
Ammonium Formate-Pd/C as a New Reducing System for 1,2,4-Oxadiazoles. Synthesis of Guanidine Derivatives and Reductive Rearrangement to Quinazolin-4-Ones with Potential Anti-Diabetic Activity

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Results
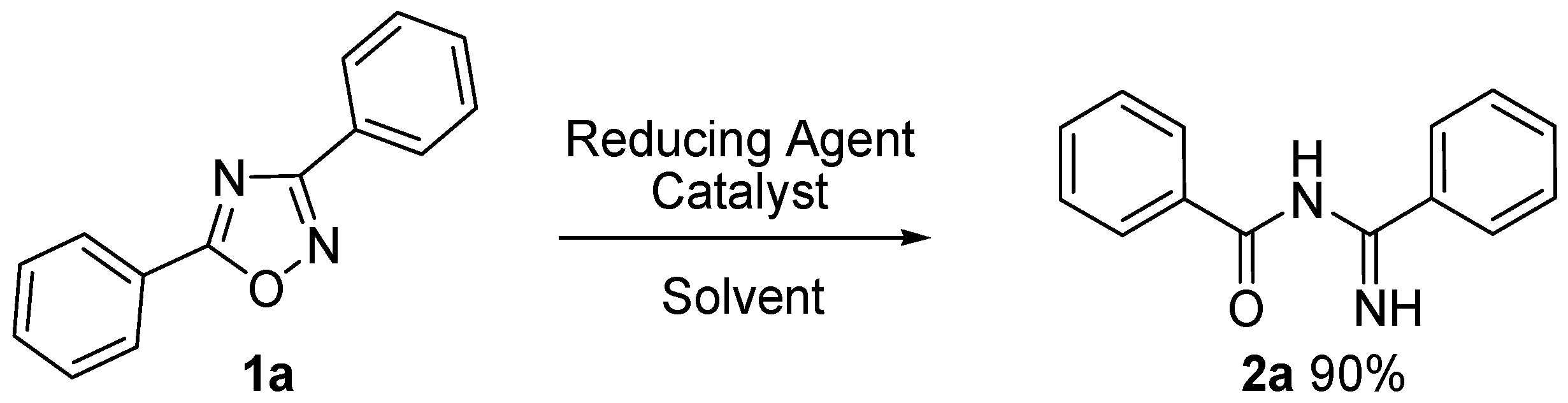
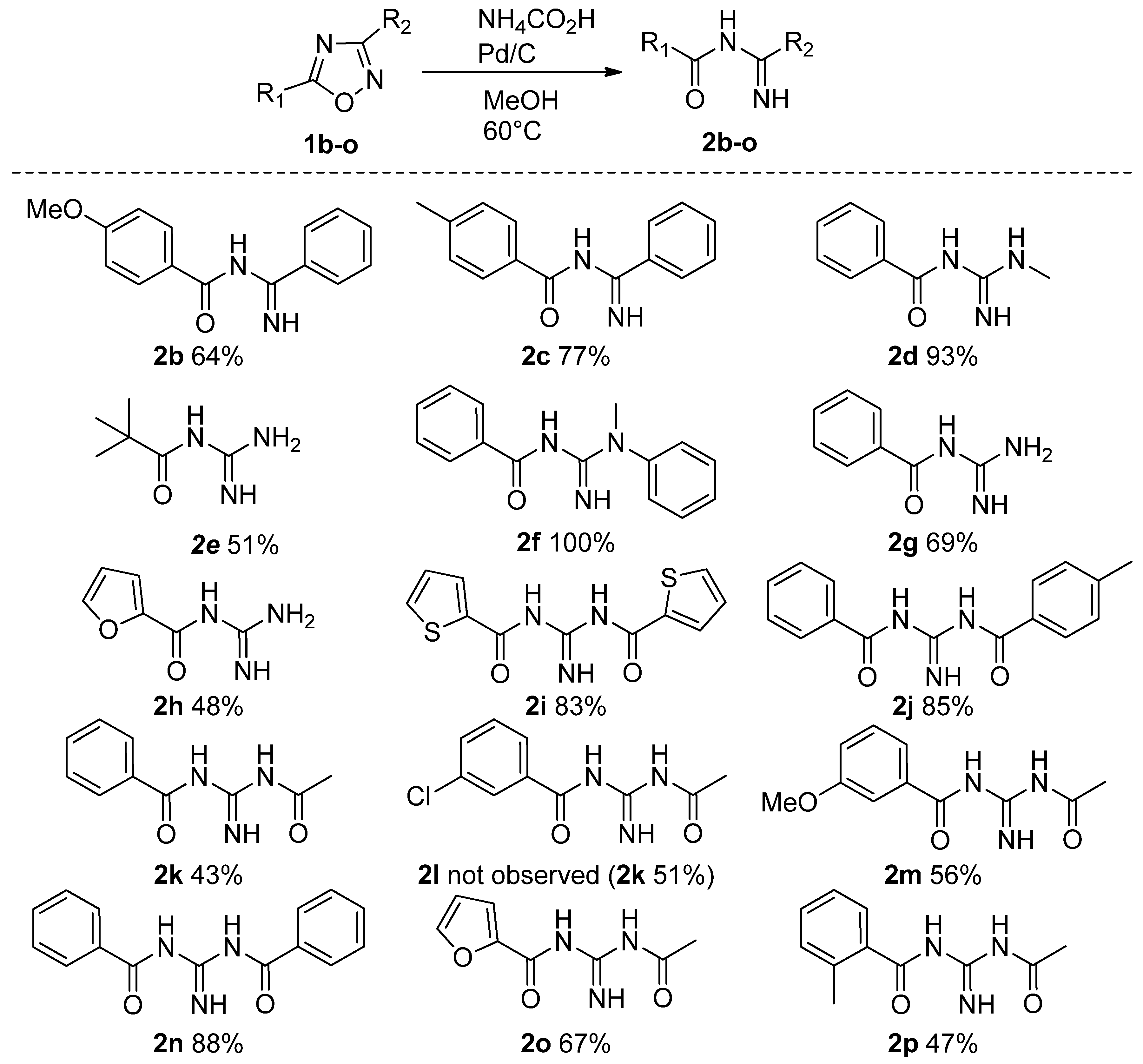
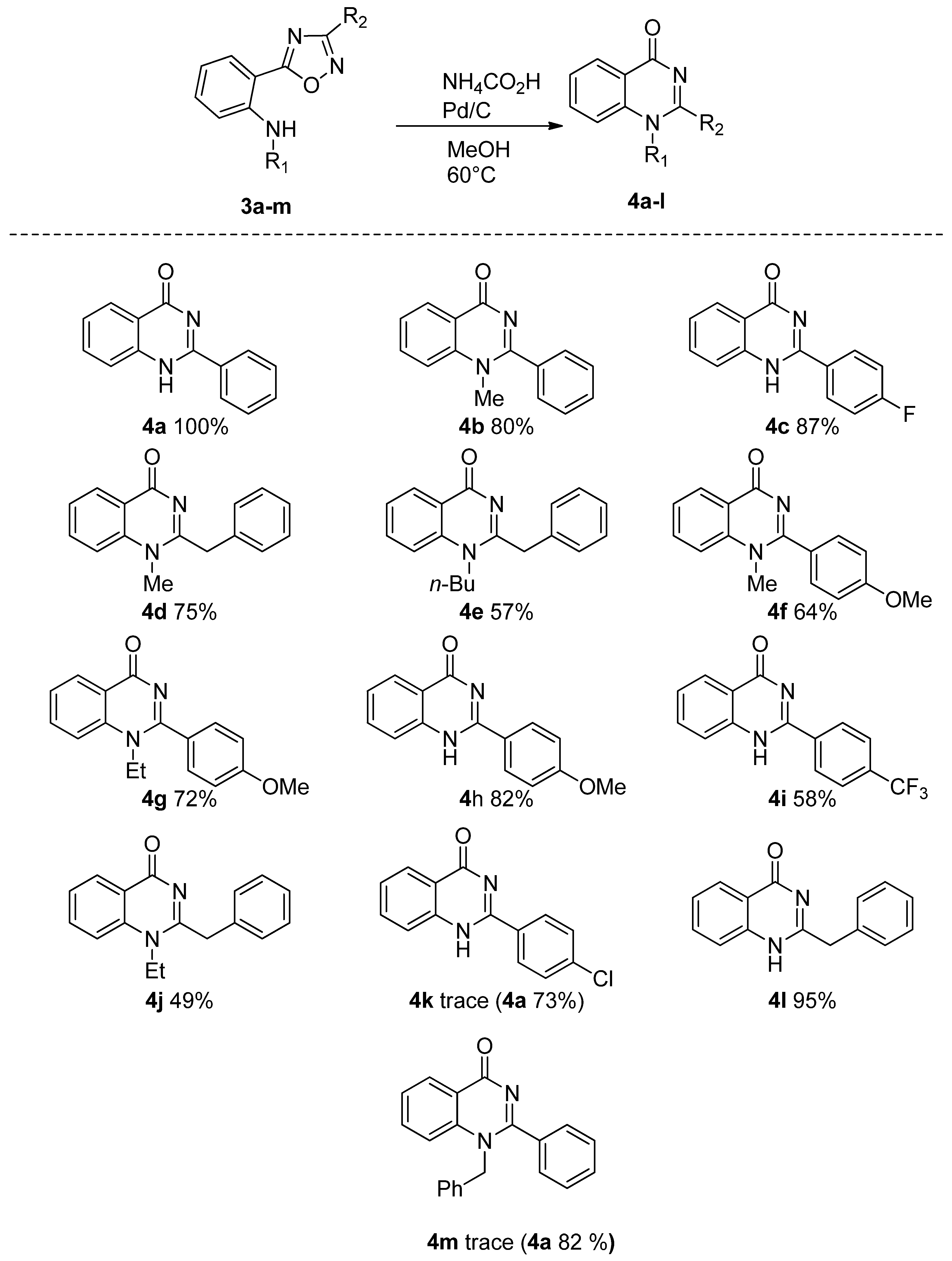
2.1. Reduction Reactions
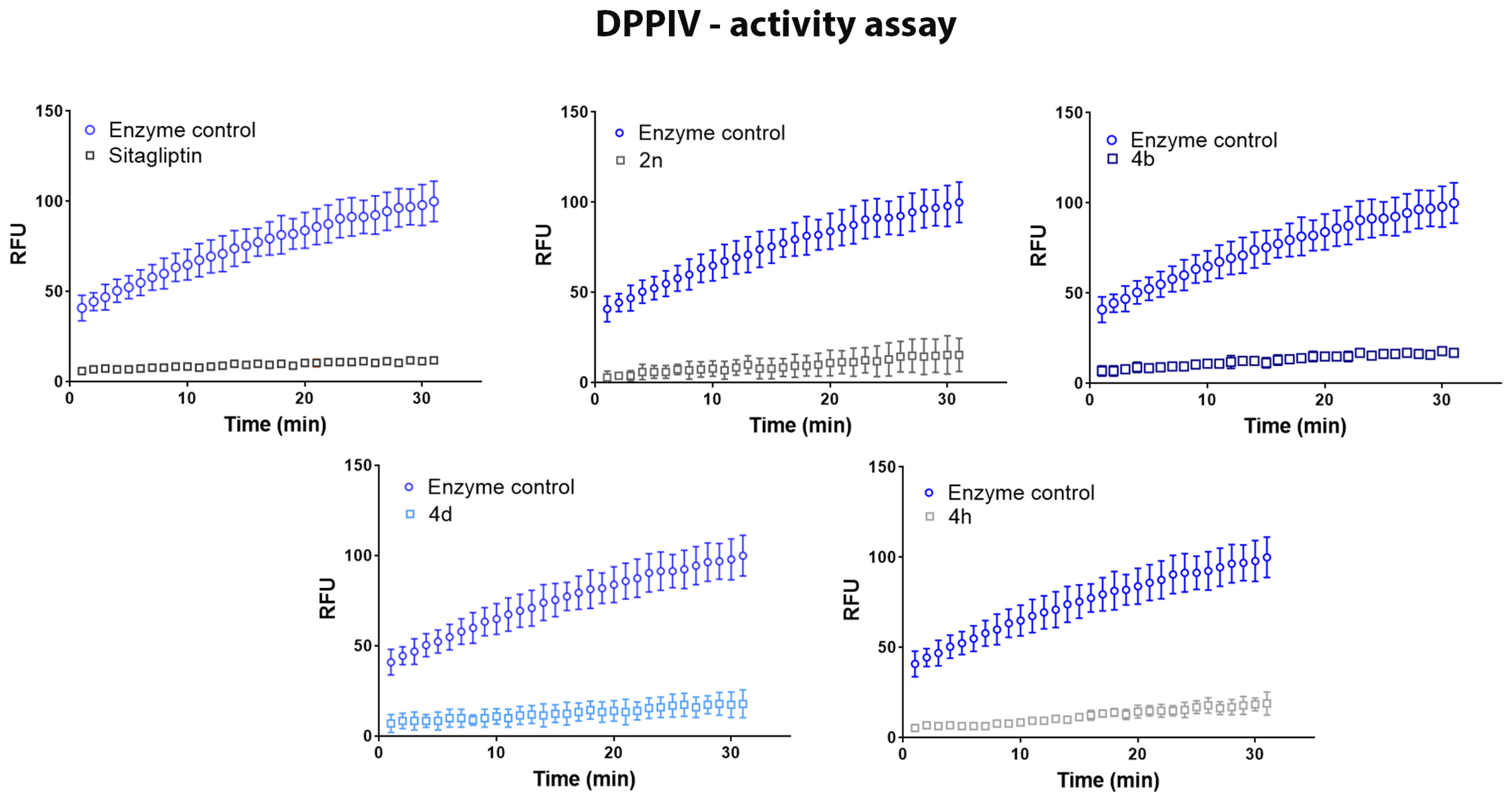
2.2. Inhibition Test of Dipeptidyl-Peptidase IV (DPPIV) Enzyme
2.3. Inhibition Test of α-Glucosidase Enzyme
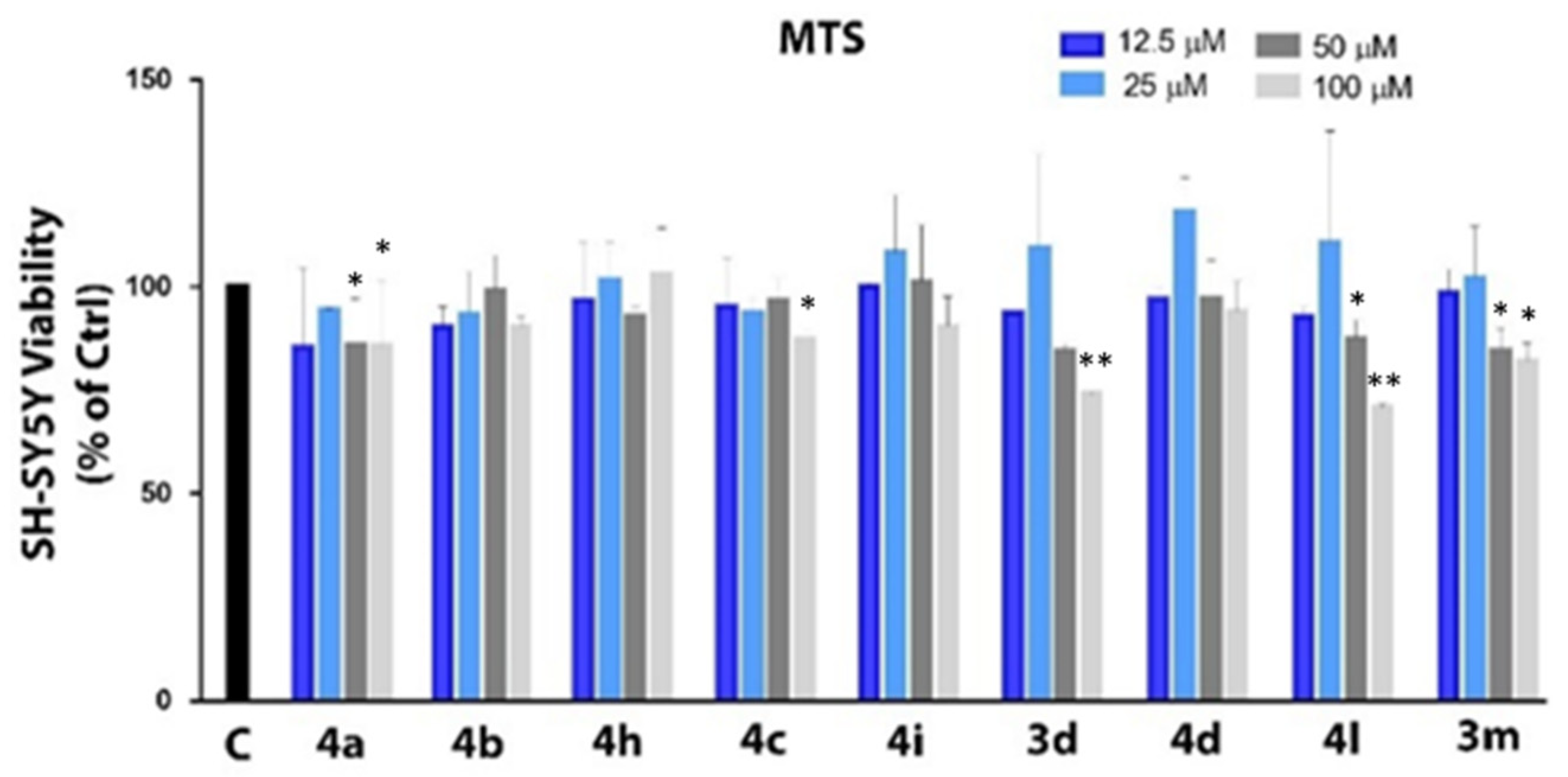
2.4. Cytotoxicity Assay and Evaluation of Intracellular Redox State
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. General Procedure for the Synthesis of N-Acylamidines, Quinazolin-4-(1H)-One, Acyl Guanidine and Diacyl Guanidine
4.3. General Procedure for the Synthesis of 2-(1,2,4 Oxadiazol-5-yl)Benzenamine 3
4.4. General Procedure for the Synthesis of N-Benzyl-2-(3-Phenyl-1,2,4-Oxadiazol-5-yl)Benzenamine 3m
4.5. DPPIV Activity Assay
4.6. α-Glucosidase Inhibition Test
4.7. MTS Assay
4.8. Analysis of Reactive Oxygen Species (ROS)
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pace, A.; Buscemi, S.; Palumbo Piccionello, A.; Pibiri, I. Recent advances in the chemistry of 1,2,4-oxadiazoles. Adv. Heterocycl. Chem. 2015, 116, 85–136. [Google Scholar] [CrossRef]
- Boström, J.; Hogner, A.; Llinàs, A.; Wellner, E.; Plowright, A.T. Oxadiazoles in Medicinal Chemistry. J. Med. Chem. 2012, 55, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
- Campofelice, A.; Lentini, L.; Di Leonardo, A.; Melfi, R.; Tutone, M.; Pace, A.; Pibiri, I. Strategies against Nonsense: Oxadiazoles as Translational Readthrough-Inducing Drugs (TRIDs). Int. J. Mol. Sci. 2019, 20, 3329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Mangione, M.R.; Palumbo Piccionello, A.; Marino, C.; Ortore, M.G.; Picone, P.; Vilasi, S.; Di Carlo, M.; Buscemi, S.; Bulone, D.; San Biagio, P.L. Photo-inhibition of Aβ-fibrillation mediated by a newly designed fluorinated oxadiazole. RSC Adv. 2015, 5, 16540–16548. [Google Scholar] [CrossRef]
- Battisti, A.; Palumbo Piccionello, A.; Sgarbossa, A.; Vilasi, S.; Ricci, C.; Ghetti, F.; Spinozzi, F.; Marino Gammazza, A.; Giacalone, V.; Martorana, A.; et al. Curcumin-like compounds designed to modify amyloid beta peptide aggregation patterns. RSC Adv. 2017, 7, 31714–31724. [Google Scholar] [CrossRef] [Green Version]
- Gillman, K.W.; Starrett, J.E.; Parker, M.F.; Xie, K.; Bronson, J.J.; Marcin, L.R.; McElhone, K.; Bergstrom, C.P.; Mate, R.A.; Williams, R.; et al. Discovery and Evaluation of BMS-708163, a potent, selective and orally bioavailable γ-Secretase Inhibitor. ACS Med. Chem. Lett. 2010, 1, 120–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Daniel, P.I.; Peng, Z.; Pi, H.; Testero, S.A.; Ding, D.; Spink, E.; Leemans, E.; Boudreau, M.A.; Yamaguchi, T.; Schroeder, V.A.; et al. Discovery of a new class of non-β-lactam inhibitors of penicillin-binding proteins with gram-positive antibacterial activity. J. Am. Chem. Soc. 2014, 136, 3664–3672. [Google Scholar] [CrossRef] [PubMed]
- Fortuna, C.G.; Bonaccorso, C.; Bulbarelli, A.; Caltabiano, G.; Rizzi, L.; Goracci, L.; Musumarra, G.; Pace, A.; Palumbo Piccionello, A.; Guarcello, A.; et al. New linezolid-like 1,2,4-oxadiazoles active against gram-positive multiresistant pathogens. Eur. J. Med. Chem. 2013, 65, 533–545. [Google Scholar] [CrossRef]
- Fortuna, C.G.; Berardozzi, R.; Bonaccorso, C.; Caltabiano, G.; Di Bari, L.; Goracci, L.; Guarcello, A.; Pace, A.; Palumbo Piccionello, A.; Pescitelli, G.; et al. New potent antibacterials against gram-positive multiresistant pathogens: Effects of side chain modification and chirality in linezolid-like 1,2,4-oxadiazoles. Bioorg. Med. Chem. 2014, 22, 6814–6825. [Google Scholar] [CrossRef]
- Shetnev, A.; Baykov, S.; Kalinin, S.; Belova, A.; Sharoyko, V.; Rozhkov, A.; Zelenkov, L.; Tarasenko, M.; Sadkov, E.; Korsakov, M.; et al. 1,2,4-Oxadiazole/2-Imidazoline hybrids: Multi-target-directed compounds for the treatment of infectious diseases and cancer. Int. J. Mol. Sci. 2019, 20, 1699. [Google Scholar] [CrossRef] [Green Version]
- Doria, F.; Pirota, V.; Petenzi, M.; Teulade-Fichou, M.P.; Verga, D.; Freccero, M. Oxadiazole/pyridine-based ligands: A structural tuning for enhancing G-quadruplex binding. Molecules 2018, 23, 2162. [Google Scholar] [CrossRef] [Green Version]
- Pathak, S.K.; Pradhan, B.; Gupta, R.K.; Gupta, M.; Pal, S.K.; Achalkumar, A.S. Aromatic Π-Π supergelation, aggregation induced emission and columnar self-assembly of star-shaped 1,2,4-oxadiazole derivatives. J. Mater. Chem. C 2016, 4, 6546–6561. [Google Scholar] [CrossRef]
- Salassa, G.; Terenzi, A. Metal Complexes of Oxadiazole Ligands: An Overview. Int. J. Mol. Sci. 2019, 20, 3483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matta, A.; Bahadur, V.; Taniike, T.; Van der Eycken, J.; Singh, B.K. Synthesis, characterisation and photophysical studies of oxadiazolyl coumarin: A new class of blue light emitting fluorescent dyes. Dyes Pigments 2017, 140, 250–260. [Google Scholar] [CrossRef]
- Tang, Y.; Imler, G.H.; Parrish, D.A.; Shreeve, J.M. Oxidative cyclization protocol for the preparation of energetic 3-amino-5-R-1,2,4-oxadiazoles. Org. Lett. 2018, 20, 8039–8042. [Google Scholar] [CrossRef] [PubMed]
- Ko, D.; Patel, H.A.; Yavuz, C.T. Synthesis of nanoporous 1,2,4-oxadiazole networks with high CO2 capture capacity. Chem. Commun. 2015, 51, 2915–2917. [Google Scholar] [CrossRef] [PubMed]
- Palumbo Piccionello, A.; Guarcello, A.; Calabrese, A.; Pibiri, I.; Pace, A.; Buscemi, S. Synthesis of fluorinated oxadiazoles with gelation and oxygen storage ability. Org. Biomol. Chem. 2012, 10, 3044–3052. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, F.S.; Di Stefano, M.; Palumbo Piccionello, A.; Fiorica, C.; Pitarresi, G.; Pibiri, I.; Buscemi, S.; Giammona, G. Perfluorocarbon functionalized hyaluronic acid derivative as oxygenating systems for cell culture. RSC Adv. 2014, 4, 22894–22901. [Google Scholar] [CrossRef]
- Palumbo Piccionello, A.; Pace, A.; Buscemi, S. Rearrangements of 1,2,4-Oxadiazole: “One Ring to Rule Them All”. Chem. Heterocycl. Compd. 2017, 53, 936–947. [Google Scholar] [CrossRef]
- Hemming, K. 1,2,4-oxadiazoles. In Comprehensive Heterocyclic Chemistry, 3rd ed.; Zhdankin, V.V., Katritzky, A.R., Ramsden, C.A., Scriven, E.F.V., Taylor, R.J.K., Eds.; Elsevier: Oxford, UK, 2008; Volume 5, pp. 243–314. [Google Scholar] [CrossRef]
- Maciejewska, D.; Zabinski, J.; Rezler, M.; Kaźmierczak, P.; Collins, M.S.; Ficker, L.; Cushion, M.T. Development of highly active anti-Pneumocystis bisbenzamidines: Insight into the influence of selected substituents on the in vitro activity. MedChemComm 2017, 8, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Portada, T.; Margetić, D.; Štrukil, V. Mechanochemical catalytic transfer hydrogenation of aromatic nitro derivatives. Molecules 2018, 23, 3163. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.Y.; Zhang, X.; Lyu, L.; Wang, S.Q.; Zhang, X.T. Pyridazino[1,6-b]quinazolinones as new anticancer scaffold: Synthesis, DNA intercalation, topoisomerase I inhibition and antitumor evaluation in vitro and in vivo. Bioorg. Chem. 2020, 99, 103814. [Google Scholar] [CrossRef] [PubMed]
- Smullen, S.; McLaughlin, N.P.; Evans, P. Chemical synthesis of febrifugine and analogues. Bioorg. Med. Chem. 2018, 26, 2199–2220. [Google Scholar] [CrossRef]
- Janardhanan, J.; Bouley, R.; Martínez-Caballero, S.; Peng, Z.; Batuecas-Mordillo, M.; Meisel, J.E.; Ding, D.; Schroeder, V.A.; Wolter, W.R.; Mahasenan, K.V.; et al. The quinazolinone allosteric inhibitor of PBP 2a synergizes with piperacillin and tazobactam against Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2019, 63, e02637-18. [Google Scholar] [CrossRef] [Green Version]
- Jain, N.; Singour, P.K. Novel 3-{4-[2-amino-4-(substitutedphenyl)-2H-[1, 3] oxazin/thiazin-6-Yl}-2-phenyl-3H-quinazolin-4-one derivatives as enhancer of GABA mediated inhibition: Synthesis, molecular modeling and pharmacological studies. Lett. Drug Des. Discov. 2020, 17, 199–213. [Google Scholar] [CrossRef]
- Abdel-Aziz, A.A.M.; Abou-Zeid, L.A.; ElTahir, K.E.H.; Mohamed, M.A.; El-Enin, M.A.A.; El-Azab, A.S. Design, synthesis of 2,3-disubstitued 4(3H)-quinazolinone derivatives as anti-inflammatory and analgesic agents: COX-1/2 inhibitory activities and molecular docking studies. Bioorg. Med. Chem. 2016, 24, 3818–3828. [Google Scholar] [CrossRef]
- Pieterse, L.; Van der Walt, M.M.; Terre’Blanche, G. C2-substituted quinazolinone derivatives exhibit A1 and/or A2A adenosine receptor affinities in the low micromolar range. Bioorg. Med. Chem. Lett. 2020, 30, 127274–127280. [Google Scholar] [CrossRef]
- Mokale, S.N.; Palkar, A.D.; Dube, P.N.; Sakle, N.S.; Miniyar, P.B. Design, synthesis and in vivo screening of some novel quinazoline analogs as anti-hyperlipidemic and hypoglycemic agents. Bioorg. Med. Chem. Lett. 2016, 26, 272–276. [Google Scholar] [CrossRef]
- Wei, M.; Chai, W.M.; Wang, R.; Yang, Q.; Deng, Z.; Peng, Y. Quinazolinone derivatives: Synthesis and comparison of inhibitory mechanisms on α-glucosidase. Bioorg. Med. Chem. 2017, 25, 1303–1308. [Google Scholar] [CrossRef]
- Nisha, C.M.; Kumar, A.; Nair, P.; Gupta, N.; Silakari, C.; Tripathi, T.; Kumar, A. Molecular Docking and In Silico ADMET study reveals acylguanidine 7a as a Potential Inhibitor of β-Secretase. Adv. Bionform. 2016, 2016, 9258578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogister, F.; Laeckmann, D.; Plasman, P.O.; Van Eylen, F.; Ghyoot, M.; Maggetto, C.; Liégeois, J.F.; Géczy, J.; Herchuelz, A.; Delarge, J.; et al. Novel inhibitors of sodium-calcium exchanger: Benzene ring analogues of N-guanidino substituted amiloride derivatives. Eur. J. Med. Chem. 2001, 36, 597–614. [Google Scholar] [CrossRef]
- Mwimanzi, P.; Tietjen, I.; Miller, S.C.; Shahid, A.; Cobarrubias, K.; Kinloch, N.N.; Baraki, B.; Richard, J.; Finzi, A.; Fedida, D.; et al. Novel Acylguanidine-Based Inhibitor of HIV-1. J. Virol. 2016, 90, 9495–9508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Liang, E.; Shi, J.; Wu, Y.; Wen, K.; Yao, X.; Tang, X. Metal-free synthesis of 1,4-benzodiazepines and quinazolinones from hexafluoroisopropyl 2- aminobenzoates at room temperature. RSC Adv. 2021, 11, 4966–4970. [Google Scholar] [CrossRef]
- Ali, Z.; Akhtar, M.J.; Siddiqui, A.A.; Khan, A.A.; Haider, M.R.; Yar, M.S. Design, synthesis, and biological evaluation of novel quinazoline clubbed thiazoline derivatives. Arch. Pharm. Chem. Life Sci. 2017, 350, e1600298. [Google Scholar] [CrossRef]
- Selveraj, G.; Kaliamurthi, S.; Thirugnasambandan, R. Effect of Glycosin alkaloid from Rhizophora apiculata in non-insulin dependent diabetic rats and its mechanism of action: In vivo and in silico studies. Phytomedicine 2016, 23, 632–640. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Lv, M.; Liu, J.; Xu, Q.; Zhang, X.; Cao, H. Efficient Synthesis of quinazolinones by transition-metal-free direct aerobic oxidative cascade annulation of alcohols with o-aminoarylnitriles. ChemSusChem 2019, 12, 3043–3048. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, G.; Shen, J.; Kuai, C.; Cui, X. Cleavage of the C_C triple bond of ketoalkynes: Synthesis of 4(3H)-quinazolinones. Org. Chem. Front. 2015, 2, 366–368. [Google Scholar] [CrossRef]
- Picone, P.; Nuzzo, D.; Caruana, L.; Messina, E.; Barera, A.; Vasto, S.; Di Carlo, M. Metformin increases APP expression and processing via oxidative stress, mitochondrial dysfunction and NF-kB activation: Use of insulin to attenuate metformin’s effect. Biochim. Biophys. Acta 2015, 1853, 1046–1059. [Google Scholar] [CrossRef] [Green Version]
- Buscemi, S.; Cicero, M.G.; Vivona, N.; Caronna, T. Photochemical behaviour of some 1,2,4-oxadiazole derivatives. J. Chem. Soc. Perkin Trans. 1 1988, 6, 1313–1315. [Google Scholar] [CrossRef]
- Baykov, S.; Sharonova, T.; Shetnev, A.; Rozhkov, S.; Kalinin, S.; Smirnov, A.V. The first one-pot ambient-temperature synthesis of 1,2,4-oxadiazoles from amidoximes and carboxylic acid esters. Tetrahedron 2017, 73, 945–951. [Google Scholar] [CrossRef]
- Li, E.; Wang, M.; Whang, Z.; Wenquan, Y.; Chang, J. NBS-mediated practical cyclization of N-acylamidines to 1,2,4-oxadiazoles via oxidative N-O bond formation. Tetrahedron 2018, 74, 4613–4618. [Google Scholar] [CrossRef]
- Palumbo Piccionello, A.; Pace, A.; Buscemi, S.; Vivona, N. 1,2,4-Oxadiazole rearrangements involving an NNC side -chain sequence. Org. Lett. 2009, 11, 4018–4020. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, S.; Pace, A.; Frenna, V.; Vivona, N. A generalized synthesis of 3-amino-5-aryl-3-amino-5-polyfluorophenyl- and 3-amino-5-alkyl-1,2,4-oxadiazoles through ring-degenerate rearrangements. Heterocycles 2002, 57, 811–823. [Google Scholar] [CrossRef]
- Buscemi, S.; Cicero, M.G.; Vivona, N.; Caronna, T. Heterocyclic photorearrangements. Photochemical behaviour of some 3,5-disubstituted 1,2,4-oxadiazoles in methanol at 254 nm. J. Heterocycl. Chem. 1988, 25, 931–935. [Google Scholar] [CrossRef]
- Palumbo Piccionello, A.; Guarcello, A.; Buscemi, S.; Vivona, N.; Pace, A. Synthesis of amino-1,2,4-triazoles by reductive ANRORC rearrangements of 1,2,4-oxadiazoles. J. Org. Chem. 2010, 75, 8724–8727. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, S.; Vivona, N.; Pibiri, I.; Palumbo Piccionello, A.; Barone, G.; Pace, A. Experimental and DFT studies on competitive heterocyclic rearrangements. Part 2: A one-atom side-chain versus the classic three-atom side chain (Boulton-Katritzky) ring rearrangement of 3-acylamino-1,2,4-oxadiazoles. J. Org. Chem. 2007, 72, 7656–7666. [Google Scholar] [CrossRef]
- Buscemi, S.; Frenna, V.; Vivona, N.; Petrillo, G.; Spinelli, D. Studies on azole-to-azole interconversions. Substituent effects on the ring-degenerate equilibration between 3-aroylamino-5-methyl-1,2,4-oxadiazoles and 3-acetylamino-5-aryl-1,2,4-oxadiazoles. Tetrahedron 1995, 51, 5133–5142. [Google Scholar] [CrossRef]
- Buscemi, S.; Frenna, V.; Pace, A.; Vivona, N.; Cosimelli, B.; Spinelli, D. Studies on azole-to-azole interconversion-an interesting case of absence of a “primary steric effect” in the ring-degenerate equilibration between ortho-substituted 3-aroylamino-5-methyl-1,2,4-oxadiazoles and 3-acetylamino-5-aryl-1,2,4-oxadiazoles in methanol. Eur. J. Org. Chem. 2002, 8, 1417–1423. [Google Scholar] [CrossRef]
- Guzman, A.; Romero, M.; Xavier Talamas, F.X.; Villena, R.; Greenhouse, R.; Muchowski, J.M. 1,3-Diaza-1,3-butadienes. Synthesis and conversion into pyrimidines by [4Π+2Π] cycloaddition with electron deficient acetylenes. Synthetic utility of 2-(trichloromethyl)pyrimidines. J. Org. Chem. 1996, 61, 2470–2483. [Google Scholar] [CrossRef]
- Buscemi, S.; Pace, A.; Vivona, N.; Caronna, T. Photoinduced molecular rearrangements. Some comments on the ring-photoisomerization of 1,2,4-oxadiazoles into 1,3,4-oxadiazoles. J. Heterocycl. Chem. 2001, 38, 777–780. [Google Scholar] [CrossRef]
- Lu, K.; Duan, L.; Xu, B.; Yin, W.; Wu, D.; Zhao, Y.; Gong, P. Facile synthesis of 3-amino-5-aryl-1,2,4-oxadiazoles via PIDA-mediated intramolecular oxidative cyclization. RSC Adv. 2016, 6, 54277–54280. [Google Scholar] [CrossRef]
- Hanusek, J.; Sedlák, M.; Šimůnek, P.; Štĕrba, V. Kinetics and Mechanism of the Base-Catalysed Cyclisation of 2-(Substituted benzoylamino)benzamides Giving Quinazolin-4-one and Quinazolin-4-thione Derivatives. Eur. J. Org. Chem. 2002, 2002, 1855–1863. [Google Scholar] [CrossRef]
- Sayahi, H.M.; Bahadorikhalili, S.; Mahdavi, M.; Banghshirin, L. Synthesis of quinazolin-4(3H)-ones via the reaction of isatoic anhydride with benzyl azides in the presence of potassium tert-butoxide in DMSO. Chem. Heterocycl. Compounds 2019, 55, 964–967. [Google Scholar] [CrossRef]
- Bhattacharryya, J. Carbon-13 NMR Analysis of Some 4-Quinazolinone Alkaloids and Related Compounds. Heterocycles 1980, 14, 1469–1473. [Google Scholar] [CrossRef]
- Kidwai, M.; Priya. Synthesis of quinazolinone analogues using sodium perborate as catalyst. Indian J. Chem. Sect. B Org. Med. Chem. 2007, 47, 1876–1881. [Google Scholar]
- Bie, Z.; Li, G.; Wang, L.; Lv, Y.; Niu, J.; Gao, S. A facile vanadium-catalyzed aerobic oxidative synthesis of quinazolinones from 2-aminobenzamides with aldehydes or alcohols. Tetrahedron Lett. 2016, 57, 4935–4938. [Google Scholar] [CrossRef]
- Chakrabarty, M.; Batabyal, A.; Morales-Ríos, M.S.; Joseph-Nathan, P. NMR studies of 4(3H)-quinazolinones and 4(3H)-quinazolinethiones. Monatsh. Chem. 1995, 126, 789–794. [Google Scholar] [CrossRef]
- Palumbo Piccionello, A.; Pace, A.; Pierro, P.; Pibiri, I.; Buscemi, S.; Vivona, N. On the reaction of some 5-polyfluoroaryl-1,2,4-oxadiazoles with methylhydrazine: Synthesis of fluorinated indazoles. Tetrahedron 2009, 65, 119–127. [Google Scholar] [CrossRef]
- Farooq, A.; Shahazadi, L.; Bajda, M.; Ullah, N.; Rauf, A.; Shahzad, S.A.; Farooq Khan, A.; Ashraf, M.; Yar, M. Organocatalyzed Novel Synthetic Methodology for Highly Functionalized Piperidines as Potent a-Glucosidase Inhibitors. Arch. Pharm. Chem. Life Sci. 2016, 349, 724–732. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvent | Reducing Agent | Catalyst | T (°C) | Time | 2a Yield % |
|---|---|---|---|---|---|---|
| 1 | THF | NH4CO2H | Pd/C (5%) | 25 °C | 48 h | NR 1 |
| 2 | Toluene | NH4CO2H | Pd/C (5%) | 25 °C | 48 h | NR 1 |
| 3 | AcOEt | NH4CO2H | Pd/C (5%) | 25 °C | 48 h | NR 1 |
| 4 | CHCl3 | NH4CO2H | Pd/C (5%) | 25 °C | 48 h | NR 1 |
| 5 | CH3CN | NH4CO2H | Pd/C (5%) | 25 °C | 48 h | NR 1 |
| 6 | MeOH | NH4CO2H | none | 25 °C | 48 h | NR 1 |
| 7 | MeOH | NH4CO2H | Zn | 25 °C | 48 h | NR 1 |
| 8 | MeOH | NH4CO2H | Pd/C (5%) | 25 °C | 48 h | 15% 2 |
| 9 | EtOH | NH4CO2H | Pd/C (5%) | 25 °C | 48 h | 10% 2 |
| 10 | MeOH | HCO2H | Pd/C (5%) | 25 °C | 48 h | 13% 2 |
| 11 | MeOH | HCO2H | Pd/C (5%) | 60 °C | 1 h | 78% 2 |
| 12 | MeOH | NH4CO2H | Pd/C (5%) | 60 °C | 1 h | 90% 2 |
| Compound | Inhibition% (100 µM) | IC50 (µM) |
|---|---|---|
| 2e | 37.3 | ND 1 |
| 2k | 79.3 | ND 1 |
| 2n | 82.7 | ND 1 |
| 4b | 83.2 | 0.157 |
| 4d | 83.9 | 0.038 |
| 4e | 87.1 | 0.619 |
| 4f | 82.4 | ND 1 |
| 4h | 79.1 | ND 1 |
| 4i | 72.5 | ND 1 |
| 4l | 98.0 | 0.436 |
| Sitagliptin | 92.1 | 0.026 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marzullo, P.; Vasto, S.; Buscemi, S.; Pace, A.; Nuzzo, D.; Palumbo Piccionello, A. Ammonium Formate-Pd/C as a New Reducing System for 1,2,4-Oxadiazoles. Synthesis of Guanidine Derivatives and Reductive Rearrangement to Quinazolin-4-Ones with Potential Anti-Diabetic Activity. Int. J. Mol. Sci. 2021, 22, 12301. https://doi.org/10.3390/ijms222212301
Marzullo P, Vasto S, Buscemi S, Pace A, Nuzzo D, Palumbo Piccionello A. Ammonium Formate-Pd/C as a New Reducing System for 1,2,4-Oxadiazoles. Synthesis of Guanidine Derivatives and Reductive Rearrangement to Quinazolin-4-Ones with Potential Anti-Diabetic Activity. International Journal of Molecular Sciences. 2021; 22(22):12301. https://doi.org/10.3390/ijms222212301
Chicago/Turabian StyleMarzullo, Paola, Sonya Vasto, Silvestre Buscemi, Andrea Pace, Domenico Nuzzo, and Antonio Palumbo Piccionello. 2021. "Ammonium Formate-Pd/C as a New Reducing System for 1,2,4-Oxadiazoles. Synthesis of Guanidine Derivatives and Reductive Rearrangement to Quinazolin-4-Ones with Potential Anti-Diabetic Activity" International Journal of Molecular Sciences 22, no. 22: 12301. https://doi.org/10.3390/ijms222212301