Inhibition of InsP3R with Xestospongin B Reduces Mitochondrial Respiration and Induces Selective Cell Death in T Cell Acute Lymphoblastic Leukemia Cells

 ,
,  , and
, and 
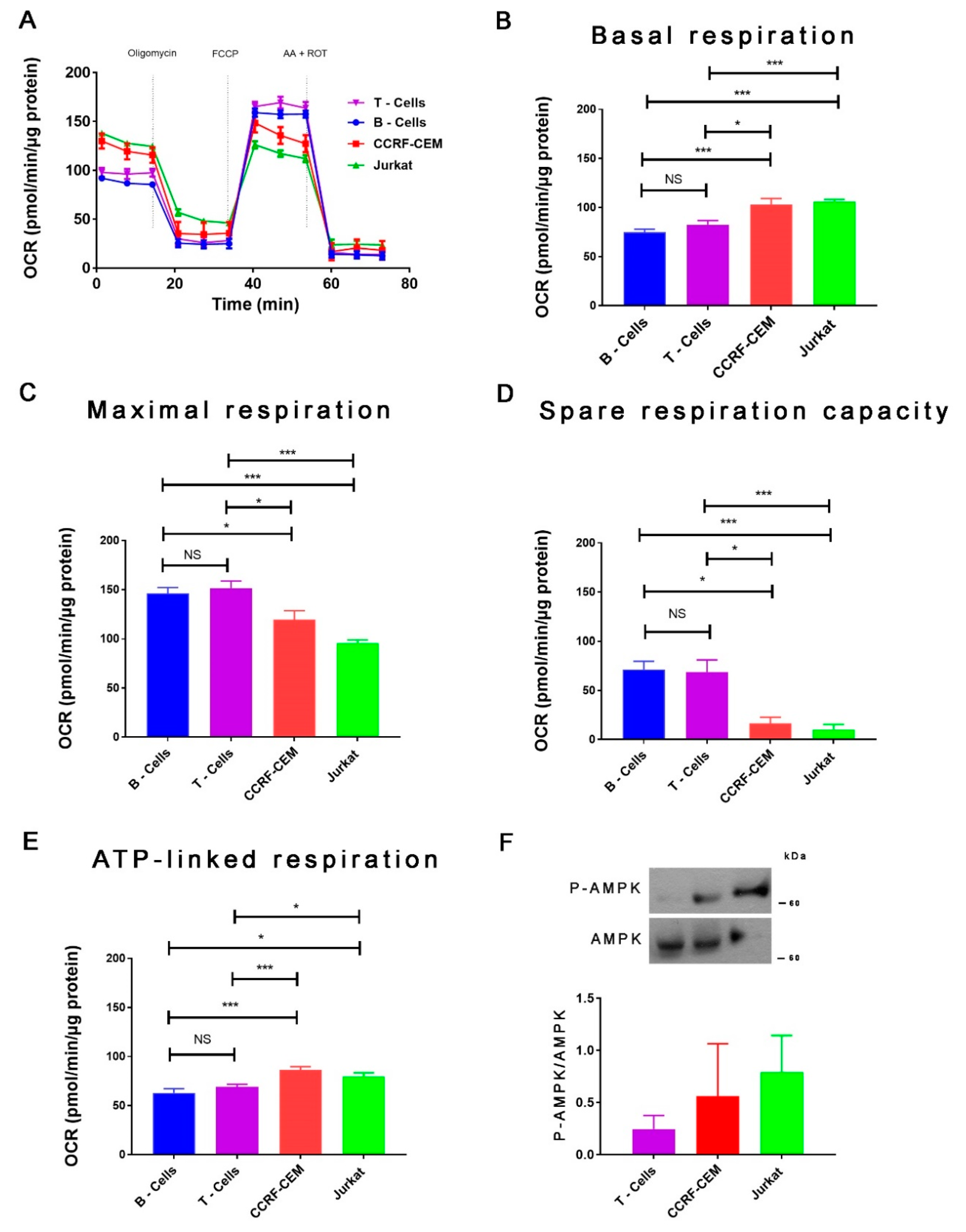{kind=link}
{kind=link}
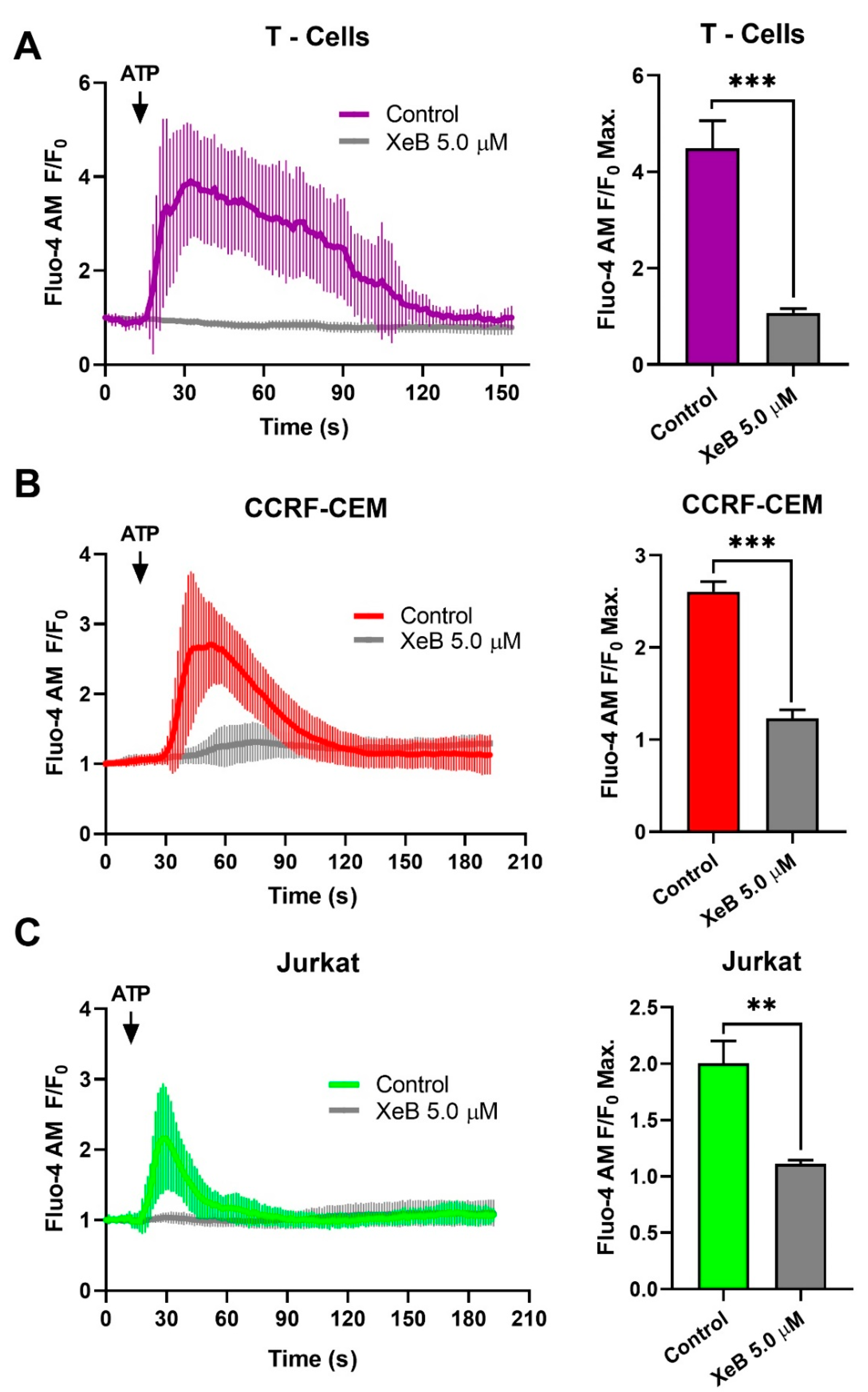{kind=link}
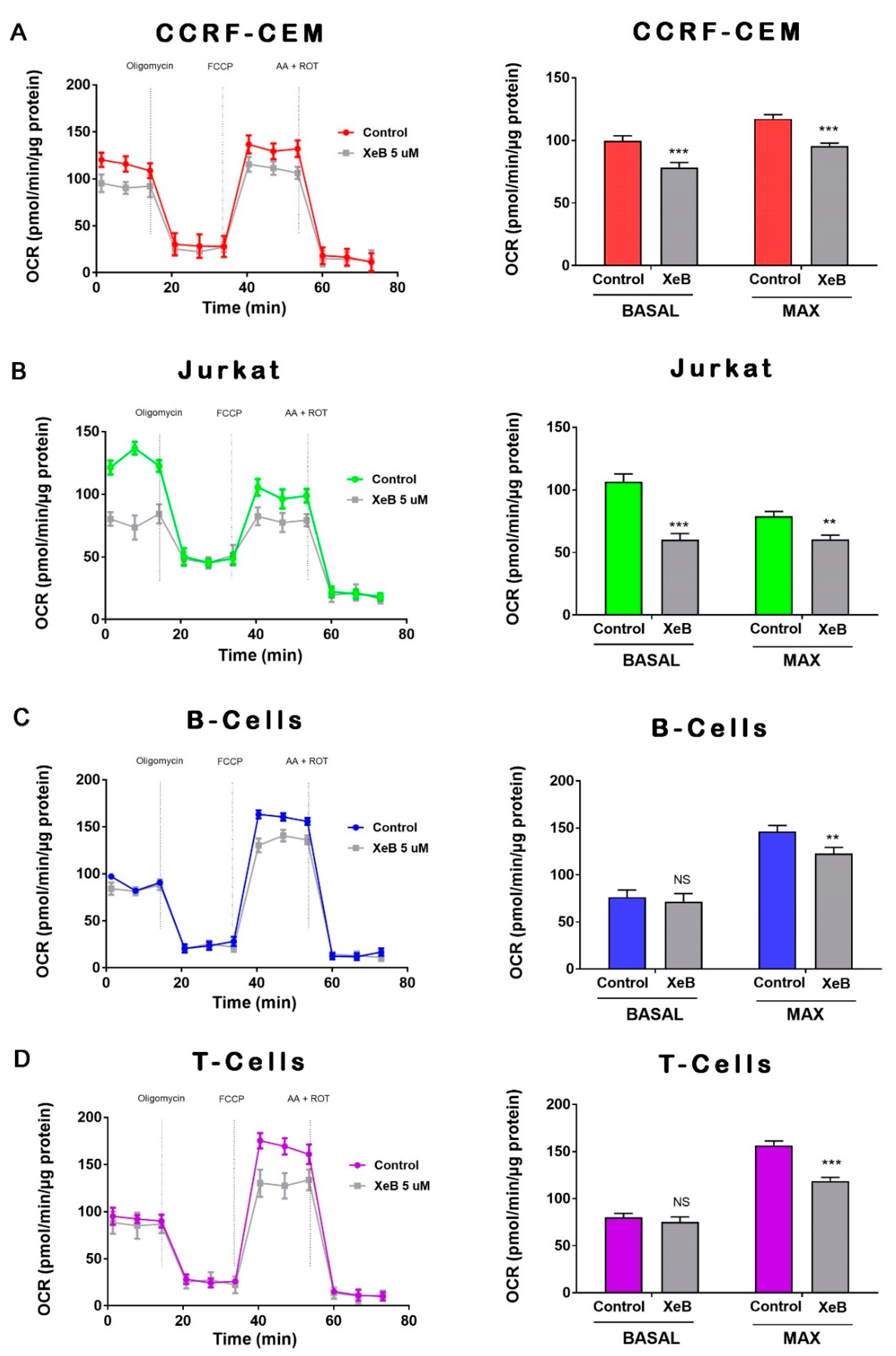{kind=link}
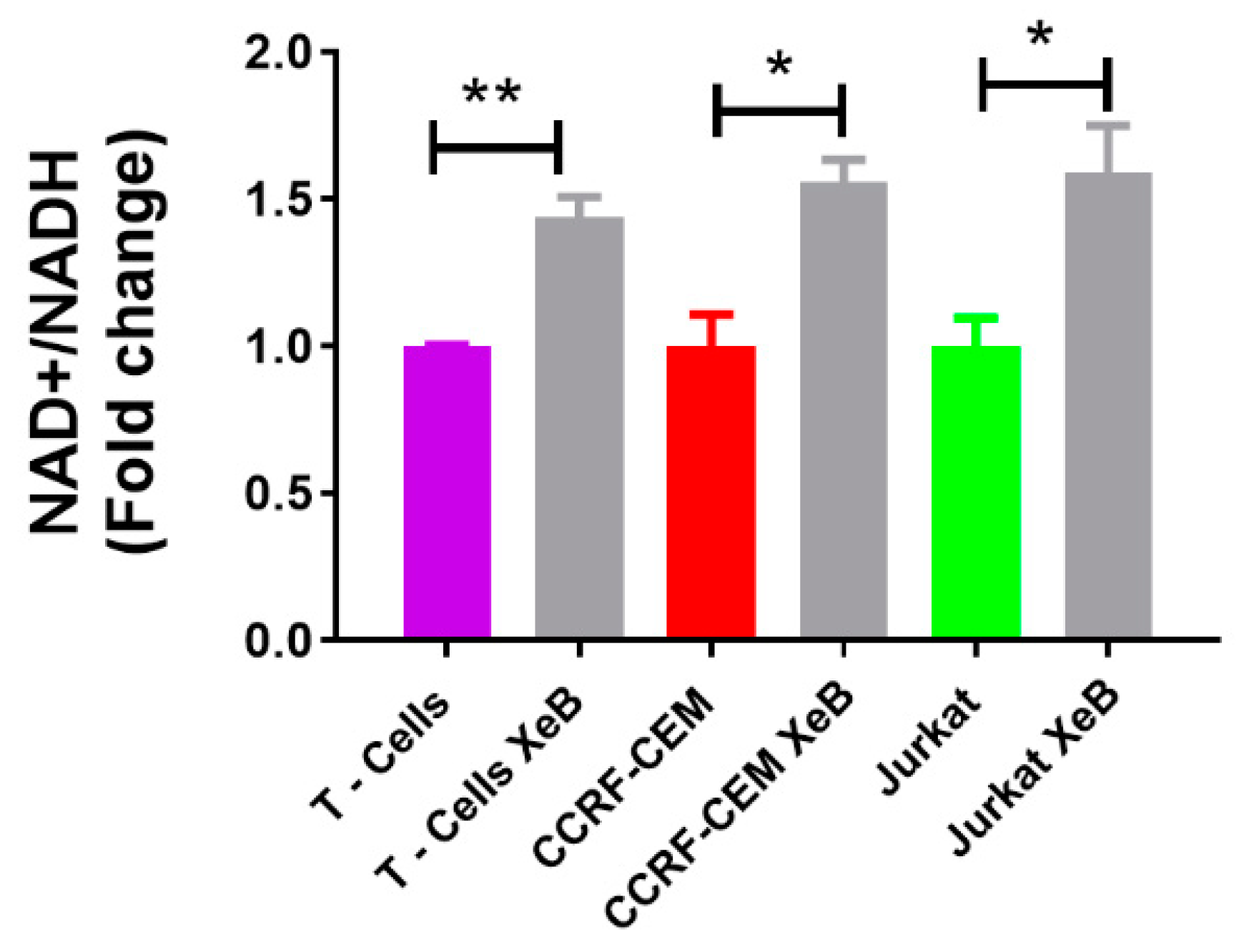{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
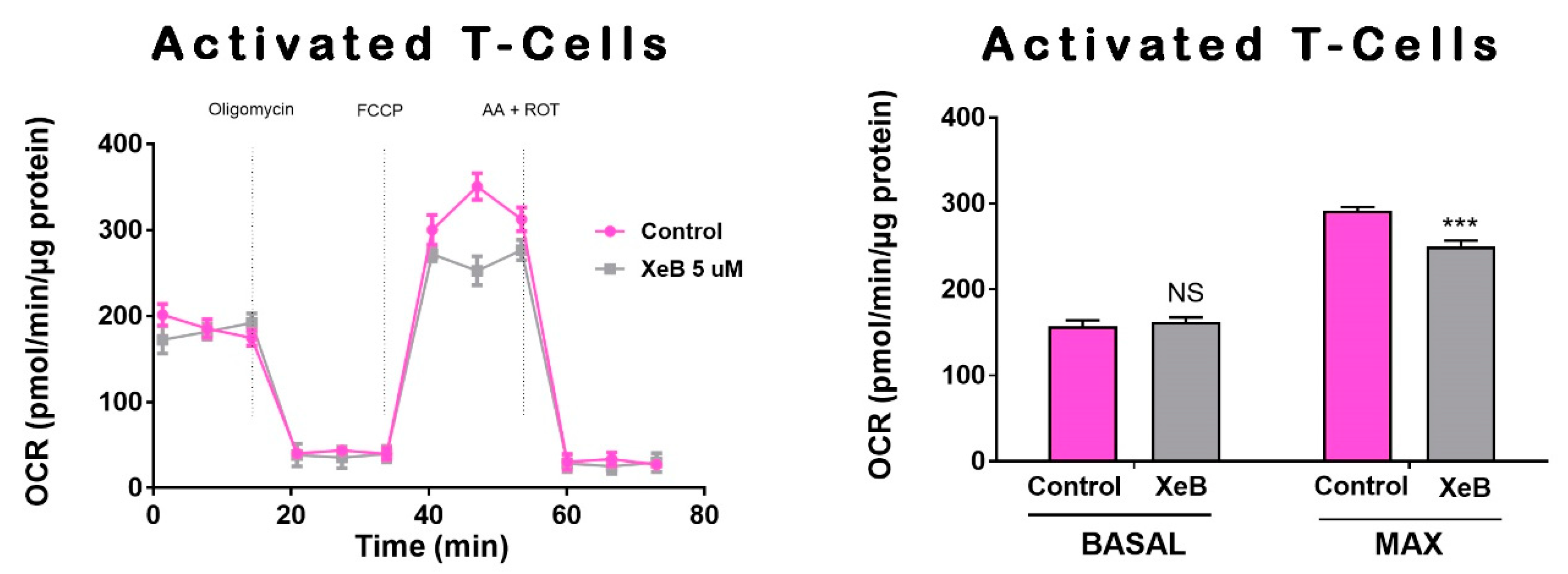
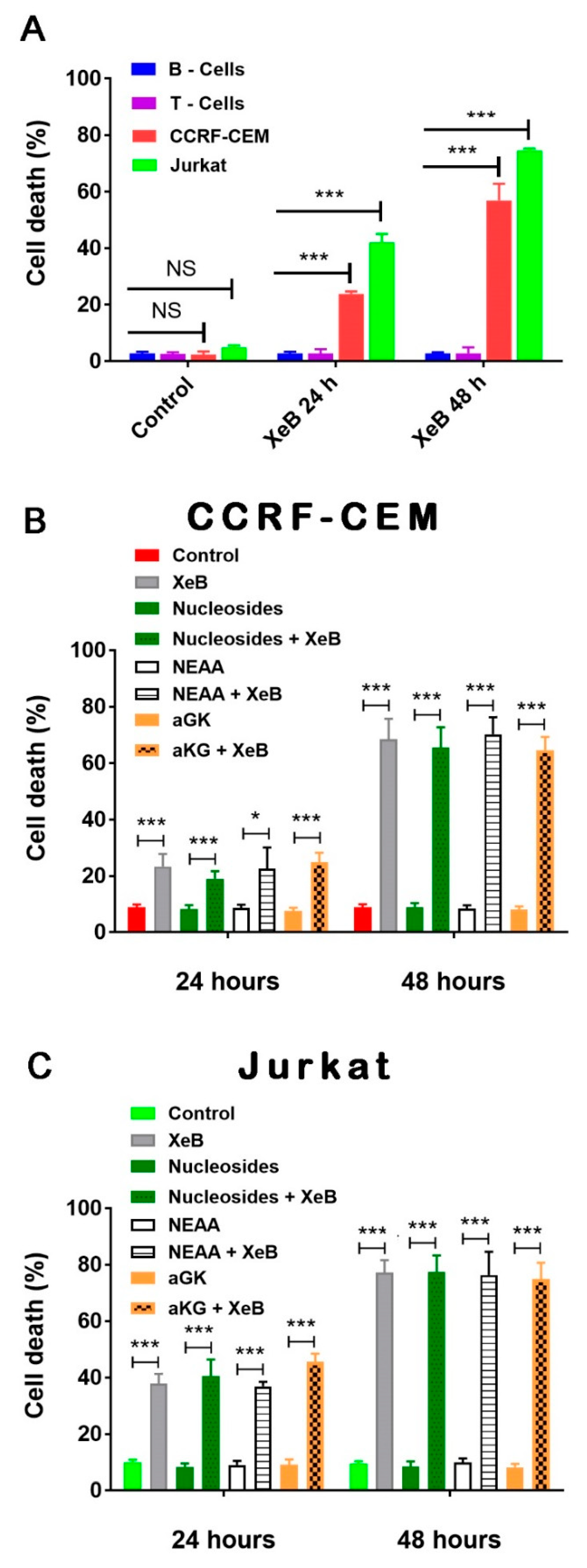
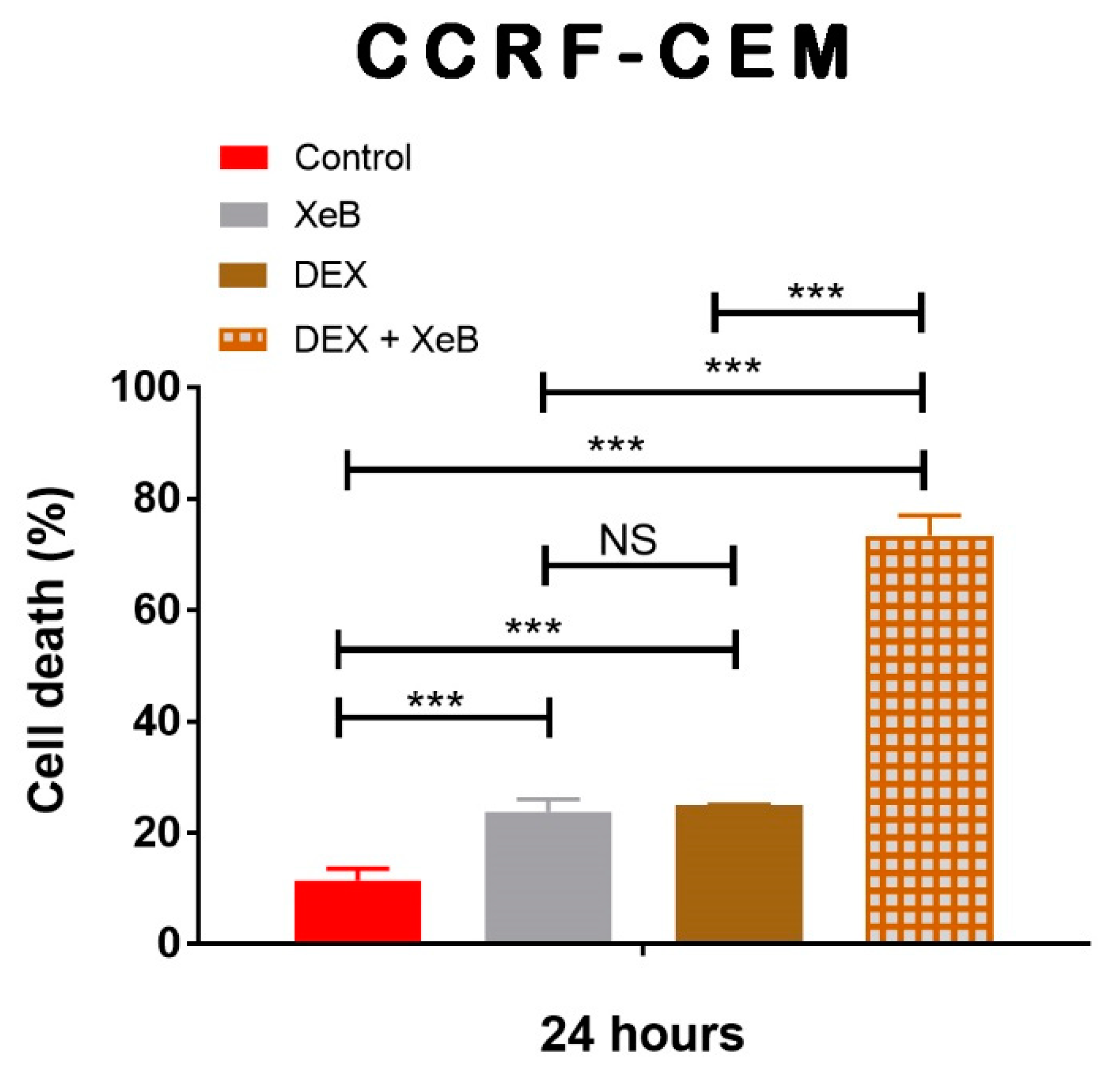
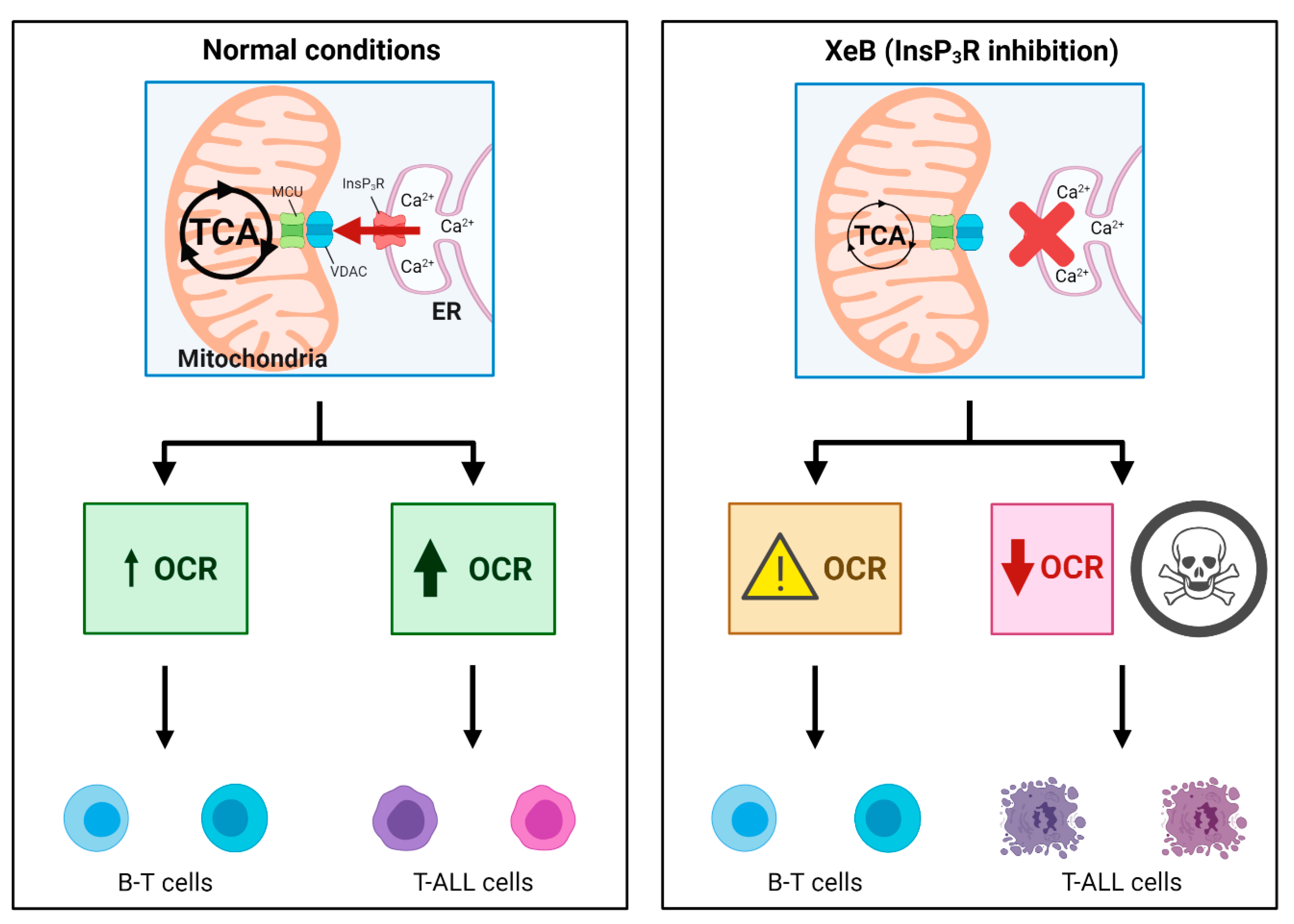
2. Results
3. Discussion
4. Materials and Methods
4.1. Chemical and Reagents
4.2. Cell Culture
4.3. Isolation of Peripheral Blood Mononuclear Cells
4.4. Activation of T Cells
4.5. Bioenergetic Profile
4.6. Ca2+ Measures
4.7. Immunoblots
4.8. NAD+/NADH Measures
4.9. Cell Death
4.10. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| XeB | Xestospongin B |
| T-ALL | Acute T-lymphoblastic leukemia |
| OCR | Oxygen Consumption Rate |
| OXPHOS | Oxidative phosphorylation |
| DEX | Dexamethasone |
| PBMC | Peripheral blood mononuclear cells |
References
- Pui, C.H.; Relling, M.V.; Downing, J.R. Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2004, 350, 1535–1548. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Evans, W.E. Treatment of Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2006, 354, 166–178. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Robison, L.L.; Hudson, M.M. Survivors of Childhood and Adolescent Cancer: Life-Long Risks and Responsibilities. Nat. Rev. Cancer 2014, 14, 61–70. [Google Scholar] [CrossRef]
- Jones, C.L.; Gearheart, C.M.; Fosmire, S.; Delgado-Martin, C.; Evensen, N.A.; Bride, K.; Waanders, A.J.; Pais, F.; Wang, J.; Bhatla, T.; et al. MAPK Signaling Cascades Mediate Distinct Glucocorticoid Resistance Mechanisms in Pediatric Leukemia. Blood 2015, 126, 2202–2212. [Google Scholar] [CrossRef]
- Chonghaile, T.N.; Roderick, J.E.; Glenfield, C.; Ryan, J.; Sallan, S.E.; Silverman, L.B.; Loh, M.L.; Hunger, S.P.; Wood, B.; DeAngelo, D.J.; et al. Maturation Stage of T-Cell Acute Lymphoblastic Leukemia Determines BCL-2 versus BCL-XL Dependence and Sensitivity to ABT-199. Cancer Discov. 2014, 4, 1074–1087. [Google Scholar] [CrossRef]
- Peirs, S.; Matthijssens, F.; Goossens, S.; Van de Walle, I.; Ruggero, K.; de Bock, C.E.; Degryse, S.; Canté-Barrett, K.; Briot, D.; Clappier, E.; et al. ABT-199 Mediated Inhibition of BCL-2 as a Novel Therapeutic Strategy in T-Cell Acute Lymphoblastic Leukemia. Blood 2014, 124, 3738–3747. [Google Scholar] [CrossRef]
- Deenik, W.; Beverloo, H.B.; van der Poel-van de Luytgaarde, S.C.P.A.M.; Wattel, M.M.; van Esser, J.W.J.; Valk, P.J.M.; Cornelissen, J.J. Rapid Complete Cytogenetic Remission after Upfront Dasatinib Monotherapy in a Patient with a NUP214-ABL1-Positive T-Cell Acute Lymphoblastic Leukemia. Leukemia 2009, 23, 627–629. [Google Scholar] [CrossRef]
- Pikman, Y.; Alexe, G.; Roti, G.; Conway, A.S.; Furman, A.; Lee, E.S.; Place, A.E.; Kim, S.; Saran, C.; Modiste, R.; et al. Synergistic Drug Combinations with a CDK4/6 Inhibitor in T-Cell Acute Lymphoblastic Leukemia. Clin. Cancer Res. 2017, 23, 1012–1024. [Google Scholar] [CrossRef]
- Gomes-Silva, D.; Srinivasan, M.; Sharma, S.; Lee, C.M.; Wagner, D.L.; Davis, T.H.; Rouce, R.H.; Bao, G.; Brenner, M.K.; Mamonkin, M. CD7-Edited T Cells Expressing a CD7-Specific CAR for the Therapy of T-Cell Malignancies. Blood 2017, 130, 285–296. [Google Scholar] [CrossRef]
- Subramaniam, P.S.; Whye, D.W.; Efimenko, E.; Chen, J.; Tosello, V.; De Keersmaecker, K.; Kashishian, A.; Thompson, M.A.; Castillo, M.; Cordon-Cardo, C.; et al. Targeting Nonclassical Oncogenes for Therapy in T-ALL. Cancer Cell 2012, 21, 459–472. [Google Scholar] [CrossRef] [PubMed]
- Bride, K.L.; Vincent, T.L.; Im, S.-Y.; Aplenc, R.; Barrett, D.M.; Carroll, W.L.; Carson, R.; Dai, Y.; Devidas, M.; Dunsmore, K.P.; et al. Preclinical Efficacy of Daratumumab in T-Cell Acute Lymphoblastic Leukemia. Blood 2018, 131, 995–999. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Sayed, N.; Ditsworth, D.; Thompson, C.B. Brick by Brick: Metabolism and Tumor Cell Growth. Curr. Opin. Genet. Dev. 2008, 18, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the Origin of Cancer Cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Fan, Y.; Dickman, K.G.; Zong, W.X. Akt and C-Myc Differentially Activate Cellular Metabolic Programs and Prime Cells to Bioenergetic Inhibition. J. Biol. Chem. 2010, 285, 7324–7333. [Google Scholar] [CrossRef]
- Moreno-Sánchez, R.; Marín-Hernández, A.; Saavedra, E.; Pardo, J.P.; Ralph, S.J.; Rodríguez-Enríquez, S. Who Controls the ATP Supply in Cancer Cells? Biochemistry Lessons to Understand Cancer Energy Metabolism. Int. J. Biochem. Cell Biol. 2014, 50, 10–23. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic Reprogramming and Cancer Progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.-N.A.; et al. An Inhibitor of Oxidative Phosphorylation Exploits Cancer Vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef]
- McGuirk, S.; Audet-Delage, Y.; St-Pierre, J. Metabolic Fitness and Plasticity in Cancer Progression. Trends Cancer 2020, 6, 49–61. [Google Scholar] [CrossRef]
- Sanchez-Martin, M.; Ferrando, A. The NOTCH1-MYC Highway toward T-Cell Acute Lymphoblastic Leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Kishton, R.J.; Barnes, C.E.; Nichols, A.G.; Cohen, S.; Gerriets, V.A.; Siska, P.J.; Macintyre, A.N.; Goraksha-Hicks, P.; de Cubas, A.A.; Liu, T.; et al. AMPK Is Essential to Balance Glycolysis and Mitochondrial Metabolism to Control T-ALL Cell Stress and Survival. Cell Metab. 2016, 23, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential Regulation of Cell Bioenergetics by Constitutive InsP3 Receptor Ca2+ Transfer to Mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, C.; Müller, M.; McNeal, A.; Lovy, A.; Jaňa, F.; Bustos, G.; Urra, F.; Smith, N.; Molgó, J.; Diehl, J.A.; et al. Selective Vulnerability of Cancer Cells by Inhibition of Ca(2+) Transfer from Endoplasmic Reticulum to Mitochondria. Cell Rep. 2016, 15, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, C.; Lovy, A.; Silva-Pavez, E.; Urra, F.; Mizzoni, C.; Ahumada-Castro, U.; Bustos, G.; Jaňa, F.; Cruz, P.; Farias, P.; et al. Cancer Cells with Defective Oxidative Phosphorylation Require Endoplasmic Reticulum-to-Mitochondria Ca. Sci. Signal. 2020, 13, eaay1212. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Yi, Q.; Xu, B.; Li, S.; Wang, T.; Liu, F.; Zhu, B.; Hoffmann, P.R.; Ji, G.; Lei, P.; et al. ORP4L Is Essential for T-Cell Acute Lymphoblastic Leukemia Cell Survival. Nat. Commun. 2016, 7, 12702. [Google Scholar] [CrossRef]
- Orabi, K.Y.; El Sayed, K.A.; Hamann, M.T.; Dunbar, D.C.; Al-Said, M.S.; Higa, T.; Kelly, M. Araguspongines K and L, New Bioactive Bis-1-Oxaquinolizidine N-Oxide Alkaloids from Red Sea Specimens of Xestospongia Exigua. J. Nat. Prod. 2002, 65, 1782–1785. [Google Scholar] [CrossRef]
- Jaimovich, E.; Mattei, C.; Liberona, J.L.; Cardenas, C.; Estrada, M.; Barbier, J.; Debitus, C.; Laurent, D.; Molgó, J. Xestospongin B, a Competitive Inhibitor of IP3-Mediated Ca2+ Signalling in Cultured Rat Myotubes, Isolated Myonuclei, and Neuroblastoma (NG108-15) Cells. FEBS Lett. 2005, 579, 2051–2057. [Google Scholar] [CrossRef]
- Lovy, A.; Ahumada-Castro, U.; Bustos, G.; Farias, P.; Gonzalez-Billault, C.; Molgó, J.; Cardenas, C. Concerted Action of AMPK and Sirtuin-1 Induces Mitochondrial Fragmentation Upon Inhibition of Ca(2+) Transfer to Mitochondria. Front. Cell Dev. Biol. 2020, 8, 378. [Google Scholar] [CrossRef]
- Sinha, R.A.; Singh, B.K.; Zhou, J.; Wu, Y.; Farah, B.L.; Ohba, K.; Lesmana, R.; Gooding, J.; Bay, B.H.; Yen, P.M. Thyroid Hormone Induction of Mitochondrial Activity Is Coupled to Mitophagy via ROS-AMPK-ULK1 Signaling. Autophagy 2015, 11, 1341–1357. [Google Scholar] [CrossRef]
- Nicholas, D.; Proctor, E.A.; Raval, F.M.; Ip, B.C.; Habib, C.; Ritou, E.; Grammatopoulos, T.N.; Steenkamp, D.; Dooms, H.; Apovian, C.M.; et al. Advances in the Quantification of Mitochondrial Function in Primary Human Immune Cells through Extracellular Flux Analysis. PLoS ONE 2017, 12, e0170975. [Google Scholar] [CrossRef] [PubMed]
- Waters, L.R.; Ahsan, F.M.; Wolf, D.M.; Shirihai, O.; Teitell, M.A. Initial B Cell Activation Induces Metabolic Reprogramming and Mitochondrial Remodeling. iScience 2018, 5, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sánchez, R.; Rodríguez-Enríquez, S.; Marín-Hernández, A.; Saavedra, E. Energy Metabolism in Tumor Cells. FEBS J. 2007, 274, 1393–1418. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and Cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between Glycolysis and Oxidative Phosphorylation: A Tumor’s Dilemma? Biochim. Biophys. Acta Bioenerg. 2011, 1807, 552–561. [Google Scholar] [CrossRef]
- Urra, F.A.; Muñoz, F.; Córdova-Delgado, M.; Ramírez, M.P.; Peña-Ahumada, B.; Rios, M.; Cruz, P.; Ahumada-Castro, U.; Bustos, G.; Silva-Pavez, E.; et al. FR58P1a; a New Uncoupler of OXPHOS That Inhibits Migration in Triple-Negative Breast Cancer Cells via Sirt1/AMPK/Β1-Integrin Pathway. Sci. Rep. 2018, 8, 13190. [Google Scholar] [CrossRef]
- Jana, F.; Bustos, G.; Rivas, J.; Cruz, P.; Urra, F.; Basualto-Alarcon, C.; Sagredo, E.; Rios, M.; Lovy, A.; Dong, Z.; et al. Complex I and II Are Required for Normal Mitochondrial Ca(2+) Homeostasis. Mitochondrion 2019, 49, 73–82. [Google Scholar] [CrossRef]
- Ashton, T.M.; Gillies McKenna, W.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef]
- Ganapathy-kanniappan, S.; Geschwind, J.-F. Tumor Glycolysis as a Target for Cancer Therapy. Mol. Cancer 2013, 12, 152. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Glycolysis in Cancer: A Potential Target for Therapy. Int. J. Biochem. Cell Biol. 2007, 39, 1358–1366. [Google Scholar] [CrossRef]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 Inhibition Targets Oxidative Phosphorylation and Selectively Eradicates Quiescent Human Leukemia Stem Cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Martinez Marignac, V.L.; Smith, S.; Toban, N.; Bazile, M.; Aloyz, R. Resistance to Dasatinib in Primary Chronic Lymphocytic Leukemia Lymphocytes Involves AMPK-Mediated Energetic Re-Programming. Oncotarget 2013, 4, 2550–2566. [Google Scholar] [CrossRef] [PubMed]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic Signatures Uncover Distinct Targets in Molecular Subsets of Diffuse Large B Cell Lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, E.; Cioffi, M.; Sancho, P.; Sanchez-Ripoll, Y.; Trabulo, S.M.; Dorado, J.; Balic, A.; Hidalgo, M.; Heeschen, C. Metformin Targets the Metabolic Achilles Heel of Human Pancreatic Cancer Stem Cells. PLoS ONE 2013, 8, e76518. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic Determinants of Cancer Cell Sensitivity to Glucose Limitation and Biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1α Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef]
- Yuan, P.; Ito, K.; Perez-Lorenzo, R.; Del Guzzo, C.; Lee, J.H.; Shen, C.-H.; Bosenberg, M.W.; McMahon, M.; Cantley, L.C.; Zheng, B. Phenformin Enhances the Therapeutic Benefit of BRAF(V600E) Inhibition in Melanoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18226–18231. [Google Scholar] [CrossRef]
- Hlozkova, K.; Pecinova, A.; Alquezar-Artieda, N.; Pajuelo-Reguera, D.; Simcikova, M.; Hovorkova, L.; Rejlova, K.; Zaliova, M.; Mracek, T.; Kolenova, A.; et al. Metabolic Profile of Leukemia Cells Influences Treatment Efficacy of L-Asparaginase. BMC Cancer 2020, 20, 526. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Goncalves, R.L.S.; Gerencser, A.A.; Nicholls, D.G.; Brand, M.D. The Contributions of Respiration and Glycolysis to Extracellular Acid Production. Biochim. Biophys. Acta 2015, 1847, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Marquardt, C.; Foker, J. Aerobic Glycolysis during Lymphocyte Proliferation. Nature 1976, 261, 702–705. [Google Scholar] [CrossRef]
- Ahn, C.S.; Metallo, C.M. Mitochondria as Biosynthetic Factories for Cancer Proliferation. Cancer Metab. 2015, 3, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; Deberardinis, R.J. Reductive Carboxylation Supports Growth in Tumour Cells with Defective Mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; Thompson, C.B. Glutamine Addiction: A New Therapeutic Target in Cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef]
- Shanware, N.P.; Mullen, A.R.; DeBerardinis, R.J.; Abraham, R.T. Glutamine: Pleiotropic Roles in Tumor Growth and Stress Resistance. J. Mol. Med. 2011, 89, 229–236. [Google Scholar] [CrossRef]
- McCormack, J.G.; Denton, R.M. The Effects of Calcium Ions and Adenine Nucleotides on the Activity of Pig Heart 2-Oxoglutarate Dehydrogenase Complex. Biochem. J. 1979, 180, 533–544. [Google Scholar] [CrossRef]
- Denton, R.M.; Randle, P.J.; Martin, B.R. Stimulation by Calcium Ions of Pyruvate Dehydrogenase Phosphate Phosphatase. Biochem. J. 1972, 128, 161–163. [Google Scholar] [CrossRef]
- Glancy, B.; Balaban, R.S. Role of Mitochondrial Ca2+ in the Regulation of Cellular Energetics. Biochemistry 2012, 51, 2959–2973. [Google Scholar] [CrossRef]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging Interorganelle Contacts and Local Calcium Dynamics at the ER-Mitochondrial Interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-F.; Yin, J.-H.; Hwang, C.-S.; Tang, C.-M.; Yang, D.-I. NAD Attenuates Oxidative DNA Damages Induced by Amyloid Beta-Peptide in Primary Rat Cortical Neurons. Free Radic. Res. 2014, 48, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhao, K.-K.; Tong, Y.; Zhou, Y.-L.; Wang, Y.-X.; Zhao, P.-Q.; Wang, Z.-Y. Exogenous NAD(+) Decreases Oxidative Stress and Protects H2O2-Treated RPE Cells against Necrotic Death through the up-Regulation of Autophagy. Sci. Rep. 2016, 6, 26322. [Google Scholar] [CrossRef] [PubMed]
- Campàs, C.; López, J.M.; Santidrián, A.F.; Barragán, M.; Bellosillo, B.; Colomer, D.; Gil, J. Acadesine Activates AMPK and Induces Apoptosis in B-Cell Chronic Lymphocytic Leukemia Cells but Not in T Lymphocytes. Blood 2003, 101, 3674–3680. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Xu, M.; Li, C.; Zhu, B.; Cao, X.; Li, D.; Chen, H.; Hu, C.; Li, R.; Luo, C.; et al. ORP4L Extracts and Presents PIP. Cell Rep. 2019, 26, 2166–2177. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, C.; Liberona, J.L.; Molgó, J.; Colasante, C.; Mignery, G.A.; Jaimovich, E. Nuclear Inositol 1,4,5-Trisphosphate Receptors Regulate Local Ca2+ Transients and Modulate CAMP Response Element Binding Protein Phosphorylation. J. Cell Sci. 2005, 118 Pt 14, 3131–3140. [Google Scholar] [CrossRef]
- Zhong, F.; Harr, M.W.; Bultynck, G.; Monaco, G.; Parys, J.B.; De Smedt, H.; Rong, Y.P.; Molitoris, J.K.; Lam, M.; Ryder, C.; et al. Induction of Ca2+-Driven Apoptosis in Chronic Lymphocytic Leukemia Cells by Peptide-Mediated Disruption of Bcl-2-IP3 Receptor Interaction. Blood 2011, 117, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Akl, H.; Monaco, G.; La Rovere, R.; Welkenhuyzen, K.; Kiviluoto, S.; Vervliet, T.; Molgó, J.; Distelhorst, C.W.; Missiaen, L.; Mikoshiba, K.; et al. IP3R2 Levels Dictate the Apoptotic Sensitivity of Diffuse Large B-Cell Lymphoma Cells to an IP3R-Derived Peptide Targeting the BH4 Domain of Bcl-2. Cell Death Dis. 2013, 4, e632. [Google Scholar] [CrossRef]
- Lavik, A.R.; Zhong, F.; Chang, M.J.; Greenberg, E.; Choudhary, Y.; Smith, M.R.; McColl, K.S.; Pink, J.; Reu, F.J.; Matsuyama, S.; et al. A Synthetic Peptide Targeting the BH4 Domain of Bcl-2 Induces Apoptosis in Multiple Myeloma and Follicular Lymphoma Cells Alone or in Combination with Agents Targeting the BH3-Binding Pocket of Bcl-2. Oncotarget 2015, 6, 27388–27402. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz, P.; Ahumada-Castro, U.; Bustos, G.; Molgó, J.; Sauma, D.; Lovy, A.; Cárdenas, C. Inhibition of InsP3R with Xestospongin B Reduces Mitochondrial Respiration and Induces Selective Cell Death in T Cell Acute Lymphoblastic Leukemia Cells. Int. J. Mol. Sci. 2021, 22, 651. https://doi.org/10.3390/ijms22020651
Cruz P, Ahumada-Castro U, Bustos G, Molgó J, Sauma D, Lovy A, Cárdenas C. Inhibition of InsP3R with Xestospongin B Reduces Mitochondrial Respiration and Induces Selective Cell Death in T Cell Acute Lymphoblastic Leukemia Cells. International Journal of Molecular Sciences. 2021; 22(2):651. https://doi.org/10.3390/ijms22020651
Chicago/Turabian StyleCruz, Pablo, Ulises Ahumada-Castro, Galdo Bustos, Jordi Molgó, Daniela Sauma, Alenka Lovy, and César Cárdenas. 2021. "Inhibition of InsP3R with Xestospongin B Reduces Mitochondrial Respiration and Induces Selective Cell Death in T Cell Acute Lymphoblastic Leukemia Cells" International Journal of Molecular Sciences 22, no. 2: 651. https://doi.org/10.3390/ijms22020651
APA StyleCruz, P., Ahumada-Castro, U., Bustos, G., Molgó, J., Sauma, D., Lovy, A., & Cárdenas, C. (2021). Inhibition of InsP3R with Xestospongin B Reduces Mitochondrial Respiration and Induces Selective Cell Death in T Cell Acute Lymphoblastic Leukemia Cells. International Journal of Molecular Sciences, 22(2), 651. https://doi.org/10.3390/ijms22020651