GPR119 and GPR55 as Receptors for Fatty Acid Ethanolamides, Oleoylethanolamide and Palmitoylethanolamide


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. OEA and GPR119
2.1. Physiological Actions of OEA
2.1.1. Anorectic Action of OEA
2.1.2. Analgesic and Other Actions of OEA
2.2. Pharmacology of GPR119
2.2.1. GPR119 Expression
2.2.2. GPR119 Ligands
2.2.3. GPR119 Functions
Pancreas
GI Tract
2.3. Drug Development
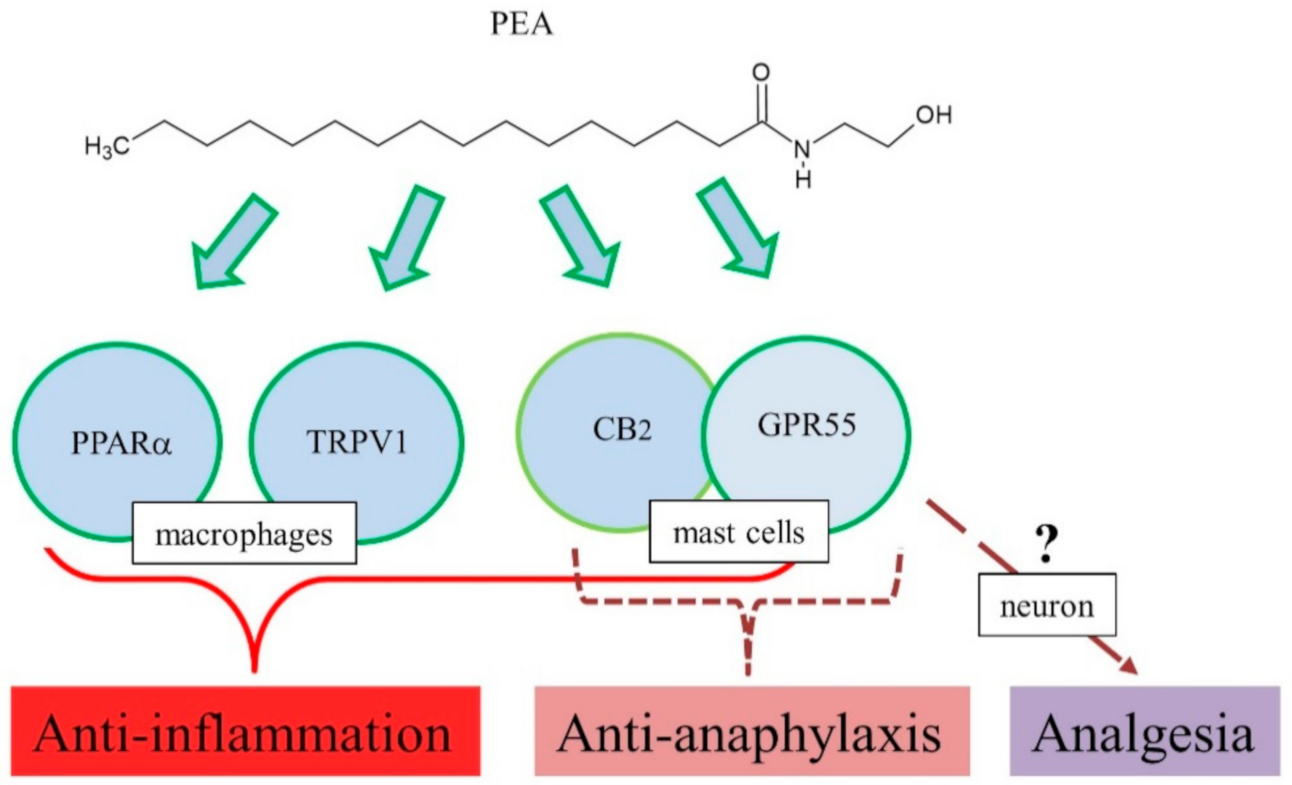
3. PEA and GPR55
3.1. Pharmacological Actions of PEA
3.1.1. Implications of PEA in Anti-Anaphylactic Activity
3.1.2. Implications of PEA in Anti-Inflammatory Effects
3.1.3. Implications of PEA in Analgesic and Neurologic Activities
3.1.4. Other Effects
3.2. Pharmacology of GPR55
4. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| OEA | Oleoylethanolamide |
| PEA | Palmitoylethanolmide |
| PPARα | Peroxisome proliferator-activated receptor-α |
| TRPV1 | Transient receptor potential vanilloid 1 |
| DAGL | Diacylglycerol lipase |
References
- Wellner, N.; Diep, T.A.; Janfelt, C.; Hansen, H.S. N-acylation of phosphatidylethanolamine and its biological functions in mammals. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2013, 1831, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Aziz, M.; Wang, X.; Tripathi, A.; Bankaitis, V.A.; Chapman, K.D. Structural analysis of a plant fatty acid amide hydrolase provides insights into the evolutionary diversity of bioactive acylethanolamides. J. Biol. Chem. 2019, 294, 7419–7432. [Google Scholar] [CrossRef] [PubMed]
- Godlewski, G.; Offertáler, L.; Wagner, J.A.; Kunos, G. Receptors for acylethanolamides—GPR55 and GPR119. Prostaglans Other Lipid Mediat. 2009, 89, 105–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrelli, F.; Izzo, A.A. Role of acylethanolamides in the gastrointestinal tract with special reference to food intake and energy balance. Best Pract. Res. Clin. Endocrinol. Metab. 2009, 23, 33–49. [Google Scholar] [CrossRef]
- Sugiura, T.; Kishimoto, S.; Oka, S.; Gokoh, M. Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog. Lipid Res. 2006, 45, 405–446. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Overton, H.A.; Fyfe, M.C.; Reynet, C. GPR119, a novel G protein-coupled receptor target for the treatment of type 2 diabetes and obesity. Br. J. Pharmacol. 2008, 153 (Suppl. 1), S76–S81. [Google Scholar] [CrossRef] [Green Version]
- De Fonseca, F.R.; Navarro, M.; Gomez, R.; Escuredo, L.; Nava, F.; Fu, J.; Murillo-Rodriguez, E.; Giuffrida, A.; LoVerme, J.; Gaetani, S. An anorexic lipid mediator regulated by feeding. Nature 2001, 414, 209–212. [Google Scholar] [CrossRef] [Green Version]
- Gaetani, S.; Oveisi, F.; Piomelli, D. Modulation of meal pattern in the rat by the anorexic lipid mediator oleoylethanolamide. Neuropsychopharmacology 2003, 28, 1311–1316. [Google Scholar] [CrossRef]
- Proulx, K.; Cota, D.; Castaneda, T.R.; Tschop, M.H.; D’Alessio, D.A.; Tso, P.; Woods, S.C.; Seeley, R.J. Mechanisms of oleoylethanolamide-induced changes in feeding behavior and motor activity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R729–R737. [Google Scholar] [CrossRef] [Green Version]
- Laleh, P.; Yaser, K.; Alireza, O. Oleoylethanolamide: A novel pharmaceutical agent in the management of obesity-an updated review. J. Cell Physiol. 2019, 234, 7893–7902. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Kim, J.; Oveisi, F.; Astarita, G.; Piomelli, D. Targeted enhancement of oleoylethanolamide production in proximal small intestine induces across-meal satiety in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R45–R50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, H.S.; Diep, T.A. N-acylethanolamines, anandamide and food intake. Biochem. Pharmacol. 2009, 78, 553–560. [Google Scholar] [CrossRef] [Green Version]
- Ahern, G.P. Activation of TRPV1 by the satiety factor oleoylethanolamide. J. Biol. Chem. 2003, 278, 30429–30434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Miyares, R.L.; Ahern, G.P. Oleoylethanolamide excites vagal sensory neurones, induces visceral pain and reduces short-term food intake in mice via capsaicin receptor TRPV1. J. Physiol. 2005, 564, 541–547. [Google Scholar] [CrossRef]
- Fu, J.; Gaetani, S.; Oveisi, F.; Verme, J.L.; Serrano, A.; De Fonseca, F.R.; Rosengarth, A.; Luecke, H.; Di Giacomo, B.; Tarzia, G. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature 2003, 425, 90–93. [Google Scholar] [CrossRef]
- Verme, J.L.; Gaetani, S.; Fu, J.; Oveisi, F.; Burton, K.; Piomelli, D. Regulation of food intake by oleoylethanolamide. Cell Mol. Life Sci. 2005, 62, 708. [Google Scholar] [CrossRef] [Green Version]
- Overton, H.A.; Babbs, A.J.; Doel, S.M.; Fyfe, M.C.; Gardner, L.S.; Griffin, G.; Jackson, H.C.; Procter, M.J.; Rasamison, C.M.; Tang-Christensen, M.; et al. Deorphanization of a G protein-coupled receptor for oleoylethanolamide and its use in the discovery of small-molecule hypophagic agents. Cell Metab. 2006, 3, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.H.; Choi, H.H.; Lee, D.H.; Chung, S.Y.; Yang, N.Y.; Kim, D.H.; Ju, M.K.; Han, T.D.; Nam, S.Y.; Kim, K.-W. YH18421, a novel GPR119 agonist exerts sustained glucose lowering and weight loss in diabetic mouse model. Arch. Pharm. Res. 2017, 40, 772–782. [Google Scholar] [CrossRef]
- Gao, J.; Tian, L.; Weng, G.; Bhagroo, N.V.; Sorenson, R.L.; O’Brien, T.D.; Luo, J.; Guo, Z. Stimulating beta cell replication and improving islet graft function by GPR119 agonists. Transp. Int. 2011, 24, 1124–1134. [Google Scholar] [CrossRef]
- Chu, Z.-L.; Jones, R.M.; He, H.; Carroll, C.; Gutierrez, V.; Lucman, A.; Moloney, M.; Gao, H.; Mondala, H.; Bagnol, D. A role for β-cell-expressed G protein-coupled receptor 119 in glycemic control by enhancing glucose-dependent insulin release. Endocrinology 2007, 148, 2601–2609. [Google Scholar] [CrossRef] [PubMed]
- Lan, H.; Vassileva, G.; Corona, A.; Liu, L.; Baker, H.; Golovko, A.; Abbondanzo, S.J.; Hu, W.; Yang, S.; Ning, Y.; et al. GPR119 is required for physiological regulation of glucagon-like peptide-1 secretion but not for metabolic homeostasis. J. Endocrinol. 2009, 201, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Ning, Y.; O’Neill, K.; Lan, H.; Pang, L.; Shan, L.X.; Hawes, B.E.; Hedrick, J.A. Endogenous and synthetic agonists of GPR119 differ in signalling pathways and their effects on insulin secretion in MIN6c4 insulinoma cells. Br. J. Pharmacol. 2008, 155, 1056–1065. [Google Scholar] [CrossRef] [Green Version]
- Gaetani, S.; Fu, J.; Cassano, T.; Dipasquale, P.; Romano, A.; Righetti, L.; Cianci, S.; Laconca, L.; Giannini, E.; Scaccianoce, S. The fat-induced satiety factor oleoylethanolamide suppresses feeding through central release of oxytocin. J. Neurosci. 2010, 30, 8096–8101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleberg, K.; Hassing, H.A.; Hansen, H.S. Classical endocannabinoid-like compounds and their regulation by nutrients. Biofactors 2014, 40, 363–372. [Google Scholar] [CrossRef]
- Suardíaz, M.; Estivill-Torrús, G.; Goicoechea, C.; Bilbao, A.; Rodríguez de Fonseca, F. Analgesic properties of oleoylethanolamide (OEA) in visceral and inflammatory pain. Pain 2007, 133, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Tough, I.R.; Forbes, S.; Herzog, H.; Jones, R.M.; Schwartz, T.W.; Cox, H.M. Bidirectional GPR119 Agonism Requires Peptide YY and Glucose for Activity in Mouse and Human Colon Mucosa. Endocrinology 2018, 159, 1704–1717. [Google Scholar] [CrossRef]
- Grill, M.; Högenauer, C.; Blesl, A.; Haybaeck, J.; Golob-Schwarzl, N.; Ferreirós, N.; Thomas, D.; Gurke, R.; Trötzmüller, M.; Köfeler, H.C.; et al. Members of the endocannabinoid system are distinctly regulated in inflammatory bowel disease and colorectal cancer. Sci. Rep. 2019, 9, 2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payahoo, L.; Khajebishak, Y.; Alivand, M.R.; Soleimanzade, H.; Alipour, S.; Barzegari, A.; Ostadrahimi, A. Investigation the effect of oleoylethanolamide supplementation on the abundance of Akkermansia uciniphila bacterium and the dietary intakes in people with obesity: A randomized clinical trial. Appetite 2019, 141, 104301. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, D.K.; Jin, X.; Che, X.; Choi, J.Y. Oleoylethanolamide Exhibits GPR119-Dependent Inhibition of Osteoclast Function and GPR119-Independent Promotion of Osteoclast Apoptosis. Mol. Cells 2020, 43, 340–349. [Google Scholar] [PubMed]
- Markovics, A.; Angyal, Á.; Tóth, K.F.; Ádám, D.; Pénzes, Z.; Magi, J.; Pór, Á.; Kovács, I.; Törőcsik, D.; Zouboulis, C.C.; et al. GPR119 Is a Potent Regulator of Human Sebocyte Biology. J. Investig. Dermatol. 2020, 140, 1909–1918.e8. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Hoglund, P.J.; Gloriam, D.E.; Lagerstrom, M.C.; Schioth, H.B. Seven evolutionarily conserved human rhodopsin G protein-coupled receptors lacking close relatives. FEBS Lett 2003, 554, 381–388. [Google Scholar] [CrossRef]
- Soga, T.; Ohishi, T.; Matsui, T.; Saito, T.; Matsumoto, M.; Takasaki, J.; Matsumoto, S.-I.; Kamohara, M.; Hiyama, H.; Yoshida, S. Lysophosphatidylcholine enhances glucose-dependent insulin secretion via an orphan G-protein-coupled receptor. Biochem. Biophys. Res. Commun. 2005, 326, 744–751. [Google Scholar] [CrossRef] [PubMed]
- Chu, Z.-L.; Carroll, C.; Alfonso, J.; Gutierrez, V.; He, H.; Lucman, A.; Pedraza, M.; Mondala, H.; Gao, H.; Bagnol, D. A role for intestinal endocrine cell-expressed g protein-coupled receptor 119 in glycemic control by enhancing glucagon-like Peptide-1 and glucose-dependent insulinotropic Peptide release. Endocrinology 2008, 149, 2038–2047. [Google Scholar] [CrossRef] [Green Version]
- Lauffer, L.M.; Iakoubov, R.; Brubaker, P.L. GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes 2009, 58, 1058–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odori, S.; Hosoda, K.; Tomita, T.; Fujikura, J.; Kusakabe, T.; Kawaguchi, Y.; Doi, R.; Takaori, K.; Ebihara, K.; Sakai, Y. GPR119 expression in normal human tissues and islet cell tumors: Evidence for its islet-gastrointestinal distribution, expression in pancreatic beta and alpha cells, and involvement in islet function. Metabolism 2013, 62, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.; Harada, N.; Ikeguchi, E.; Sankoda, A.; Hatoko, T.; Lu, X.; Yasuda, T.; Yamane, S.; Inagaki, N. Gene expression of nutrient-sensing molecules in I cells of CCK reporter male mice. J. Mol. Endocrinol. 2020, 66, 11–22. [Google Scholar] [CrossRef]
- Parker, H.; Habib, A.; Rogers, G.; Gribble, F.; Reimann, F. Nutrient-dependent secretion of glucose-dependent insulinotropic polypeptide from primary murine K cells. Diabetologia 2009, 52, 289. [Google Scholar] [CrossRef] [Green Version]
- Sykaras, A.G.; Demenis, C.; Case, R.M.; McLaughlin, J.T.; Smith, C.P. Duodenal enteroendocrine I-cells contain mRNA transcripts encoding key endocannabinoid and fatty acid receptors. PLoS ONE 2012, 7, e42373. [Google Scholar] [CrossRef] [Green Version]
- Bonini, J.A.; Borowsky, B.E.; Adham, N.; Boyle, N.; Thompson, T.O. Methods of Identifying Compounds That Bind to SNORF25 Receptors. U.S. Patent US6468756B1, 22 October 2002. [Google Scholar]
- Sakamoto, Y.; Inoue, H.; Kawakami, S.; Miyawaki, K.; Miyamoto, T.; Mizuta, K.; Itakura, M. Expression and distribution of Gpr119 in the pancreatic islets of mice and rats: Predominant localization in pancreatic polypeptide-secreting PP-cells. Biochem. Biophys. Res. Commun. 2006, 351, 474–480. [Google Scholar] [CrossRef]
- Li, N.X.; Brown, S.; Kowalski, T.; Wu, M.; Yang, L.; Dai, G.; Petrov, A.; Ding, Y.; Dlugos, T.; Wood, H.B. GPR119 agonism increases glucagon secretion during insulin-induced hypoglycemia. Diabetes 2018, 67, 1401–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Z.-L.; Carroll, C.; Chen, R.; Alfonso, J.; Gutierrez, V.; He, H.; Lucman, A.; Xing, C.; Sebring, K.; Zhou, J. N-oleoyldopamine enhances glucose homeostasis through the activation of GPR119. Mol. Endocrinol. 2010, 24, 161–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kogure, R.; Toyama, K.; Hiyamuta, S.; Kojima, I.; Takeda, S. 5-Hydroxy-eicosapentaenoic acid is an endogenous GPR119 agonist and enhances glucose-dependent insulin secretion. Biochem. Biophys. Res. Commun. 2011, 416, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.B.; Rosenkilde, M.M.; Knop, F.K.; Wellner, N.; Diep, T.A.; Rehfeld, J.F.; Andersen, U.B.; Holst, J.J.; Hansen, H.S. 2-Oleoyl glycerol is a GPR119 agonist and signals GLP-1 release in humans. J. Clin. Endocrinol. Metab. 2011, 96, E1409–E1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, H.S.; Rosenkilde, M.M.; Holst, J.J.; Schwartz, T.W. GPR119 as a fat sensor. Trends Pharmacol. Sci. 2012, 33, 374–381. [Google Scholar] [CrossRef]
- Stone, V.M.; Dhayal, S.; Smith, D.M.; Lenaghan, C.; Brocklehurst, K.J.; Morgan, N.G. The cytoprotective effects of oleoylethanolamide in insulin-secreting cells do not require activation of GPR119. Br. J. Pharmacol. 2012, 165, 2758–2770. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Fang, Y.; Park, H. Synthesis and biological evaluation of pyrimidine derivatives with diverse azabicyclic ether/amine as novel GPR119 agonist. Bioorg. Med. Chem. Lett. 2017, 27, 2515–2519. [Google Scholar] [CrossRef]
- Moran, B.M.; Abdel-Wahab, Y.H.; Flatt, P.R.; McKillop, A.M. Activation of GPR119 by fatty acid agonists augments insulin release from clonal β-cells and isolated pancreatic islets and improves glucose tolerance in mice. Biol. Chem. 2014, 395, 453–464. [Google Scholar] [CrossRef]
- Tadaki, H.; Ogawa, N.; Yamanaka, M.; Motohashi, Y.; Sasase, T.; Kawai, T.; Toriniwa, Y.; Fukuda, S.; Ogawa, N.; Harada, K.; et al. JTP-109192, a novel G protein-coupled receptor 119 agonist, prevents atherosclerosis by improving hypercholesterolaemia in congenic spontaneously hyperlipidaemic mice. Clin. Exp. Pharmacol. Physiol. 2020. [Google Scholar] [CrossRef]
- Drzazga, A.; Kristinsson, H.; Sałaga, M.; Zatorski, H.; Koziołkiewicz, M.; Gendaszewska-Darmach, E.; Bergsten, P. Lysophosphatidylcholine and its phosphorothioate analogues potentiate insulin secretion via GPR40 (FFAR1), GPR55 and GPR119 receptors in a different manner. Mol. Cell Endocrinol. 2018, 472, 117–125. [Google Scholar] [CrossRef]
- Fyfe, M.C.; McCormack, J.G.; Overton, H.A.; Procter, M.J.; Reynet, C. GPR119 agonists as potential new oral agents for the treatment of type 2 diabetes and obesity. Expert Opin. Drug Discov. 2008, 3, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Ritter, K.; Buning, C.; Halland, N.; Pöverlein, C.; Schwink, L. G protein-coupled receptor 119 (GPR119) agonists for the treatment of diabetes: Recent progress and prevailing challenges. J. Med. Chem. 2016, 59, 3579–3592. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.M.; Leonard, J.N.; Buzard, D.J.; Lehmann, J. GPR119 agonists for the treatment of type 2 diabetes. Expert Opin. Ther. Patents 2009, 19, 1339–1359. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.-U. GPR119 agonists: A promising approach for T2DM treatment? A SWOT analysis of GPR119. Drug Discov. Today 2013, 18, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Nunez, D.J.; Bush, M.A.; Collins, D.A.; McMullen, S.L.; Gillmor, D.; Apseloff, G.; Atiee, G.; Corsino, L.; Morrow, L.; Feldman, P.L. Gut hormone pharmacology of a novel GPR119 agonist (GSK1292263), metformin, and sitagliptin in type 2 diabetes mellitus: Results from two randomized studies. PLoS ONE 2014, 9, e92494. [Google Scholar] [CrossRef] [PubMed]
- Semple, G.; Ren, A.; Fioravanti, B.; Pereira, G.; Calderon, I.; Choi, K.; Xiong, Y.; Shin, Y.-J.; Gharbaoui, T.; Sage, C.R. Discovery of fused bicyclic agonists of the orphan G-protein coupled receptor GPR119 with in vivo activity in rodent models of glucose control. Bioorg. Med. Chem. Lett. 2011, 21, 3134–3141. [Google Scholar] [CrossRef]
- Hassing, H.A.; Fares, S.; Larsen, O.; Pad, H.; Hauge, M.; Jones, R.M.; Schwartz, T.W.; Hansen, H.S.; Rosenkilde, M.M. Biased signaling of lipids and allosteric actions of synthetic molecules for GPR119. Biochem. Pharmacol. 2016, 119, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Katz, L.; Gambale, J.; Rothenberg, P.; Vanapalli, S.; Vaccaro, N.; Xi, L.; Sarich, T.; Stein, P. Effects of JNJ-38431055, a novel GPR119 receptor agonist, in randomized, double-blind, placebo-controlled studies in subjects with type 2 diabetes. Diabetes Obes. Metab. 2012, 14, 709–716. [Google Scholar] [CrossRef]
- Engelstoft, M.S.; Norn, C.; Hauge, M.; Holliday, N.D.; Elster, L.; Lehmann, J.; Jones, R.M.; Frimurer, T.M.; Schwartz, T.W. Structural basis for constitutive activity and agonist-induced activation of the enteroendocrine fat sensor GPR119. Br. J. Pharmacol. 2014, 171, 5774–5789. [Google Scholar] [CrossRef] [Green Version]
- Han, T.; Lee, B.M.; Park, Y.H.; Lee, D.H.; Choi, H.H.; Lee, T.; Kim, H. YH18968, a novel 1, 2, 4-triazolone G-protein coupled receptor 119 agonist for the treatment of type 2 diabetes mellitus. Biomol. Ther. 2018, 26, 201. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, K.; Yoshitomi, T.; Ishimoto, Y.; Tanaka, N.; Takahashi, K.; Watanabe, A.; Chiba, K. DS-8500a, an orally available G protein-coupled receptor 119 agonist, upregulates glucagon-like peptide-1 and enhances glucose-dependent insulin secretion and improves glucose homeostasis in type 2 diabetic rats. J. Pharmacol. Exp. Ther. 2018, 367, 509–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Terauchi, Y.; Watada, H.; Nakatsuka, Y.; Shiosakai, K.; Washio, T.; Taguchi, T. Efficacy and safety of GPR119 agonist DS-8500a in Japanese patients with type 2 diabetes: A randomized, double-blind, placebo-controlled, 12-week study. Adv. Ther. 2018, 35, 367–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, M.; Dow, J.; Van Vliet, A.; Pleszko, A.; Lockton, J. Orally administered GPR119 agonist PSN821 shows clinically significant glucose lowering and other potential cardiometabolic benefits in patients with type 2 diabetes. In Diabetologia; Springer: New York, NY, USA, 2011. [Google Scholar]
- Nunez, D.J.; Bush, M.A.; Collins, D.A.; Mcmullen, S.L.; Apseloff, G.; Atiee, G.; Cosino, L.; Morrow, L.; Feldman, P.L. Novel Effects on Lipids of GSK1292263, a GPR119 Agonist, in Type 2 Diabetics. In Diabetes; American Diabetes Association: Alexandria, VA, USA, 2012. [Google Scholar]
- Thabuis, C.; Destaillats, F.; Landrier, J.F.; Tissot-Favre, D.; Martin, J.C. Analysis of gene expression pattern reveals potential targets of dietary oleoylethanolamide in reducing body fat gain in C3H mice. J. Nutr. Biochem. 2010, 21, 922–928. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.W.; Kim, H.S.; Im, J.H.; Kim, J.W.; Jun, D.W.; Lim, S.C.; Lee, K.; Choi, J.M.; Kim, S.K.; Kang, K.W. GPR119: A promising target for nonalcoholic fatty liver disease. FASEB J. 2016, 30, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Ganley, O.H.; Graessle, O.E.; Robinson, H.J. Anti-inflammatory activity of compounds obtained from egg yolk, peanut oil, and soybean lecithin. J. Lab. Clin. Med. 1958, 51, 709–714. [Google Scholar]
- Ganley, O.H.; Robinson, H.J. Antianaphylactic and antiserotonin activity of a compound obtained from egg yolk, peanut oil, and soybean lecithin. J. Allergy 1959, 30, 415–419. [Google Scholar] [CrossRef]
- Perlik, F.; Raskova, H.; Elis, J. Anti-inflammatory properties of N (2-hydroxyethyl) palmitamide. Acta Physiol. Acad. Sci. Hung. 1971, 39, 395. [Google Scholar]
- Aloe, L.; Leon, A.; Levi-Montalcini, R. A proposed autacoid mechanism controlling mastocyte behaviour. Agents Actions 1993, 39, C145–C147. [Google Scholar] [CrossRef]
- Facci, L.; Dal Toso, R.; Romanello, S.; Buriani, A.; Skaper, S.; Leon, A. Mast cells express a peripheral cannabinoid receptor with differential sensitivity to anandamide and palmitoylethanolamide. Proc. Natl. Acad. Sci. USA 1995, 92, 3376–3380. [Google Scholar] [CrossRef] [Green Version]
- Galiègue, S.; Mary, S.; Marchand, J.; Dussossoy, D.; Carrière, D.; Carayon, P.; Bouaboula, M.; Shire, D.; LE Fur, G.; Casellas, P. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur. J. Biochem. 1995, 232, 54–61. [Google Scholar] [CrossRef]
- Skaper, S.D.; Buriani, A.; Dal Toso, R.; Petrelli, L.; Romanello, S.; Facci, L.; Leon, A. The ALIAmide palmitoylethanolamide and cannabinoids, but not anandamide, are protective in a delayed postglutamate paradigm of excitotoxic death in cerebellar granule neurons. Proc. Natl. Acad. Sci. USA 1996, 93, 3984–3989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, V.; Reddy, P.V.; Schmid, P.C.; Schmid, H.H. N-Acylation of ethanolamine phospholipids in canine myocardium. Biochim. Biophys. Acta Lipids Lipid Metab. 1982, 712, 342–355. [Google Scholar] [CrossRef]
- Mazzari, S.; Canella, R.; Petrelli, L.; Marcolongo, G.; Leon, A. N-(2-hydroxyethyl) hexadecanamide is orally active in reducing edema formation and inflammatory hyperalgesia by down-modulating mast cell activation. Eur. J. Pharmacol. 1996, 300, 227–236. [Google Scholar] [CrossRef]
- Roviezzo, F.; Rossi, A.; Caiazzo, E.; Orlando, P.; Riemma, M.A.; Iacono, V.M.; Guarino, A.; Ialenti, A.; Cicala, C.; Peritore, A. Palmitoylethanolamide supplementation during sensitization prevents airway allergic symptoms in the mouse. Front. Pharmacol. 2017, 8, 857. [Google Scholar] [CrossRef] [Green Version]
- Lambert, D.M.; Di, V.M. The palmitoylethanolamide and oleamide enigmas: Are these two fatty acid amides cannabimimetic? Cur. Med. Chem. 1999, 6, 757–773. [Google Scholar]
- Petrosino, S.; Moriello, A.S.; Verde, R.; Allarà, M.; Imperatore, R.; Ligresti, A.; Mahmoud, A.M.; Peritore, A.F.; Iannotti, F.A.; Di Marzo, V. Palmitoylethanolamide counteracts substance P-induced mast cell activation in vitro by stimulating diacylglycerol lipase activity. J. Neuroinflamm. 2019, 16, 274. [Google Scholar] [CrossRef] [Green Version]
- Showalter, V.M.; Compton, D.R.; Martin, B.R.; Abood, M.E. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): Identification of cannabinoid receptor subtype selective ligands. J. Pharmacol. Exp. Ther. 1996, 278, 989–999. [Google Scholar]
- Conti, S.; Costa, B.; Colleoni, M.; Parolaro, D.; Giagnoni, G. Antiinflammatory action of endocannabinoid palmitoylethanolamide and the synthetic cannabinoid nabilone in a model of acute inflammation in the rat. Br. J. Pharmacol. 2002, 135, 181–187. [Google Scholar] [CrossRef] [Green Version]
- Devane, W.A.; Hanus, L.; Breuer, A.; Pertwee, R.G.; Stevenson, L.A.; Griffin, G.; Gibson, D.; Mandelbaum, A.; Etinger, A.; Mechoulam, R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 1992, 258, 1946–1949. [Google Scholar] [CrossRef]
- Felder, C.C.; Briley, E.M.; Axelrod, J.; Simpson, J.T.; Mackie, K.; Devane, W.A. Anandamide, an endogenous cannabimimetic eicosanoid, binds to the cloned human cannabinoid receptor and stimulates receptor-mediated signal transduction. Proc. Natl. Acad. Sci. USA 1993, 90, 7656–7660. [Google Scholar] [CrossRef] [Green Version]
- Couch, D.G.; Cook, H.; Ortori, C.; Barrett, D.; Lund, J.N.; O’Sullivan, S.E. Palmitoylethanolamide and cannabidiol prevent inflammation-induced hyperpermeability of the human gut in vitro and in vivo—A randomized, placebo-controlled, double-blind controlled trial. Inflamm. Bowel Dis. 2019, 25, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Galiazzo, G.; Giancola, F.; Stanzani, A.; Fracassi, F.; Bernardini, C.; Forni, M.; Pietra, M.; Chiocchetti, R. Localization of cannabinoid receptors CB1, CB2, GPR55, and PPARα in the canine gastrointestinal tract. Histochem. Cell Biol. 2018, 150, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Rinne, P.; Guillamat-Prats, R.; Rami, M.; Bindila, L.; Ring, L.; Lyytikäinen, L.-P.; Raitoharju, E.; Oksala, N.; Lehtimäki, T.; Weber, C. Palmitoylethanolamide promotes a proresolving macrophage phenotype and attenuates atherosclerotic plaque formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2562–2575. [Google Scholar] [CrossRef] [PubMed]
- Borrelli, F.; Romano, B.; Petrosino, S.; Pagano, E.; Capasso, R.; Coppola, D.; Battista, G.; Orlando, P.; Di Marzo, V.; Izzo, A.A. Palmitoylethanolamide, a naturally occurring lipid, is an orally effective intestinal anti-inflammatory agent. Br. J. Pharmacol. 2015, 172, 142–158. [Google Scholar] [CrossRef] [Green Version]
- Naderi, N.; Majidi, M.; Mousavi, Z.; Tusi, S.K.; Mansouri, Z.; Khodagholi, F. The interaction between intrathecal administration of low doses of palmitoylethanolamide and AM251 in formalin-induced pain related behavior and spinal cord IL1-β expression in rats. Neurochem. Res. 2012, 37, 778–785. [Google Scholar] [CrossRef]
- Kramar, C.; Loureiro, M.; Renard, J.; Laviolette, S.R. Palmitoylethanolamide modulates GPR55 receptor signaling in the ventral hippocampus to regulate mesolimbic dopamine activity, social interaction, and memory processing. Cannabis Cannabinoid Res. 2017, 2, 8–20. [Google Scholar] [CrossRef]
- Kerr, D.; Downey, L.; Conboy, M.; Finn, D.; Roche, M. Alterations in the endocannabinoid system in the rat valproic acid model of autism. Behav. Brain Res. 2013, 249, 124–132. [Google Scholar] [CrossRef]
- Musella, A.; Fresegna, D.; Rizzo, F.; Gentile, A.; Bullitta, S.; De Vito, F.; Guadalupi, L.; Centonze, D.; Mandolesi, G. A novel crosstalk within the endocannabinoid system controls GABA transmission in the striatum. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Henry, R.J.; Kerr, D.M.; Flannery, L.E.; Killilea, M.; Hughes, E.M.; Corcoran, L.; Finn, D.P.; Roche, M. Pharmacological inhibition of FAAH modulates TLR-induced neuroinflammation, but not sickness behaviour: An effect partially mediated by central TRPV1. Brain Behav. Immun. 2017, 62, 318–331. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Qiao, Z.; Kumar, P.; Song, Z.-H. Effects of palmitoylethanolamide on aqueous humor outflow. Investig. Ophthalmol. Vis. Sci. 2012, 53, 4416–4425. [Google Scholar] [CrossRef] [Green Version]
- Marichal-Cancino, B.A.; González-Hernández, A.; MaassenVanDenBrink, A.; Ramírez-San Juan, E.; Villalón, C.M. Potential Mechanisms Involved in Palmitoylethanolamide-Induced Vasodepressor Effects in Rats. J. Vasc. Res. 2020, 57, 152–163. [Google Scholar] [CrossRef]
- Kapur, A.; Zhao, P.; Sharir, H.; Bai, Y.; Caron, M.G.; Barak, L.S.; Abood, M.E. Atypical responsiveness of the orphan receptor GPR55 to cannabinoid ligands. J. Biol. Chem. 2009, 284, 29817–29827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, S.; Nakajima, K.; Yamashita, A.; Kishimoto, S.; Sugiura, T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem. Biophys. Res. Commun. 2007, 362, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Lauckner, J.E.; Jensen, J.B.; Chen, H.Y.; Lu, H.C.; Hille, B.; Mackie, K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc. Natl. Acad. Sci. USA 2008, 105, 2699–2704. [Google Scholar] [CrossRef] [Green Version]
- Oka, S.; Toshida, T.; Maruyama, K.; Nakajima, K.; Yamashita, A.; Sugiura, T. 2-Arachidonoyl-sn-glycero-3-phosphoinositol: A possible natural ligand for GPR55. J. Biochem. 2009, 145, 13–20. [Google Scholar] [CrossRef]
- Henstridge, C.M.; Balenga, N.A.; Ford, L.A.; Ross, R.A.; Waldhoer, M.; Irving, A.J. The GPR55 ligand L-alpha-lysophosphatidylinositol promotes RhoA-dependent Ca2+ signaling and NFAT activation. FASEB J. 2009, 23, 183–193. [Google Scholar] [CrossRef]
- Guida, F.; Luongo, L.; Boccella, S.; Giordano, M.; Romano, R.; Bellini, G.; Manzo, I.; Furiano, A.; Rizzo, A.; Imperatore, R. Palmitoylethanolamide induces microglia changes associated with increased migration and phagocytic activity: Involvement of the CB2 receptor. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Balenga, N.A.; Martinez-Pinilla, E.; Kargl, J.; Schroder, R.; Peinhaupt, M.; Platzer, W.; Balint, Z.; Zamarbide, M.; Dopeso-Reyes, I.G.; Ricobaraza, A.; et al. Heteromerization of GPR55 and cannabinoid CB2 receptors modulates signalling. Br. J. Pharmacol. 2014, 171, 5387–5406. [Google Scholar] [CrossRef] [Green Version]
- Kargl, J.; Balenga, N.; Parzmair, G.P.; Brown, A.J.; Heinemann, A.; Waldhoer, M. The cannabinoid receptor CB1 modulates the signaling properties of the lysophosphatidylinositol receptor GPR55. J. Biol. Chem. 2012, 287, 44234–44248. [Google Scholar] [CrossRef] [Green Version]
- Johns, D.G.; Behm, D.J.; Walker, D.J.; Ao, Z.; Shapland, E.M.; Daniels, D.A.; Riddick, M.; Dowell, S.; Staton, P.C.; Green, P.; et al. The novel endocannabinoid receptor GPR55 is activated by atypical cannabinoids but does not mediate their vasodilator effects. Br. J. Pharmacol. 2007, 152, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Alhouayek, M.; Masquelier, J.; Muccioli, G.G. Lysophosphatidylinositols, from cell membrane constituents to GPR55 ligands. Trends Pharmacol. Sci. 2018, 39, 586–604. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.M.; Gutierrez, T.; Deng, L.; Lee, W.-H.; Mackie, K.; Hohmann, A.G. Inflammatory and neuropathic nociception is preserved in GPR55 knockout mice. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staton, P.C.; Hatcher, J.P.; Walker, D.J.; Morrison, A.D.; Shapland, E.M.; Hughes, J.P.; Chong, E.; Mander, P.K.; Green, P.J.; Billinton, A.; et al. The putative cannabinoid receptor GPR55 plays a role in mechanical hyperalgesia associated with inflammatory and neuropathic pain. Pain 2008, 139, 225–236. [Google Scholar] [CrossRef]
- Schicho, R.; Bashashati, M.; Bawa, M.; McHugh, D.; Saur, D.; Hu, H.M.; Zimmer, A.; Lutz, B.; Mackie, K.; Bradshaw, H.B.; et al. The atypical cannabinoid O-1602 protects against experimental colitis and inhibits neutrophil recruitment. Inflamm. Bowel Dis. 2011, 17, 1651–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stancic, A.; Jandl, K.; Hasenohrl, C.; Reichmann, F.; Marsche, G.; Schuligoi, R.; Heinemann, A.; Storr, M.; Schicho, R. The GPR55 antagonist CID16020046 protects against intestinal inflammation. Neurogastroenterol. Motil. 2015, 27, 1432–1445. [Google Scholar] [CrossRef] [Green Version]
- Ross, G.R.; Lichtman, A.; Dewey, W.L.; Akbarali, H.I. Evidence for the putative cannabinoid receptor (GPR55)-mediated inhibitory effects on intestinal contractility in mice. Pharmacology 2012, 90, 55–65. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Fichna, J.; Schicho, R.; Saur, D.; Bashashati, M.; Mackie, K.; Li, Y.; Zimmer, A.; Goke, B.; Sharkey, K.A.; et al. A role for O-1602 and G protein-coupled receptor GPR55 in the control of colonic motility in mice. Neuropharmacology 2013, 71, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Sylantyev, S.; Jensen, T.P.; Ross, R.A.; Rusakov, D.A. Cannabinoid- and lysophosphatidylinositol-sensitive receptor GPR55 boosts neurotransmitter release at central synapses. Proc. Natl. Acad. Sci. USA 2013, 110, 5193–5198. [Google Scholar] [CrossRef] [Green Version]
- Hurst, K.; Badgley, C.; Ellsworth, T.; Bell, S.; Friend, L.; Prince, B.; Welch, J.; Cowan, Z.; Williamson, R.; Lyon, C.; et al. A putative lysophosphatidylinositol receptor GPR55 modulates hippocampal synaptic plasticity. Hippocampus 2017, 27, 985–998. [Google Scholar] [CrossRef]
- Okine, B.N.; Mc Laughlin, G.; Gaspar, J.C.; Harhen, B.; Roche, M.; Finn, D.P. Antinociceptive Effects of the GPR55 Antagonist CID16020046 Injected into the Rat Anterior Cingulate Cortex. Neuroscience 2020, 443, 19–29. [Google Scholar] [CrossRef]
- Breen, C.; Brownjohn, P.W.; Ashton, J.C. The atypical cannabinoid O-1602 increases hind paw sensitisation in the chronic constriction injury model of neuropathic pain. Neurosci. Lett. 2012, 508, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Arteaga, A.; Vazquez, M.J.; Vazquez-Martinez, R.; Pulido, M.R.; Suarez, J.; Velasquez, D.A.; Lopez, M.; Ross, R.A.; de Fonseca, F.R.; Bermudez-Silva, F.J.; et al. The atypical cannabinoid O-1602 stimulates food intake and adiposity in rats. Diabetes Obes. Metab. 2012, 14, 234–243. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Im, D.-S. GPR119 and GPR55 as Receptors for Fatty Acid Ethanolamides, Oleoylethanolamide and Palmitoylethanolamide. Int. J. Mol. Sci. 2021, 22, 1034. https://doi.org/10.3390/ijms22031034
Im D-S. GPR119 and GPR55 as Receptors for Fatty Acid Ethanolamides, Oleoylethanolamide and Palmitoylethanolamide. International Journal of Molecular Sciences. 2021; 22(3):1034. https://doi.org/10.3390/ijms22031034
Chicago/Turabian StyleIm, Dong-Soon. 2021. "GPR119 and GPR55 as Receptors for Fatty Acid Ethanolamides, Oleoylethanolamide and Palmitoylethanolamide" International Journal of Molecular Sciences 22, no. 3: 1034. https://doi.org/10.3390/ijms22031034