef] [PubMed]
- Rouprêt, M.; Yates, D.R.; Comperat, E.; Cussenot, O. Upper urinary tract urothelial cell carcinomas and other urological malignancies involved in the hereditary nonpolyposis colorectal cancer (lynch syndrome) tumor spectrum. Eur. Urol. 2008, 54, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Rouprêt, M.; Fromont, G.; Azzouzi, A.-R.; Catto, J.W.; Vallancien, G.; Hamdy, F.C.; Cussenot, O. Microsatellite instability as predictor of survival in patients with invasive upper urinary tract transitional cell carcinoma. Urology 2005, 65, 1233–1237. [Google Scholar] [CrossRef] [PubMed]
- Giardiello, F.M.; Allen, J.I.; Axilbund, J.E.; Boland, C.R.; Burke, C.A.; Burt, R.W.; Church, J.M.; Dominitz, J.A.; Johnson, D.A.; Kaltenbach, T.; et al. Guidelines on genetic evaluation and management of Lynch syndrome: A consensus statement by the US Multi-Society Task Force on colorectal cancer. Gastroenterology 2014, 147, 502–526. [Google Scholar] [CrossRef] [Green Version]
- Yurgelun, M.B.; Hampel, H. Recent Advances in Lynch Syndrome: Diagnosis, Treatment, and Cancer Prevention. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 101–109. [Google Scholar] [CrossRef]
- Metcalfe, M.J.; Petros, F.G.; Rao, P.; Mork, M.E.; Xiao, L.; Broaddus, R.R.; Matin, S.F. Universal Point of Care Testing for Lynch Syndrome in Patients with Upper Tract Urothelial Carcinoma. J. Urol. 2018, 199, 60–65. [Google Scholar] [CrossRef]
- Hegde, M.; Ferber, M.; Mao, R.; Samowitz, W.; Ganguly, A. ACMG technical standards and guidelines for genetic testing for inherited colorectal cancer (Lynch syndrome, familial adenomatous polyposis, and MYH-associated polyposis). Genet. Med. 2014, 16, 101–116. [Google Scholar] [CrossRef] [Green Version]
- Urakami, S.; Inoshita, N.; Oka, S.; Miyama, Y.; Nomura, S.; Arai, M.; Sakaguchi, K.; Kurosawa, K.; Okaneya, T. Clinicopathological characteristics of patients with upper urinary tract urothelial cancer with loss of immunohistochemical expression of the DNA mismatch repair proteins in universal screening. Int. J. Urol. 2018, 25, 151–156. [Google Scholar] [CrossRef] [Green Version]
- Watkins, J.C.; Yang, E.J.; Muto, M.G.; Feltmate, C.M.; Berkowitz, R.S.; Horowitz, N.S.; Syngal, S.; Yurgelun, M.B.; Chittenden, A.; Hornick, J.L.; et al. Universal Screening for Mismatch-Repair Deficiency in Endometrial Cancers to Identify Patients With Lynch Syndrome and Lynch-like Syndrome. Int. J. Gynecol. Pathol. 2017, 36, 115–127. [Google Scholar] [CrossRef]
- O’Kane, G.M.; Ryan, É.; McVeigh, T.P.; Creavin, B.; Hyland, J.M.; O’Donoghue, D.P.; Keegan, D.; Geraghty, R.; Flannery, D.; Nolan, C.; et al. Screening for mismatch repair deficiency in colorectal cancer: Data from three academic medical centers. Cancer Med. 2017, 6, 1465–1472. [Google Scholar] [CrossRef] [PubMed]
- Mills, A.M.; Longacre, T.A. Lynch Syndrome Screening in the Gynecologic Tract: Current State of the Art. Am. J. Surg. Pathol. 2016, 40, e35–e44. [Google Scholar] [CrossRef] [PubMed]
- Pasche, B.; Pennison, M.J.; DeYoung, B. Lynch Syndrome Testing: A Missed Opportunity in the Era of Precision Medicine. JAMA 2016, 316, 38–39. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.Y.; Mills, A.M.; Mahadevan, M.S.; Fan, J.; Culp, S.H.; Thomas, M.H.; Cathro, H.P. Universal Lynch Syndrome Screening should be Performed in All Upper Tract Urothelial Carcinomas. Am. J. Surg. Pathol. 2018, 42, 1549–1555. [Google Scholar] [CrossRef]
- Vasen, H.F.A.; Möslein, G.; Alonso, A.; Bernstein, I.; Bertario, L.; Blanco, I.; Burn, J.; Capella, G.; Engel, C.; Frayling, I.; et al. Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J. Med. Genet. 2007, 44, 353–362. [Google Scholar] [CrossRef]
- Myrhøj, T.; Andersen, M.-B.; Bernstein, I. Screening for urinary tract cancer with urine cytology in Lynch syndrome and familial colorectal cancer. Fam. Cancer 2008, 7, 303–307. [Google Scholar] [CrossRef]
- Lindor, N.M.; Petersen, G.M.; Hadley, D.W.; Kinney, A.Y.; Miesfeldt, S.; Lu, K.H.; Lynch, P.; Burke, W.; Press, N. Recommendations for the care of individuals with an inherited predisposition to Lynch syndrome: A systematic review. JAMA 2006, 296, 1507–1517. [Google Scholar] [CrossRef]
- Koornstra, J.J.; Mourits, M.J.; Sijmons, R.H.; Leliveld, A.M.; Hollema, H.; Kleibeuker, J.H. Management of extracolonic tumours in patients with Lynch syndrome. Lancet Oncol. 2009, 10, 400–408. [Google Scholar] [CrossRef]
- Lim, A.; Rao, P.; Matin, S.F. Lynch syndrome and urologic malignancies: A contemporary review. Curr. Opin. Urol. 2019, 29, 357–363. [Google Scholar] [CrossRef]
- Saita, C.; Yamaguchi, T.; Horiguchi, S.-I.; Yamada, R.; Takao, M.; Iijima, T.; Wakaume, R.; Aruga, T.; Tabata, T.; Koizumi, K. Tumor development in Japanese patients with Lynch syndrome. PLoS ONE 2018, 13, e0195572. [Google Scholar] [CrossRef]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, N.; Zheng, Y.; Kelly, V.; Katz, S.S.; Chou, J.; Do, R.K.G.; Capanu, M.; Zamarin, D.; Saltz, L.B.; Ariyan, C.E. PD-1 Blockade in Advanced Adrenocortical Carcinoma. J. Clin. Oncol. 2020, 38, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.E.; Puccini, A.; Grothey, A.; Raghavan, D.; Goldberg, R.M.; Xiu, J.; Korn, W.M.; Weinberg, B.A.; Hwang, J.J.; Shields, A.F.; et al. Landscape of Tumor Mutation Load, Mismatch Repair Deficiency, and PD-L1 Expression in a Large Patient Cohort of Gastrointestinal Cancers. Mol. Cancer Res. 2018, 16, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korehisa, S.; Oki, E.; Iimori, M.; Nakaji, Y.; Shimokawa, M.; Saeki, H.; Okano, S.; Oda, Y.; Maehara, Y. Clinical significance of programmed cell death-ligand 1 expression and the immune microenvironment at the invasive front of colorectal cancers with high microsatellite instability. Int. J. Cancer 2018, 142, 822–832. [Google Scholar] [CrossRef] [Green Version]
- Hamanishi, J.; Mandai, M.; Matsumura, N.; Abiko, K.; Baba, T.; Konishi, I. PD-1/PD-L1 blockade in cancer treatment: Perspectives and issues. Int. J. Clin. Oncol. 2016, 21, 462–473. [Google Scholar] [CrossRef] [Green Version]
- Sclafani, F. PD-1 inhibition in metastatic dMMR/MSI-H colorectal cancer. Lancet Oncol. 2017, 18, 1141–1142. [Google Scholar] [CrossRef]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.-J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit with Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Feng, Y.; Cao, Y.; Yuan, M.; Chen, R.; Ji, X.; Hu, X. Different responses to anti-programmed cell death protein 1 (PD-1) immunotherapy in a patient with Lynch syndrome and metachronous urothelial and colon cancer: A case report. Oncol. Lett. 2019, 18, 5085–5090. [Google Scholar] [CrossRef] [Green Version]
- Keating, M.; Giscombe, L.; Tannous, T.; Hartshorn, K. Prolonged Treatment Response to Pembrolizumab in a Patient with Pretreated Metastatic Colon Cancer and Lynch Syndrome. Case Rep. Oncol. Med. 2019, 2019, 3847672. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA Approval Agnostic of Cancer Site—When a Biomarker Defines the Indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Demircan, N.C.; Boussios, S.; Tasci, T.; Öztürk, M.A. Current and future immunotherapy approaches in ovarian cancer. Ann. Transl. Med. 2020. [Google Scholar] [CrossRef]
- PD-1 Inhibitor Bests Chemo for Colorectal Cancer. Cancer Discov. 2020, 10. [CrossRef]
- Alfred Witjes, J.; Lebret, T.; Compérat, E.M.; Cowan, N.C.; de Santis, M.; Bruins, H.M.; Hernández, V.; Espinós, E.L.; Dunn, J.; Rouanne, M.; et al. Updated 2016 EAU Guidelines on Muscle-invasive and Metastatic Bladder Cancer. Eur. Urol. 2017, 71, 462–475. [Google Scholar] [CrossRef]
- Pearlman, R.; Markow, M.; Knight, D.; Chen, W.; Arnold, C.A.; Pritchard, C.C.; Hampel, H.; Frankel, W.L. Two-stain immunohistochemical screening for Lynch syndrome in colorectal cancer may fail to detect mismatch repair deficiency. Mod. Pathol. 2018, 31, 1891–1900. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amsterdam Criteria I |
|---|
| There should be at least three relatives with a CRC (colorectal cancer). |
| One should be a first-degree relative of the other two. |
| At least two successive generations should be affected. |
| At least one should be diagnosed before the age of 50 years. |
| Familial adenomatous polyposis (FAP) should be excluded. |
| Tumors should be verified by pathological examination. |
| Revised Bethesda Guidelines |
|---|
| Patients meeting any one of the following should undergo microsatellite instability (MSI) testing: |
| CRC diagnosed in an individual under age 50 years. |
| Presence of synchronous, metachronous colorectal, or other LS-associated tumors *, regardless of age. |
| CRC with the MSI-H (high-frequency MSI) histology ‡, in a patient <60 years of age. |
| CRC diagnosed in 2 or more first- or second-degree relatives with LS-related tumors *, regardless of age. |
| CRC in 1 or more first-degree relatives with a LS-related tumor *, with 1 of the cancers being diagnosed under age 50 years. |
| MIPA Criteria |
|---|
| Patients meeting any one of the following should undergo MSI analysis: |
| CRC before the age of 50 years. |
| Two LS-associated tumors, including synchronous or metachronous CRCs or LS-associated tumors. |
| Adenoma before the age of 40 years. |
| Gene | MLH1 | MSH2 | MSH6 | PMS2 |
|---|---|---|---|---|
| MLH | − * | + | + | + |
| MSH2 | + | − | + | + |
| MSH6 | + | − | − | + |
| PMS2 | − | + | + | − |
| Cluster 1 | No PIK3CA mutation, non-smokers, high-grade < pT2 tumors, high recurrence |
| Cluster 2 | 100% FGFR3 mutation, tobacco use, low-grade tumors, non-invasive disease, no bladder recurrences |
| Cluster 3 | 100% FGFR3 mutations, 71% PIK3CA, no TP53 mutations, tobacco use, tumors all <pT2, five bladder recurrences |
| Cluster 4 | KMT2D (62.5%), FGFR3 (50%), TP53 (50%) mutations, no PIK3CA mutations, tobacco use, high-grade pT2+ disease, carcinoma in situ, shorter survival |
| Agents | Targets | Comparator | Study | Study Phase | Status | Patient Enrollment | Study Number | Primary Outcome Measures | Secondary Outcome Measures |
|---|---|---|---|---|---|---|---|---|---|
| Pembrolizumab | PD-1 | - | MK-3475-016 | II | completed | 113 | NCT01876511 | irPFS 20 w irORR PFS 20 w | OS irPFS 28 w ORR AE PFS 28 w DCR MSI as marker |
| Nivolumab/ Nivolumab + Ipilimumab/ Nivolumab + Ipilimumab + Cobimetinib/ Nivolumab + Daratumumab | PD-1 CTLA-4 MEK CD38 | - | Checkmate 142 | II | active, not recruiting | 340 | NCT02060188 | ORR | ORR |
| Combination Chemotherapy + Atezolizumab | PD-L1 | Combination Chemotherapy | NCI-2016-01417 | III | recruiting | 700 | NCT02912559 | DFS | OS AE |
| Pembrolizumab | PD-1 | Standard of Care | Keynote-177 | III | active, not recruiting | 308 | NCT02563002 | PFS OS | ORR |
| Nivolumab | PD-1 | - | NCI-2018-01491 | II | active, not recruiting | 3 | NCT03631641 | Adenoma incidence | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindner, A.K.; Schachtner, G.; Tulchiner, G.; Thurnher, M.; Untergasser, G.; Obrist, P.; Pipp, I.; Steinkohl, F.; Horninger, W.; Culig, Z.; et al. Lynch Syndrome: Its Impact on Urothelial Carcinoma. Int. J. Mol. Sci. 2021, 22, 531. https://doi.org/10.3390/ijms22020531
Lindner AK, Schachtner G, Tulchiner G, Thurnher M, Untergasser G, Obrist P, Pipp I, Steinkohl F, Horninger W, Culig Z, et al. Lynch Syndrome: Its Impact on Urothelial Carcinoma. International Journal of Molecular Sciences. 2021; 22(2):531. https://doi.org/10.3390/ijms22020531
Chicago/Turabian StyleLindner, Andrea Katharina, Gert Schachtner, Gennadi Tulchiner, Martin Thurnher, Gerold Untergasser, Peter Obrist, Iris Pipp, Fabian Steinkohl, Wolfgang Horninger, Zoran Culig, and et al. 2021. "Lynch Syndrome: Its Impact on Urothelial Carcinoma" International Journal of Molecular Sciences 22, no. 2: 531. https://doi.org/10.3390/ijms22020531
APA StyleLindner, A. K., Schachtner, G., Tulchiner, G., Thurnher, M., Untergasser, G., Obrist, P., Pipp, I., Steinkohl, F., Horninger, W., Culig, Z., & Pichler, R. (2021). Lynch Syndrome: Its Impact on Urothelial Carcinoma. International Journal of Molecular Sciences, 22(2), 531. https://doi.org/10.3390/ijms22020531