The Landscape of microRNAs in βCell: Between Phenotype Maintenance and Protection

 ,
, 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. β Cell Identity and Function in Health and Disease
2.1. What Does Define the Path to βCell Identity?
2.2. How Do β Cells Maintain Their Identity?
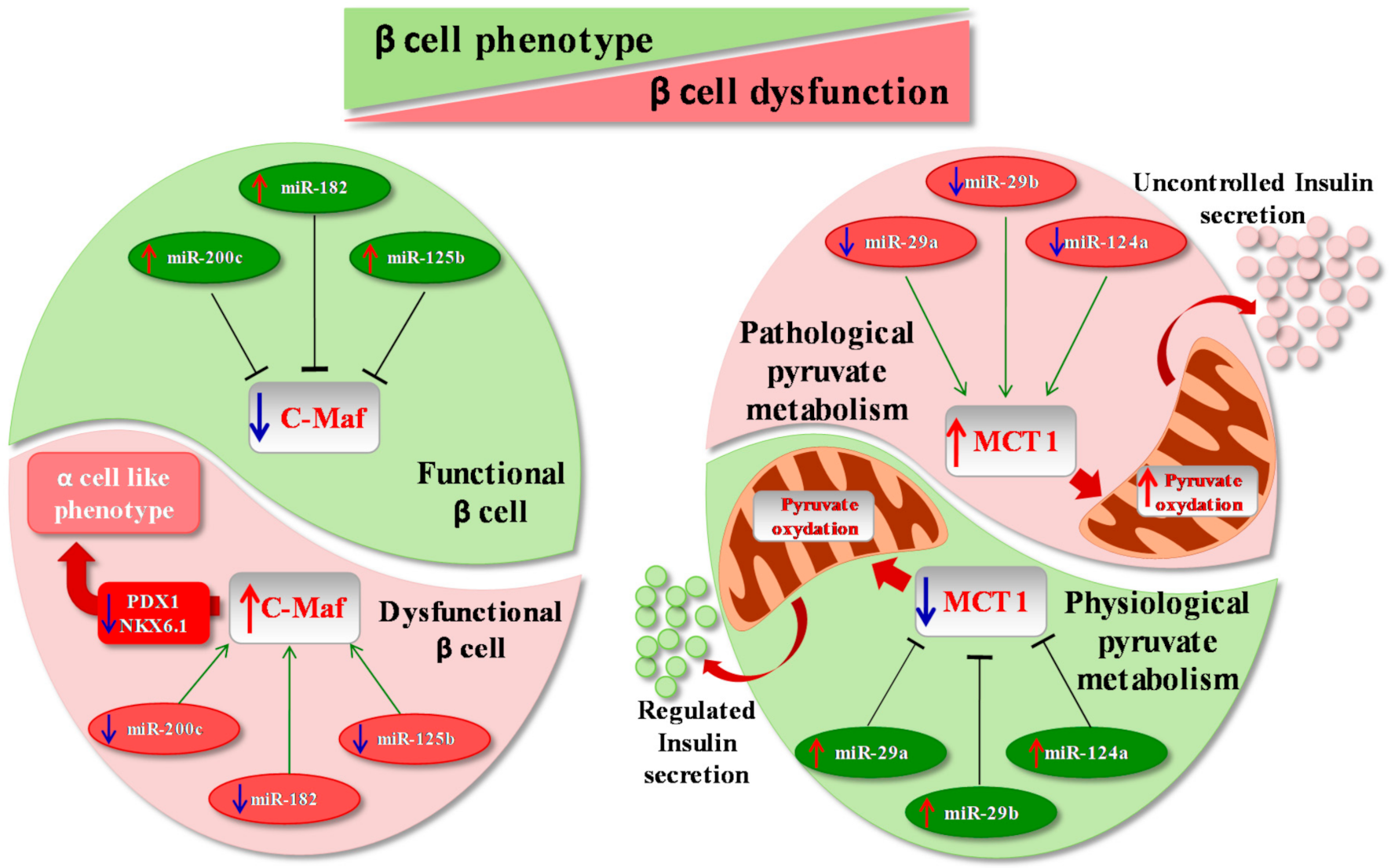
2.3. β Cell Phenotype Loss in Diabetes Mellitus
3. microRNAs, β Cells and Disallowed Genes
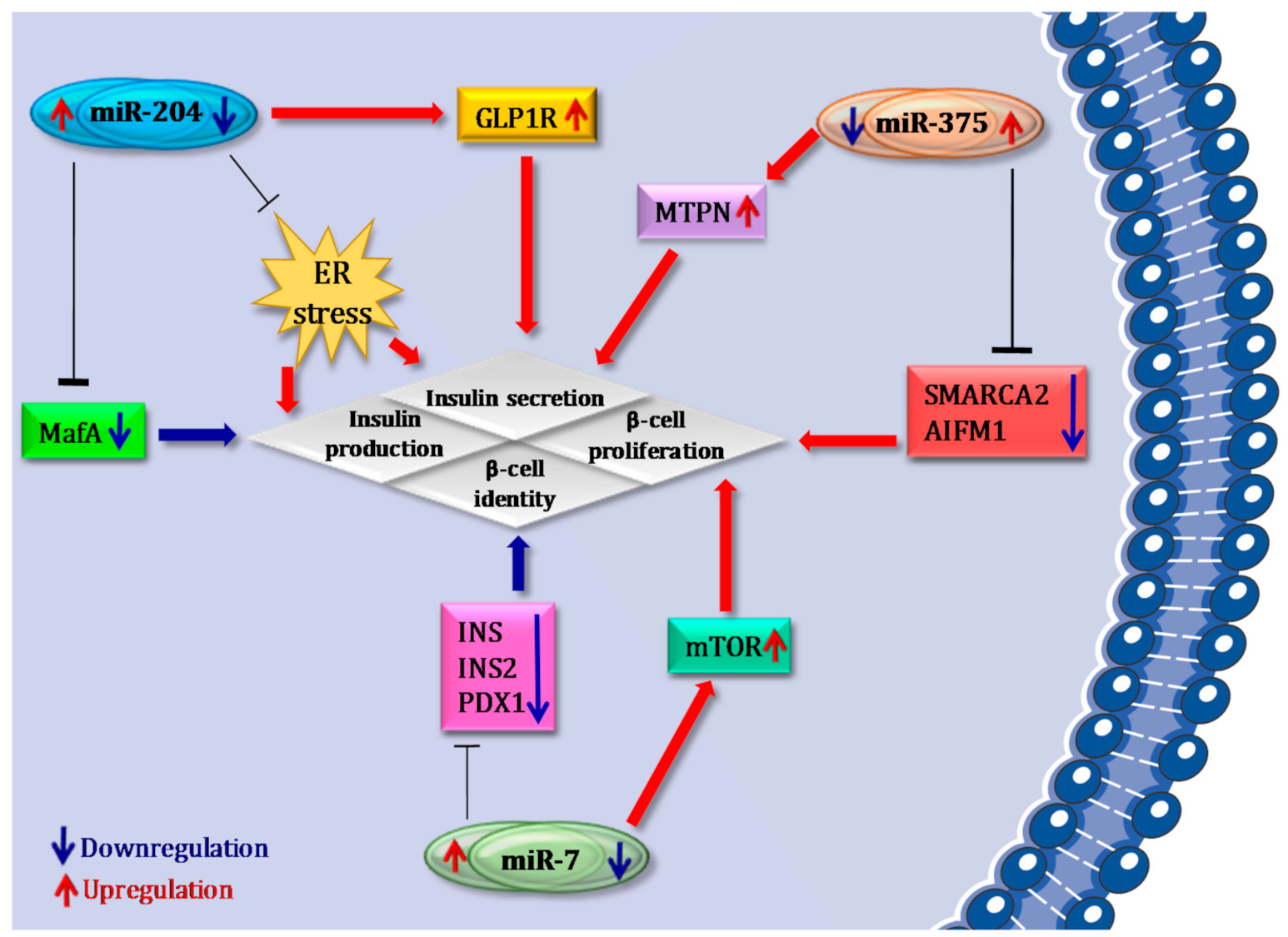
4. microRNAs that Confer Robustness to β Cell Identity: Focus on miR-375, miR-7 and miR-204
4.1. miR-375
4.2. miR-7
4.3. miR-204
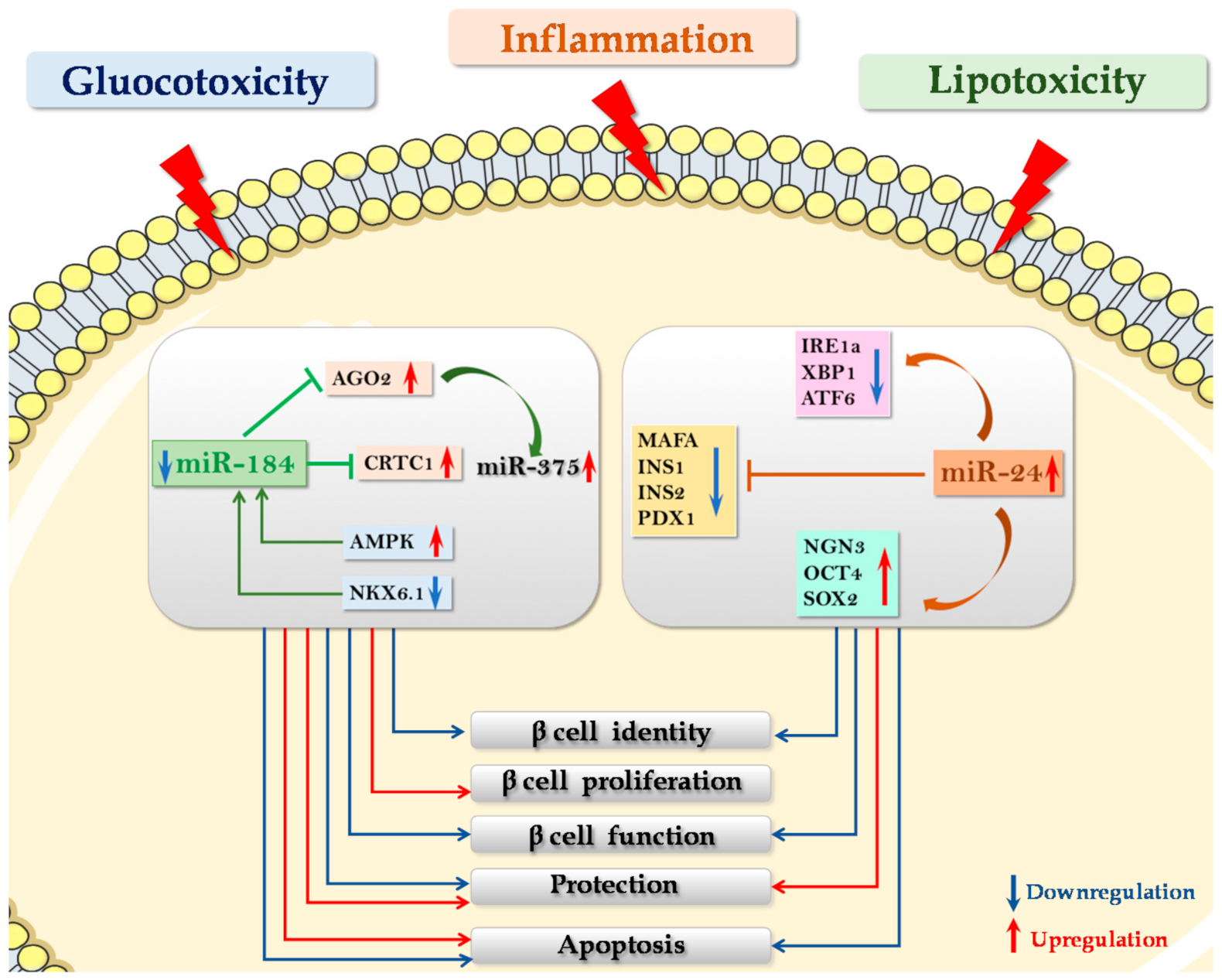
5. microRNAs in the Maintenance of β Cell Function: Protective and Compensatory Mechanisms in T2D
5.1. miR-24
5.2. miR-184
5.3. miR-200 Family
6. Can We Protect or Restore β Cell Function by Mimicking or Antagonizing Key Groups of miRNAs?
- 1.
- Generating in vitro functional β cells by modulating differentiation mechanisms of iPSCs and ESCs or by inducing transdifferentiation of other adult cell types into mature β cells. Substitution of highly expressed β cell miRNAs and/or silencing of dominant non-β cell miRNAs such as liver miR-122 [84] or neuronal miR-124 [85] have been demonstrated to affect the ability of multipotent stem cells to differentiate into insulin-positive cells. Indeed, virus-mediated overexpression of miR-375 in human skin fibroblast-derived iPSCs was sufficient to trigger their differentiation into insulin-expressing cells and to allow glucose-dependent insulin secretion in vitro [86]. Overexpression of miR-186 and miR-375 by chemical transfection of human iPSCs promoted the generation of islet-like cell clusters and induced the expression of β cell-specific markers [87].
- 2.
- Protecting β cells from gluco-lipotoxic and/or inflammatory stress. A recent work by Zhu and colleagues reported that microRNA miR-24, which is upregulated in pancreatic islets of diabetic db/db mice [73] as well as in islet cells subjected to palmitate-induced lipotoxicity [74], directly binds and regulates the expression of Ire1α in MIN6 murine β cell line (see above). Importantly, downregulation of Ire1α secondary to the overexpression of miR-24 was able to protect β cells from both palmitate- and thapsigargin- induced apoptosis [74]. Accordingly, it was demonstrated that Ire1α deletion restricted to β cells was able to inhibit β cell apoptosis, thus preventing disease onset in NOD mice [30]. Therefore, it is conceivable that the regulation of Ire1α -XBP1 by miR-24 is part of the molecular mechanisms involved in β cell protection from inflammatory-stress induced apoptosis. Altogether, these data indicate that modulation of miR-24 expression could be essential to protect β cells from apoptosis induced by different types of ER stress e.g., inflammatory and lipotoxic, although impairing β cell identity and function. Importantly, a peculiar miRNA that could link protection from stress and proliferative/compensatory events is miR-184, an interesting potential therapeutic target in diabetes mellitus. As a matter of fact, miR-184 downregulation protects β cells from both gluco/lipotoxicity- and inflammation- induced apoptosis [75,77].
- 3.
- Proliferation and/or regeneration of β cell. Interestingly, miR-184 inhibition leads to increased β cell mass, mainly by enhancing β cell proliferative capacity, as described by Tattikota et al., (see above) [78]. Finally, miR-7 is an additional candidate to be targeted in order to generate a therapy aiming at inducing β cell proliferation [66].
7. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zaccardi, F.; Webb, D.R.; Yates, T.; Davies, M.J. Pathophysiology of type 1 and type 2 diabetes mellitus: A 90-year perspective. Postgrad. Med. J. 2015, 92, 63–69. [Google Scholar] [CrossRef]
- Weir, G.C.; Aguayo-Mazzucato, C.; Bonner-Weir, S. β-cell dedifferentiation in diabetes is important, but what is it? Islets 2013, 5, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Sabouri, E.; Rajabzaseh, A.; Enderami, S.E.; Saburi, E.; Soleimanifar, F.; Barati, G.; Rahmati, M.; Khamisipour, G.; Enderami, S.E. The Role of MicroRNAs in the Induction of Pancreatic Differentiation. Curr. Stem Cell Res. Ther. 2020, 15, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kaspi, H.; Pasvolsky, R.; Hornstein, E. Could microRNAs contribute to the maintenance of β cell identity? Trends Endocrinol. Metab. 2014, 25, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, O.; Fabris, G.; Van Obberghen, E. Shaping and preserving β-cell identity with microRNAs. Diabetes Obes. Metab. 2016, 18 (Suppl. S1), 51–57. [Google Scholar] [CrossRef] [Green Version]
- Plaisance, V.; Waeber, G.; Regazzi, R.; Abderrahmani, A. Role of MicroRNAs in Islet Beta-Cell Compensation and Failure during Diabetes. J. Diabetes Res. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Chen, Y.; Tian, L.; Wan, S.; Xie, Y.; Chen, X.; Ji, X.; Zhao, Q.; Wang, C.; Zhang, K.; Hock, J.M.; et al. MicroRNA-17-92 cluster regulates pancreatic beta-cell proliferation and adaptation. Mol. Cell. Endocrinol. 2016, 437, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Brereton, M.F.; Iberl, M.; Shimomura, K.; Zhang, Q.; Adriaenssens, A.E.; Proks, P.; Spiliotis, I.I.; Dace, W.; Mattis, K.K.; Ramracheya, R.; et al. Reversible changes in pancreatic islet structure and function produced by elevated blood glucose. Nat. Commun. 2014, 5, 4639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarthy, H.; Gu, X.; Enge, M.; Dai, X.; Wang, Y.; Damond, N.; Downie, C.; Liu, K.; Wang, J.; Xing, Y.; et al. Converting Adult Pancreatic Islet α Cells into β Cells by Targeting Both Dnmt1 and Arx. Cell Metab. 2017, 25, 622–634. [Google Scholar] [CrossRef] [Green Version]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar] [PubMed]
- Collombat, P.; Xu, X.; Ravassard, P.; Sosa-Pineda, B.; Dussaud, S.; Billestrup, N.; Madsen, O.D.; Serup, P.; Heimberg, H.; Mansouri, A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell 2009, 138, 449–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradwohl, G.; Dierich, A.; LeMeur, M.; Guillemot, F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci.USA 2000, 97, 1607–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, T.; Avolio, F.; Courtney, M.; Vieira, A.; Druelle, N.; Ben-Othman, N.; Hadzic, B.; Navarro, S.; Collombat, P. Pax4 acts as a key player in pancreas development and plasticity. Semin. Cell Dev. Biol. 2015, 44, 107–114. [Google Scholar] [CrossRef]
- Zhong, F.; Jiang, Y. Endogenous Pancreatic β Cell Regeneration: A Potential Strategy for the Recovery of β Cell Deficiency in Diabetes. Front. Endocrinol. 2019, 10, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salinno, C.; Cota, P.; Bastidas-Ponce, A.; Tarquis-Medina, M.; Lickert, H.; Bakhti, M. β-Cell Maturation and Identity in Health and Disease. Int. J. Mol. Sci. 2019, 20, 5417. [Google Scholar] [CrossRef] [Green Version]
- Collombat, P.; Hecksher-Sørensen, J.; Krull, J.; Berger, J.; Riedel, D.; Herrera, P.L.; Serup, P.; Mansouri, A. Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J. Clin. Investig. 2007, 117, 961–970. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T.; et al. Pdx1 maintains β cell identity and function by repressing an α cell program. Cell Metab. 2014, 19, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Brun, T.; Kataoka, K.; Sharma, A.J.; Wollheim, C.B. MAFA controls genes implicated in insulin biosynthesis and secretion. Diabetologia 2006, 50, 348–358. [Google Scholar] [CrossRef] [Green Version]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic β Cell Dedifferentiation as a Mechanism of Diabetic β Cell Failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Puri, S.; Hebrok, M. Cellular Plasticity within the Pancreas—Lessons Learned from Development. Dev. Cell 2010, 18, 342–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonas, J.C.; Sharma, A.; Hasenkamp, W.; Ilkova, H.; Patanè, G.; Laybutt, R.; Bonner-Weir, S.; Weir, G.C. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J. Biol. Chem. 1999, 274, 14112–14121. [Google Scholar] [CrossRef] [Green Version]
- Kim-Muller, J.Y.; Zhao, S.; Srivastava, S.; Mugabo, Y.; Noh, H.-L.; Kim, Y.R.; Madiraju, S.R.M.; Ferrante, A.W.; Skolnik, E.Y.; Prentki, M.; et al. Metabolic inflexibility impairs insulin secretion and results in MODY-like diabetes in triple FoxO-deficient mice. Cell Metab. 2014, 20, 593–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, A.E.; Dhawan, S.; Hoang, J.; Cory, M.; Zeng, K.; Fritsch, H.; Meier, J.J.; Rizza, R.A.; Butler, P.C. β-Cell Deficit in Obese Type 2 Diabetes, a Minor Role of β-Cell Dedifferentiation and Degranulation. J. Clin. Endocrinol. Metab. 2016, 101, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stancill, J.S.; Cartailler, J.-P.; Clayton, H.W.; O’Connor, J.T.; Dickerson, M.T.; Dadi, P.K.; Osipovich, A.B.; Jacobson, D.A.; Magnuson, M.A. Chronic β-Cell Depolarization Impairs β-Cell Identity by Disrupting a Network of Ca2+-Regulated Genes. Diabetes 2017, 66, 2175–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim-Muller, J.Y.; Fan, J.; Kim, Y.J.R.; Lee, S.-A.; Ishida, E.; Blaner, W.S.; Accili, D. Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic β cells in diabetic mice. Nat. Commun. 2016, 7, 12631. [Google Scholar] [CrossRef]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of β-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [Green Version]
- Nordmann, T.M.; Dror, E.; Schulze, F.; Traub, S.; Berishvili, E.; Barbieux, C.; Böni-Schnetzler, M.; Donath, M.Y. The Role of Inflammation in β-cell Dedifferentiation. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Rui, J.; Deng, S.; Arazi, A.; Perdigoto, A.L.; Liu, Z.; Herold, K.C. β Cells that Resist Immunological Attack Develop during Progression of Autoimmune Diabetes in NOD Mice. Cell Metab. 2017, 25, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Lee, Y.-S.; Harenda, Q.; Pietrzak, S.; Oktay, H.Z.; Schreiber, S.; Liao, Y.; Sonthalia, S.; Ciecko, A.E.; Chen, Y.-G.; et al. Beta Cell Dedifferentiation Induced by IRE1α Deletion Prevents Type 1 Diabetes. Cell Metab. 2020, 31, 822–836.e5. [Google Scholar] [CrossRef]
- Moin, A.S.M.; Dhawan, S.; Shieh, C.; Butler, P.C.; Cory, M.; Butler, A.E. Increased Hormone-Negative Endocrine Cells in the Pancreas in Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 3487–3496. [Google Scholar] [CrossRef] [Green Version]
- Moin, A.S.M.; Cory, M.; Ong, A.; Choi, J.; Dhawan, S.; Butler, P.C.; Butler, A.E. Pancreatic Nonhormone Expressing Endocrine Cells in Children With Type 1 Diabetes. J. Endocr. Soc. 2017, 1, 385–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasserfall, C.; Nick, H.S.; Campbell-Thompson, M.; Beachy, D.; Haataja, L.; Kusmartseva, I.; Posgai, A.; Beery, M.; Rhodes, C.; Bonifacio, E.; et al. Persistence of Pancreatic Insulin mRNA Expression and Proinsulin Protein in Type 1 Diabetes Pancreata. Cell Metab. 2017, 26, 568–575.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spijker, H.S.; Ravelli, R.B.G.; Mommaas-Kienhuis, A.M.; van Apeldoorn, A.A.; Engelse, M.A.; Zaldumbide, A.; Bonner-Weir, S.; Rabelink, T.J.; Hoeben, R.C.; Clevers, H.; et al. Conversion of mature human β-cells into glucagon-producing α-cells. Diabetes 2013, 62, 2471–2480. [Google Scholar] [CrossRef] [Green Version]
- Dhawan, S.; Georgia, S.; Tschen, S.-I.; Fan, G.; Bhushan, A. Pancreatic β Cell Identity Is Maintained by DNA Methylation-Mediated Repression of Arx. Dev. Cell 2011, 20, 419–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dor, Y.; Glaser, B. β-cell dedifferentiation and type 2 diabetes. N. Engl. J. Med. 2013, 368, 572–573. [Google Scholar] [CrossRef] [PubMed]
- Spijker, H.S.; Song, H.; Ellenbroek, J.H.; Roefs, M.M.; Engelse, M.A.; Bos, E.; Koster, A.J.; Rabelink, T.J.; Hansen, B.C.; Clark, A.; et al. Loss of β-Cell Identity Occurs in Type 2 Diabetes and Is Associated with Islet Amyloid Deposits. Diabetes 2015, 64, 2928–2938. [Google Scholar] [CrossRef] [Green Version]
- Mezza, T.; Cinti, F.; Cefalo, C.M.A.; Pontecorvi, A.; Kulkarni, R.N.; Giaccari, A. β-Cell Fate in Human Insulin Resistance and Type 2 Diabetes: A Perspective on Islet Plasticity. Diabetes 2019, 68, 1121–1129. [Google Scholar] [CrossRef]
- Alán, L.; Olejár, T.; Cahová, M.; Zelenka, J.; Berková, Z.; Smětáková, M.; Saudek, F.; Matěj, R.; Ježek, P. Delta Cell Hyperplasia in Adult Goto-Kakizaki (GK/MolTac) Diabetic Rats. J. Diabetes Res. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Piran, R.; Lee, S.-H.; Li, C.-R.; Charbono, A.; Bradley, L.M.; Levine, F. Pharmacological induction of pancreatic islet cell transdifferentiation: Relevance to type I diabetes. Cell Death Dis. 2014, 5, e1357. [Google Scholar] [CrossRef] [Green Version]
- Kalis, M.; Bolmeson, C.; Esguerra, J.L.S.; Gupta, S.; Edlund, A.; Tormo-Badia, N.; Speidel, D.; Holmberg, D.; Mayans, S.; Khoo, N.K.S.; et al. Beta-Cell Specific Deletion of Dicer1 Leads to Defective Insulin Secretion and Diabetes Mellitus. PLoS ONE 2011, 6, e29166. [Google Scholar] [CrossRef] [Green Version]
- Melkman-Zehavi, T.; Oren, R.; Kredo-Russo, S.; Shapira, T.; Mandelbaum, A.D.; Rivkin, N.; Nir, T.; Lennox, K.; Behlke, M.; Dor, Y.; et al. miRNAs control insulin content in pancreatic β-cells via downregulation of transcriptional repressors. EMBO J. 2011, 30, 835–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lynn, F.C.; Skewes-Cox, P.; Kosaka, Y.; McManus, M.T.; Harfe, B.D.; German, M.S. MicroRNA Expression Is Required for Pancreatic Islet Cell Genesis in the Mouse. Diabetes 2007, 56, 2938–2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Sanchez, A.; Nguyen-Tu, M.-S.; Rutter, G.A. DICER Inactivation Identifies Pancreatic β-Cell “Disallowed” Genes Targeted by MicroRNAs. Mol. Endocrinol. 2015, 29, 1067–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullen, T.J.; Khan, A.M.; Barton, G.; Butcher, S.; Sun, G.; Rutter, G.A. Identification of genes selectively disallowed in the pancreatic islet. Islets 2010, 2, 89–95. [Google Scholar] [CrossRef] [Green Version]
- Thorrez, L.; Laudadio, I.; Van Deun, K.; Quintens, R.; Hendrickx, N.; Granvik, M.; Lemaire, K.; Schraenen, A.; Van Lommel, L.; Lehnert, S.; et al. Tissue-specific disallowance of housekeeping genes: The other face of cell differentiation. Genome Res. 2010, 21, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastiani, G.; Grieco, G.E.; Brusco, N.; Ventriglia, G.; Formichi, C.; Marselli, L.; Marchetti, P.; Dotta, F. MicroRNA Expression Analysis of In Vitro Dedifferentiated Human Pancreatic Islet Cells Reveals the Activation of the Pluripotency-Related MicroRNA Cluster miR-302s. Int. J. Mol. Sci. 2018, 19, 1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebastiani, G.; Valentini, M.; Grieco, G.E.; Ventriglia, G.; Nigi, L.; Mancarella, F.; Pellegrini, S.; Martino, G.; Sordi, V.; Piemonti, L.; et al. MicroRNA expression profiles of human iPSCs differentiation into insulin-producing cells. Acta Diabetol. 2016, 54, 265–281. [Google Scholar] [CrossRef]
- Klein, D.; Misawa, R.; Bravo-Egana, V.; Vargas, N.; Rosero, S.; Piroso, J.; Ichii, H.; Umland, O.; Zhijie, J.; Tsinoremas, N.; et al. MicroRNAexpression in alpha and beta cells of human pancreaticislets. PLoS ONE 2013, 8, e55064. [Google Scholar] [CrossRef] [Green Version]
- Sebastiani, G.; Po, A.; Miele, E.; Ventriglia, G.; Ceccarelli, E.; Bugliani, M.; Marselli, L.; Marchetti, P.; Gulino, A.; Ferretti, E.; et al. MicroRNA-124a ishyperexpressed in type 2 diabetic human pancreaticislets and negativelyregulatesinsulinsecretion. Acta Diabetol. 2015, 52, 523–530. [Google Scholar] [CrossRef]
- Lemaire, K.; Thorrez, L.; Schuit, F. Disallowed and Allowed Gene Expression: Two Faces of Mature Islet Beta Cells. Annu. Rev. Nutr. 2016, 36, 45–71. [Google Scholar] [CrossRef] [PubMed]
- Pullen, T.J.; da Silva Xavier, G.; Kelsey, G.; Rutter, G.A. miR-29a and miR-29b contribute to pancreatic beta-cell-specific silencing of monocarboxylate transporter 1 (Mct1). Mol. Cell. Biol. 2011, 31, 3182–3194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krek, A.; Grün, D.; Poy, M.N.; Wolf, R.; Rosenberg, L.; Epstein, E.J.; MacMenamin, P.; Da Piedade, I.; Gunsalus, K.C.; Stoffel, M.; et al. Combinatorial microRNA target predictions. Nat. Genet. 2005, 37, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Poy, M.N.; Eliasson, L.; Krutzfeldt, J.; Kuwajima, S.; Ma, X.; Macdonald, P.E.; Pfeffer, S.; Tuschl, T.; Rajewsky, N.; Rorsman, P.; et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nat. Cell Biol. 2004, 432, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, B.R.; Wollheim, C.B. MicroRNAs: ’ribo-regulators’ of glucose homeostasis. Nat. Med. 2006, 12, 36–38. [Google Scholar] [CrossRef] [Green Version]
- Russ, H.A.; Sintov, E.; Anker-Kitai, L.; Friedman, O.; Lenz, A.; Toren, G.; Farhy, C.; Pasmanik-Chor, M.; Oron-Karni, V.; Ravassard, P.; et al. Insulin-Producing Cells Generated from Dedifferentiated Human Pancreatic Beta Cells Expanded In Vitro. PLoS ONE 2011, 6, e25566. [Google Scholar] [CrossRef] [Green Version]
- Russ, H.A.; Ravassard, P.; Kerr-Conte, J.; Pattou, F.; Efrat, S. Epithelial-Mesenchymal Transition in Cells Expanded In Vitro from Lineage-Traced Adult Human Pancreatic Beta Cells. PLoS ONE 2009, 4, e6417. [Google Scholar] [CrossRef]
- Poy, M.N.; Hausser, J.; Trajkovski, M.; Braun, M.; Collins, S.; Rorsman, P.; Zavolan, M.; Stoffel, M. miR-375 maintains normal pancreatic alpha- and beta-cell mass. Proc. Natl. Acad. Sci. USA 2009, 106, 5813–5818. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, N.; Kido, Y.; Uchida, T.; Asahara, S.; Shigeyama, Y.; Matsuda, T.; Takeda, A.; Tsuchihashi, D.; Nishizawa, A.; Ogawa, W.; et al. Ablation of PDK1 in pancreatic beta cells induces diabetes as a result of loss of beta cell mass. Nat. Genet. 2006, 38, 589–593. [Google Scholar] [CrossRef]
- El Ouaamari, A.; Baroukh, N.; Martens, G.A.; Lebrun, P.; Pipeleers, D.; van Obberghen, E. miR-375 targets 3’-phosphoinositide-dependent protein kinase-1 and regulates glucose-induced biological responses in pancreatic beta-cells. Diabetes 2008, 57, 2708–2717. [Google Scholar] [CrossRef] [Green Version]
- Dumortier, O.; Fabris, G.; Pisani, D.F.; Casamento, V.; Gautier, N.; Hinault, C.; Lebrun, P.; Duranton, C.; Tauc, M.; Dalle, S.; et al. microRNA-375 regulates glucose metabolism-related signaling for insulin secretion. J. Endocrinol. 2020, 244, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Rosero, S.; Bravo-Egana, V.; Jiang, Z.; Khuri, S.; Tsinoremas, N.F.; Klein, D.; Sabates, E.; Correa-Medina, M.; Ricordi, C.; Dominguez-Bendala, J.; et al. MicroRNA signature of the human developing pancreas. BMC Genom. 2010, 11, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, W.; Mulas, F.; Gaertner, B.; Sui, Y.; Wang, J.; Matta, I.; Zeng, C.; Vinckier, N.; Wang, A.; Nguyen-Ngoc, K.-V.; et al. A Network of microRNAs Acts to Promote Cell Cycle Exit and Differentiation of Human Pancreatic Endocrine Cells. iScience 2019, 21, 681–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latreille, M.; Hausser, J.; Stützer, I.; Zhang, Q.; Hastoy, B.; Gargani, S.; Kerr-Conte, J.; Pattou, F.; Zavolan, M.; Esguerra, J.L.S.; et al. MicroRNA-7a regulates pancreatic β cell function. J. Clin. Investig. 2014, 124, 2722–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esguerra, J.L.S.; Bolmeson, C.; Cilio, C.M.; Eliasson, L. Differential Glucose-Regulation of MicroRNAs in Pancreatic Islets of Non-Obese Type 2 Diabetes Model Goto-Kakizaki Rat. PLoS ONE 2011, 6, e18613. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, J.; Liu, C.; Naji, A.; Stoffers, D.A. MicroRNA-7 regulates the mTOR pathway and proliferation in adult pancreatic β-cells. Diabetes 2013, 62, 887–895. [Google Scholar] [CrossRef] [Green Version]
- López-Beas, J.; Capilla-González, V.; Aguilera, Y.; Mellado, N.; Lachaud, C.C.; Martín, F.; Smani, T.; Soria, B.; Hmadcha, A. miR-7 Modulates hESC Differentiation into Insulin-Producing Beta-like Cells and Contributes to Cell Maturation. Mol. Ther. Nucleic Acids 2018, 12, 463–477. [Google Scholar] [CrossRef]
- Filios, S.R.; Shalev, A. β-Cell MicroRNAs: Small but Powerful. Diabetes 2015, 64, 3631–3644. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Chen, J.; Jing, G.; Shalev, A. Thioredoxin-interacting protein regulates insulin transcription through microRNA-204. Nat. Med. 2013, 19, 1141–1146. [Google Scholar] [CrossRef] [Green Version]
- Marzinotto, I.; Pellegrini, S.; Brigatti, C.; Nano, R.; Melzi, R.; Mercalli, A.; Liberati, D.; Sordi, V.; Ferrari, M.; Falconi, M.; et al. miR-204 is associated with an endocrine phenotype in human pancreatic islets but does not regulate the insulin mRNA through MAFA. Sci. Rep. 2017, 7, 14051. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Chen, J.; Xu, G.; Grayson, T.B.; Thielen, L.A.; Shalev, A. miR-204 Controls Glucagon-Like Peptide 1 Receptor Expression and Agonist Function. Diabetes 2017, 67, 256–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaddam, R.R.; Kim, Y.-R.; Li, Q.; Jacobs, J.S.; Gabani, M.; Mishra, A.; Promes, J.A.; Imai, Y.; Irani, K.; Vikram, A. Genetic deletion of miR-204 improves glycemic control despite obesity in db/db mice. Biochem. Biophys. Res. Commun. 2020, 532, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; You, W.; Wang, H.; Li, Y.; Qiao, N.; Shi, Y.; Zhang, C.; Bleich, D.; Han, X. MicroRNA-24/MODY Gene Regulatory Pathway Mediates Pancreatic β-Cell Dysfunction. Diabetes 2013, 62, 3194–3206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Sun, Y.; Zhou, Y.; Zhang, Y.; Zhang, T.; Li, Y.; You, W.; Chang, X.; Yuan, L.; Han, X. MicroRNA-24 promotes pancreatic beta cells toward dedifferentiation to avoid endoplasmic reticulum stress-induced apoptosis. J. Mol. Cell Biol. 2019, 11, 747–760. [Google Scholar] [CrossRef] [PubMed]
- Nesca, V.; Guay, C.; Jacovetti, C.; Menoud, V.; Peyot, M.-L.; Laybutt, D.R.; Prentki, M.; Regazzi, R. Identification of particular groups of microRNAs that positively or negatively impact on beta cell function in obese models of type 2 diabetes. Diabetologia 2013, 56, 2203–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tattikota, S.G.; Rathjen, T.; Hausser, J.; Khedkar, A.; Kabra, U.D.; Pandey, V.; Sury, M.D.; Wessels, H.-H.; Mollet, I.G.; Eliasson, L.; et al. miR-184Regulates Pancreatic β-Cell Function According to Glucose Metabolism. J. Biol. Chem. 2015, 290, 20284–20294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieco, G.E.; Brusco, N.; Nigi, L.; Formichi, C.; Fignani, D.; Licata, G.; Marselli, L.; Marchetti, P.; Salvini, L.; Tinti, L.; et al. Reduced miR-184-3p expression occurring in Type 2 diabetic pancreatic islets protects β-cells from lipotoxic and proinflammatory apoptosis via a CRTC1-dependent mechanism. bioRxiv 2021, 425234. [Google Scholar] [CrossRef]
- Tattikota, S.G.; Rathjen, T.; McAnulty, S.J.; Wessels, H.-H.; Akerman, I.; Van De Bunt, M.; Hausser, J.; Esguerra, J.L.; Musahl, A.; Pandey, A.K.; et al. Argonaute2 Mediates Compensatory Expansion of the Pancreatic β Cell. Cell Metab. 2014, 19, 122–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Sanchez, A.; Nguyen-Tu, M.; Cebola, I.; Yavari, A.; Marchetti, P.; Piemonti, L.; De Koning, E.; Shapiro, A.M.J.; Johnson, P.; Sakamoto, K.; et al. MiR-184 expression is regulated by AMPK in pancreatic islets. FASEB J. 2018, 32, 2587–2600. [Google Scholar] [CrossRef] [Green Version]
- Chu, J.Y.; Chau, M.K.; Chan, C.C.; Tai, A.C.; Cheung, K.F.; Chan, T.M.; Yung, S. miR-200c Prevents TGF-β1-Induced Epithelial-to-Mesenchymal Transition and Fibrogenesis in Mesothelial Cells by Targeting ZEB2 and Notch1. Mol. Ther. Nucleic Acids 2019, 17, 78–91. [Google Scholar] [CrossRef] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Filios, S.R.; Xu, G.; Chen, J.; Hong, K.; Jing, G.; Shalev, A. MicroRNA-200 Is Induced by Thioredoxin-interacting Protein and Regulates Zeb1 Protein Signaling and Beta Cell Apoptosis. J. Biol. Chem. 2014, 289, 36275–36283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belgardt, B.-F.; Ahmed, K.; Spranger, M.; Latreille, M.; Denzler, R.; Kondratiuk, N.; Von Meyenn, F.; Villena, F.N.; Herrmanns, K.; Bosco, D.; et al. The microRNA-200 family regulates pancreatic beta cell survival in type 2 diabetes. Nat. Med. 2015, 21, 619–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, S.; Liu, Y.; Yao, Y.; Teng, J. miR-124 promotes proliferation and differentiation of neuronal stem cells through inactivating Notch pathway. Cell Biosci. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Tzur, G.; Levy, A.; Meiri, E.; Barad, O.; Spector, Y.; Bentwich, Z.; Mizrahi, L.; Katzenellenbogen, M.; Ben-Shushan, E.; Reubinoff, B.; et al. MicroRNA Expression Patterns and Function in Endodermal Differentiation of Human Embryonic Stem Cells. PLoS ONE 2008, 3, e3726. [Google Scholar] [CrossRef]
- Lahmy, R.; Soleimani, M.; Sanati, M.H.; Behmanesh, M.; Kouhkan, F.; Mobarra, N. miRNA-375 promotes beta pancreatic differentiation in human induced pluripotent stem (hiPS) cells. Mol. Biol. Rep. 2014, 41, 2055–2066. [Google Scholar] [CrossRef]
- Shaer, A.; Azarpira, N.; Karimi, M.H. Differentiation of Human Induced Pluripotent Stem Cells into Insulin-Like Cell Clusters with miR-186 and miR-375 by using chemical transfection. Appl. Biochem. Biotechnol. 2014, 174, 242–258. [Google Scholar] [CrossRef]
- Chappell, J.; Watters, K.E.; Takahashi, M.K.; Lucks, J. A renaissance in RNA synthetic biology: New mechanisms, applications and tools for the future. Curr. Opin. Chem. Biol. 2015, 28, 47–56. [Google Scholar] [CrossRef] [Green Version]
- Cullen, B.R. Viral and cellular messenger RNA targets of viral microRNAs. Nat. Cell Biol. 2009, 457, 421–425. [Google Scholar] [CrossRef] [Green Version]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Krützfeldt, J.; Kuwajima, S.; Braich, R.; Rajeev, K.G.; Pena, J.; Tuschl, T.; Manoharan, M.; Stoffel, M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007, 35, 2885–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfrum, C.; Shi, S.; Jayaprakash, K.N.; Jayaraman, M.; Wang, G.; Pandey, R.K.; Rajeev, K.G.; Nakayama, T.; Charrise, K.; Ndungo, E.M.; et al. Mechanisms and optimization of in vivo delivery of lipophilic siRNAs. Nat. Biotechnol. 2007, 25, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.; Wu, H.; Xiao, H.; Chen, X.; Willenbring, H.; Steer, C.J.; Song, G. Inhibition of microRNA-24 expression in liver prevents hepatic lipid accumulation and hyperlipidemia. Hepatology 2014, 60, 554–564. [Google Scholar] [CrossRef] [Green Version]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, M.S.; Neilson, J.R.; Sharp, P.A. MicroRNA sponges: Competitive inhibitors of small RNAs in mammalian cells. Nat. Methods 2007, 4, 721–726. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.; Chen, Y.; Leong, K.W. MicroRNA delivery for regenerative medicine. Adv. Drug Deliv. Rev. 2015, 88, 108–122. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z. Multi-miRNA Hairpins and Multi-miRNA Mimics Technologies. In MicroRNA Interference Technologies; Springer: Berlin/Heidelberg, Gremany, 2009; pp. 101–110. [Google Scholar]
- Wang, Z. The Guideline of the Design and Validation of MiRNA Mimics. Comput. Biol. 2011, 676, 211–223. [Google Scholar] [CrossRef]
- Flierl, U.; Nero, T.L.; Lim, B.; Arthur, J.F.; Yao, Y.; Jung, S.M.; Gitz, E.; Pollitt, A.Y.; Zaldivia, M.T.; Jandrot-Perrus, M.; et al. Phosphorothioate backbone modifications of nucleotide-based drugs are potent platelet activators. J. Exp. Med. 2015, 212, 129–137. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Grieco, G.E.; Brusco, N.; Licata, G.; Nigi, L.; Formichi, C.; Dotta, F.; Sebastiani, G. Targeting microRNAs as a Therapeutic Strategy to Reduce Oxidative Stress in Diabetes. Int. J. Mol. Sci. 2019, 20, 6358. [Google Scholar] [CrossRef] [Green Version]
- Regazzi, R. MicroRNAs as therapeutic targets for the treatment of diabetes mellitus and its complications. Expert Opin. Ther. Targets 2018, 22, 153–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keefe, A.D.; Pai, S.; Ellington, A.D. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Rossi, J.J. Cell-type-specific, Aptamer-functionalized Agents for Targeted Disease Therapy. Mol. Ther. Nucleic Acids 2014, 3, e169. [Google Scholar] [CrossRef] [PubMed]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nat. Cell Biol. 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Kim, D.G.; Kim, K.H.; Seo, Y.J.; Yang, H.; Marcusson, E.G.; Son, E.; Lee, K.; Sa, J.K.; Lee, H.W.; Nam, D.-H. Anti-miR delivery strategies to bypass the blood-brain barrier in glioblastoma therapy. Oncotarget 2016, 7, 29400–29411. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grieco, G.E.; Brusco, N.; Licata, G.; Fignani, D.; Formichi, C.; Nigi, L.; Sebastiani, G.; Dotta, F. The Landscape of microRNAs in βCell: Between Phenotype Maintenance and Protection. Int. J. Mol. Sci. 2021, 22, 803. https://doi.org/10.3390/ijms22020803
Grieco GE, Brusco N, Licata G, Fignani D, Formichi C, Nigi L, Sebastiani G, Dotta F. The Landscape of microRNAs in βCell: Between Phenotype Maintenance and Protection. International Journal of Molecular Sciences. 2021; 22(2):803. https://doi.org/10.3390/ijms22020803
Chicago/Turabian StyleGrieco, Giuseppina Emanuela, Noemi Brusco, Giada Licata, Daniela Fignani, Caterina Formichi, Laura Nigi, Guido Sebastiani, and Francesco Dotta. 2021. "The Landscape of microRNAs in βCell: Between Phenotype Maintenance and Protection" International Journal of Molecular Sciences 22, no. 2: 803. https://doi.org/10.3390/ijms22020803
APA StyleGrieco, G. E., Brusco, N., Licata, G., Fignani, D., Formichi, C., Nigi, L., Sebastiani, G., & Dotta, F. (2021). The Landscape of microRNAs in βCell: Between Phenotype Maintenance and Protection. International Journal of Molecular Sciences, 22(2), 803. https://doi.org/10.3390/ijms22020803