1. Introduction
Coumarins are the naturally occurring benzopyran-2-ones that are widespread between various plants in nature [
1]. It is thought that these compounds play important fungicidal and bactericidal roles in plants. Keeping in mind the physiological significance of coumarins, there has been a strong interest in the turnover and detoxification mechanisms of those compounds in nature, including microorganisms [
2]. It is known that the metabolism of coumarins starts with the reduction and hydrolysis of its lactone moiety in several bacterial strains [
3,
4]. According to literature, reduction of the activated C=C-bond in coumarin can be catalyzed by ene-reductases (ERs) [
5]. ERs are widely used in a biocatalytic nicotinamide-dependent asymmetric reduction of the activated double bonds yielding chiral α-substituted and/or β-substituted reduction products. To date, four different classes of ERs capable of reducing a broad variety of substrates including α,β-unsaturated aldehydes, ketones, carboxylic acids and derivatives (such as esters, lactones, cyclic imides), and nitro compounds have been identified [
6,
7,
8,
9,
10]. The most extensively investigated family of ERs is the FMN-containing the Old Yellow Enzyme (OYE) family of oxidoreductases [
9]. Other families of ERs are the oxygen-sensitive FAD and [4Fe-4S]-containing clostridial enoate reductases (EnoR) [
11], the leukotriene B4 dehydrogenase subfamily of medium chain dehydrogenases/reductases (MDR) [
12] and the salutaridine/menthone reductase-like subfamily of short chain dehydrogenases/reductases (SDR) [
13,
14]. The last two families contain the less characterized NAD(P)H-dependent flavin-independent ERs that have been recently studied for their biocatalytic mechanisms [
7,
15].
Many homologous ERs can be found in bacteria.
Pseudomonas spp. are ubiquitous in nature, known to metabolize a wide variety of environmental pollutants, and to harbor a large number of enzymes, including some that catalyze the reduction of α,β-unsaturated compounds [
16]. Hence, the xenobiotic reductase A (XenA), a member of OYE family, from
Pseudomonas putida 86 has the ability to reduce the C3-C4 double bond of 8-hydroxycoumarin, an intermediate of the degradation of quinoline [
17].
In a previous study, a lower pathway of degradation of 7-hydroxycoumarin in
Pseudomonas mandelli 7HK4 bacteria encoded by a
hcdABC gene cluster and responsible for the degradation of 3-(2,4-dihydroxyphenyl)propionic acid has been described [
18]. However, it is still unclear how 7-hydroxycoumarin is converted to 3-(2,4-dihydroxyphenyl)propionic acid in these bacteria.
Here, to elucidate the first step of catabolism of 7-hydroxycoumarin, xenobiotic reductases from Pseudomonas mandelli 7HK4 bacteria were tested as putative enzymes involved in a reduction of the lactone ring. Two of three xenobiotic reductases—XenA38 and XenA45—were active towards various coumarins, however, none of them was induced in the presence of 7-hydroxycoumarin. In contrast, an analysis of 7-hydroxycoumarin-inducible genes allowed the identification of a putative alcohol dehydrogenase HcdE, which could catalyze a reduction of 7-hydroxycoumarin to 7-hydroxy-3,4-dihydrocoumarin. To our best knowledge, the HcdE was the first alcohol dehydrogenase capable of the reduction of a double bound. Based on these data, a 7-hydroxy-3,4-dihydrocoumarin pathway of utilization of 7-hydroxycoumarin in P. mandelii 7HK4 cells was proposed.
2. Results
2.1. Screening for XenA Reductase Homologues in the Genome of P. mandelii 7HK4
To find out whether
P. mandelii 7HK4 cells utilized the ERs for the catabolism of 7-hydroxycoumarin, we used the amino acid sequence of the known XenA protein to search for the homologues encoded in the genome of
P. mandelii 7HK4 [
18]. In this way, three genes denoted as
xenA38,
xenA45 and
xenA205 encoding putative reductases with close similarity to the known XenA proteins were discovered. The products of the
xenA38,
xenA45 and
xenA205 genes belonged to the mycofactocin system FadH/OYE oxidoreductase family. The first two proteins bore the most resemblance to a putative NADH oxidoreductase from
Pseudomonas fluorescens SBW25 and 2,4-dienoyl-CoA reductase from
Pseudomonas fluorescens F113, respectively (
Figure 1a,b).
XenA205 protein was similar to 2,4-dienoyl-CoA reductase from
Pseudomonas sp. ok602 (
Figure 1c). All three genes were scattered throughout the genome of
P. mandelli 7HK4 and were unlikely belonging to any putative gene clusters.
2.2. Characterization of Putative Xenobiotic Reductases
E. coli BL21 cells transformed with the plasmid harboring
xenA38,
xenA45 or
xenA205 gene were used for the assay of the activity of the appropriate enzyme by time course experiments.
E. coli BL21 containing
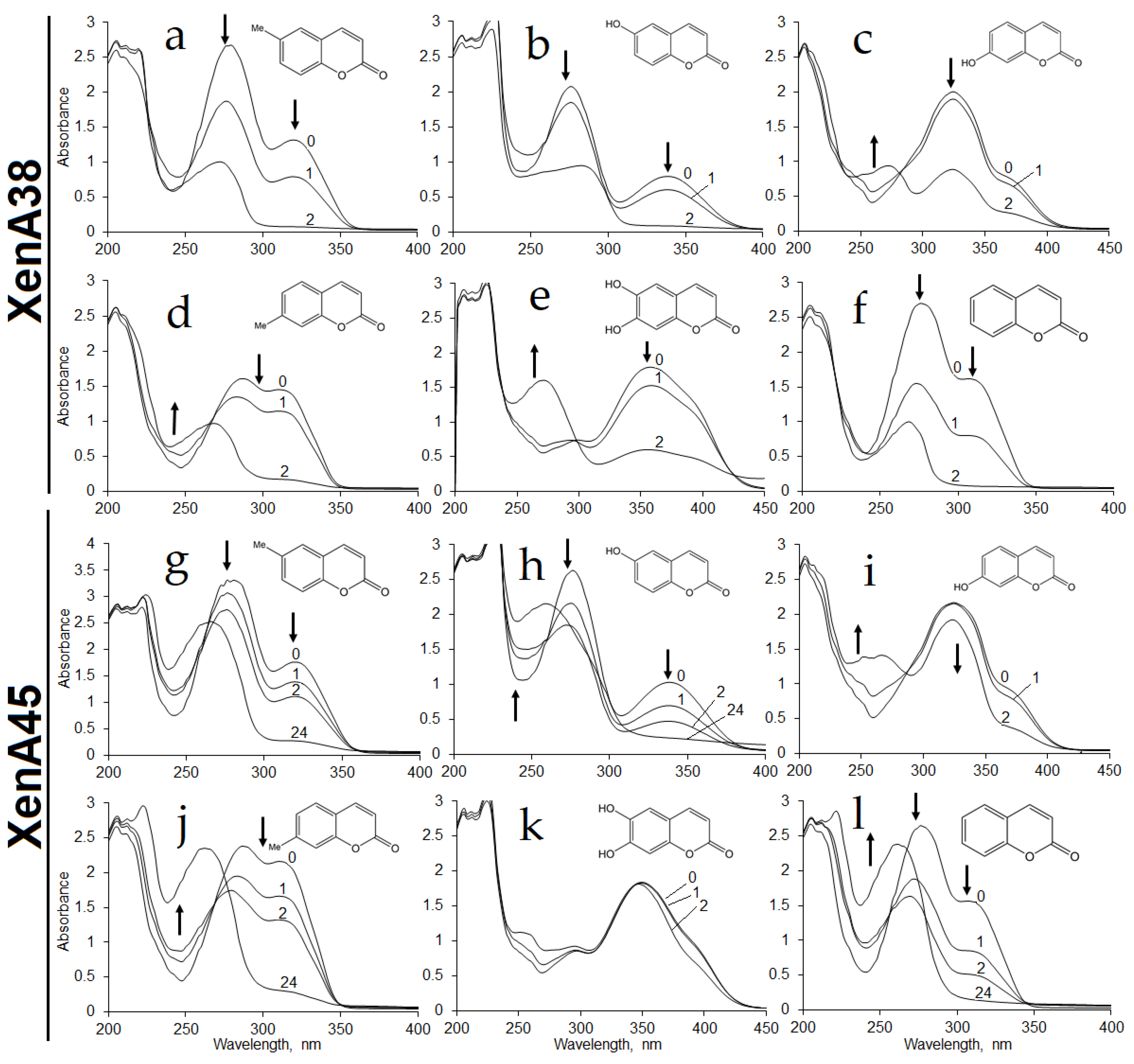
xenA38 gene showed bioconversion of coumarin, 6-hydroxycoumarin, 6-methylcoumarin, 7-hydroxycoumarin, 7-methylcoumarin and 6,7-dihydroxycoumarin according to the changes in the UV−VIS spectra (
Figure 2a–f).
Whole cells of
E. coli BL21 containing XenA45 reductase showed similar activities against these substrates except for the reaction with 6,7-dihydroxycoumarin of which UV−VIS spectra did not change over time (
Figure 2g–l). No activities were observed with
o-coumaric acid and 2,4-dihydroxycinnamic acid. The
E. coli cells with
xenA205 gene or without any additional genes showed no activity towards those compounds. These findings revealed that
P. mandelii 7HK4 encode two putative xenobiotic reductases, which could utilize a number of differently substituted coumarins.
For further characterization the C-terminal His
6-tagged XenA38 and XenA45 proteins were produced in
E. coli BL21, and purified by affinity chromatography. Both purified enzymes migrated as ~40 kDa bands in SDS-PAGE, the solution of XenA45 enzyme was colorless contrary to the XenA38 protein that had a bright yellow color inferring that the protein contained a tightly bound flavin [
19,
20]. However, all attempts to measure enzymatic activity under aerobic conditions gave no results.
2.3. Quantitative RT-PCR Analysis of the Pseudomonas mandelii 7HK4 Transcripts Induced by Coumarins
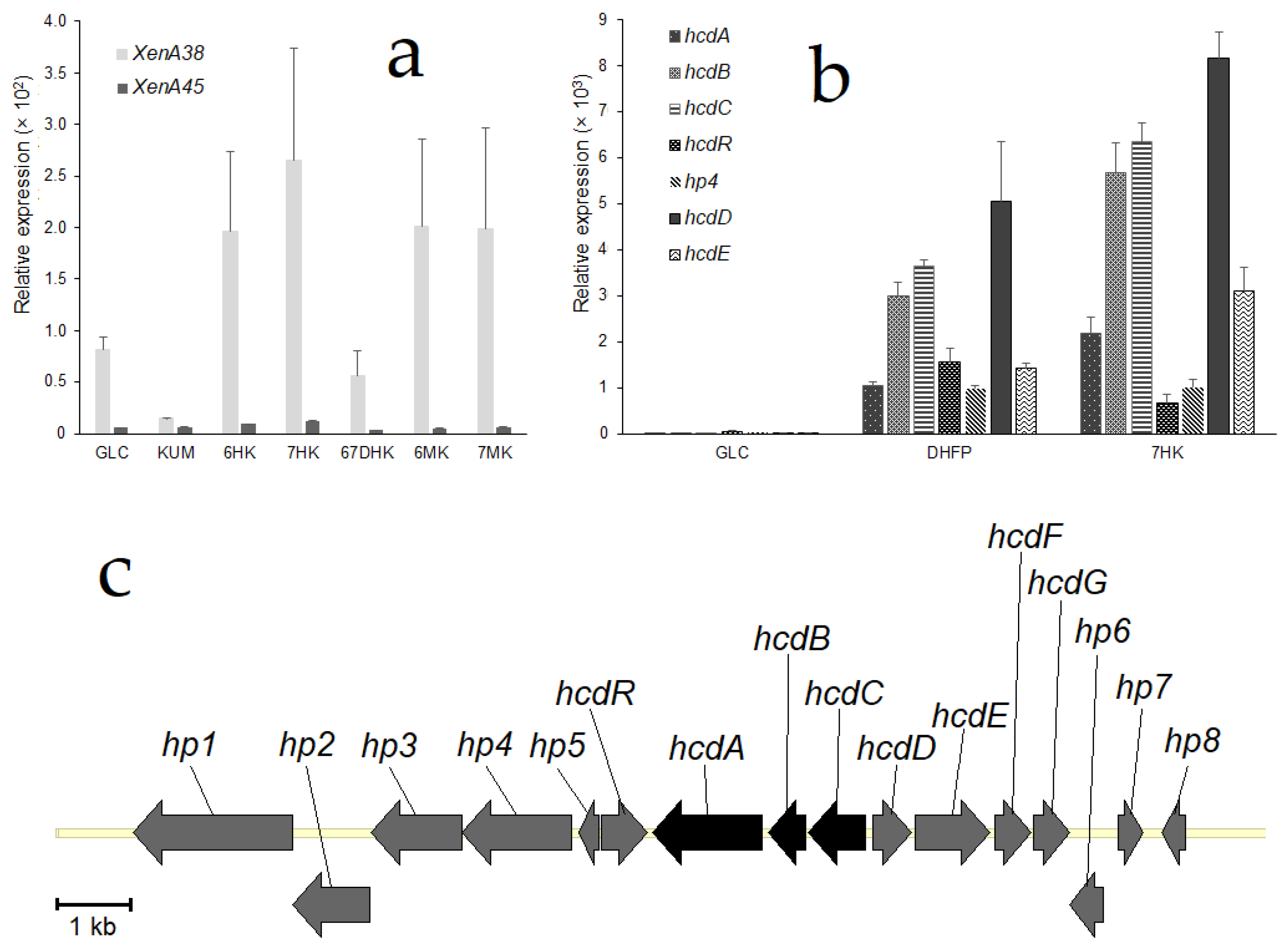
To investigate whether the expression of the putative reductase genes was dependent on 7-hydroxycoumarin, quantitative PCR analysis was performed.
Pseudomonas mandelii 7HK4 cells were induced with various coumarin derivatives and total RNA was isolated as described in Materials and Methods. Results revealed that the expression of each of the
xenA38 and
xenA45 genes were not markedly induced when
P. mandelii 7HK4 was cultivated in the presence of either 7-hydroxycoumarin, or the other coumarin derivatives (
Figure 3a). Moreover,
xenA45 gene was not expressed under any tested condition, although the expression levels of the
xenA38 gene were significantly higher than its counterpart. Hence, the levels of a
xenA38 mRNA synthesis in the hydroxycoumarin-induced 7HK4 cells were compared to the ones in the glucose-grown (noninduced) cells. Thus, it was found that the transcription of the
xenA38 gene was increased two-fold in the presence of 6-hydroxycoumarin and three-fold in the presence of 7-hydroxycoumarin.
An expression of the
xenA38 gene was also compared to the expression levels of
hcdABC genes encoding a lower part of the degradation pathway of 7-hydroxycoumarin [
18]. It was found that the levels of mRNA synthesis of
hcdABC genes were increased 1000-fold in the presence of 7-hydroxycoumarin (
Figure 3b). Therefore, we concluded that
xenA38 mRNA levels are too low to be induced by coumarin derivatives and XenA38 could be considered as a constitutive enzyme that shares a broad reducing activity in the
P. mandelii 7HK4 cells.
For further investigation, we conducted a quantitative PCR analysis of genes that are adjacent to the
hcdABC gene cluster (
Figure 3c). The results revealed that randomly chosen putative
hp4,
hcdR,
hcdD and
hcdE genes were all induced in
P. mandelii 7HK4 cells (
Figure 3b). Expression levels of
hcdR,
hp4,
hcdD and
hcdE genes were increased approximately 1000-fold, and were similar to expression levels of the
hcdABC genes under the same conditions. Each of the
hcdABCR,
hp4,
hcdD and
hcdE genes was specifically induced when
P. mandelii 7HK4 was cultivated in the presence of either 3-(2,4-dihydroxyphenyl)propionic acid or 7-hydroxycoumarin. In addition, a 100-fold increase of mRNA synthesis of these genes was also observed in
P. mandelii 7HK4 cells precultivated with 7-methylcoumarin. No expression of the analyzed genes was observed in
P. mandelii 7HK4 cells precultivated with coumarin, 6-hydroxycoumarin, 6-methylcoumarin, 6,7-dihydroxycoumarin,
o-coumaric acid,
p-coumaric acid, 2,4-dihydroxycinnamic acid, quinoline or isoquinoline.
2.4. Analysis of the Genome Locus Adjacent to 7-Hydroxycoumarin-Inducible hcd Gene Cluster
A genomic region approximately 3 kb upstream of the
hcdABC gene cluster containing
hcdD,
hcdE,
hcdF and
hcdG genes was amplified by PCR and cloned into the pACYCDuet-1 expression vector. The recombinant HcdD, HcdE, HcdF and HcdG proteins were produced together in
E. coli BL21 strain, and the expression was confirmed by SDS-PAGE. The whole cells of
E. coli BL21 transformed with the pHP4-10 plasmid (
Table S2) were used for the bioconversion of coumarin, 6-hydroxycoumarin, 6-methylcoumarin, 7-hydroxycoumarin, 7-methylcoumarin, 7-methoxycoumarin, 4-methyl-7-hydroxycoumarin and 6,7-dihydroxycoumarin. Time course experiments revealed the decrease of UV absorption maxima of these substrates over time. After the completion of bioconversions, there was a non-disappearing UV absorption maximum at 260–280 nm wavelengths similar to that of 3-(2,4-dihydroxyphenyl)propionic acid (see
Figures S1 and S7 in the Supplementary Materials). This suggested that
E. coli BL21 cells transformed with the pHP4-10 plasmid could catalyze the reduction and/or hydrolysis of the lactone moiety of coumarin derivatives. When activity rates were compared, a conversion of the 7-hydroxycoumarin was the fastest, and conversion of methyl- and methoxy-substituted coumarins proceeded at the lowest rate. In addition, no activity was observed with 3-hydroxycoumarin, 4-hydroxycoumarin, 7-ethoxycoumarin,
o-coumaric acid, 2,4-dihydroxycinnamic acid, 7-hydroxyquinoline-(1
H)-2-one, 3,4-dihydroquinoline-(1
H)-2-one, 2-hydroxyquinoline or 3,4-dihydro-7-hydroxyquinoline-(1
H)-2-one, which implied that one or all together studied hypothetical proteins were specific for coumarin derivatives with unsubstituted and non-hydrolyzed lactone moiety. The
E. coli cells without the
hcdD,
hcdE,
hcdF and
hcdG genes showed no activity towards the aforementioned compounds.
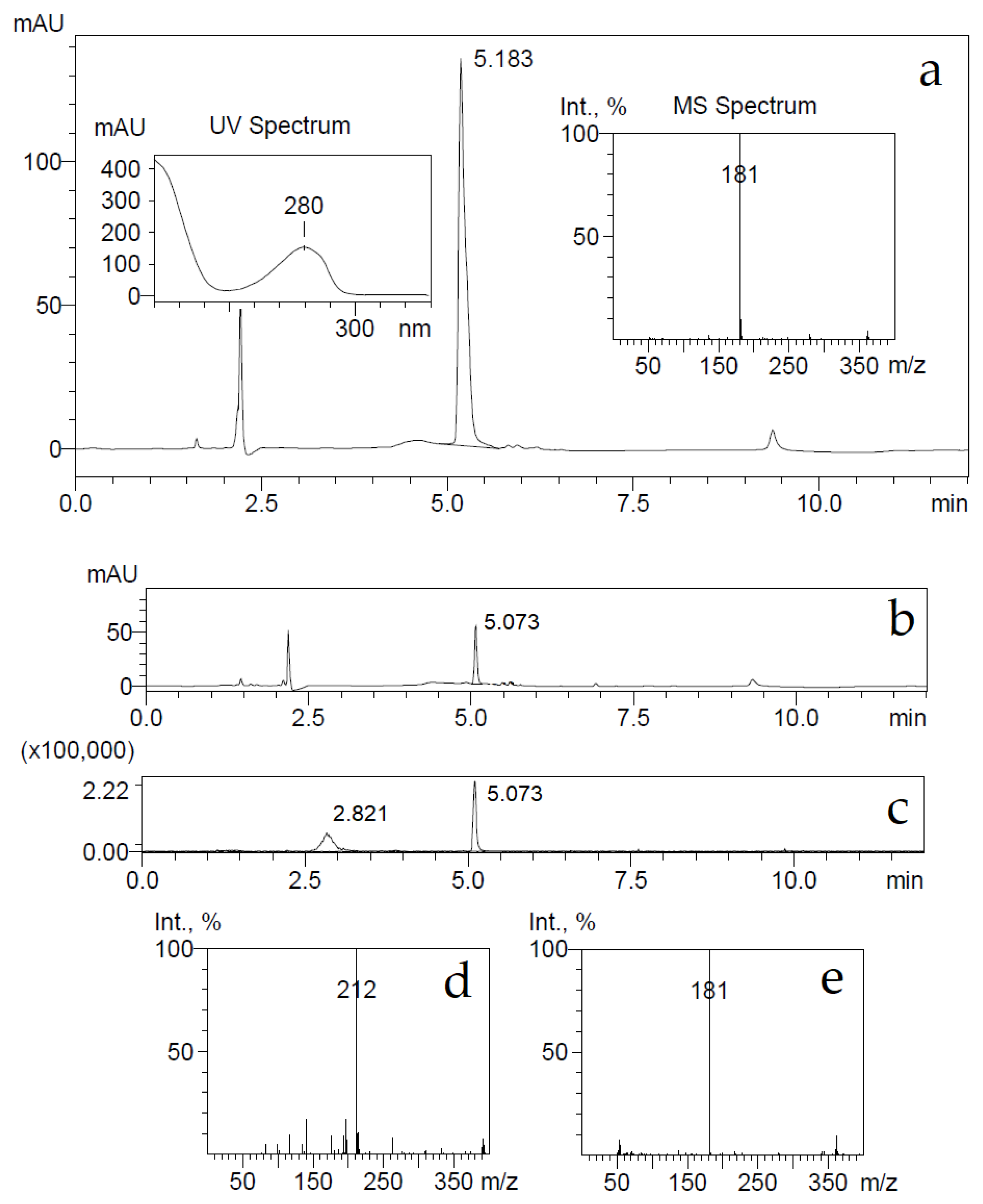
Furthermore, the recombinant HcdD, HcdE, HcdF and HcdG proteins were coexpressed with the HcdA hydroxylase and HcdB dioxygenase in
E. coli BL21 cells, which were used for bioconversion of 7-hydroxycoumarin. The conversion products were analyzed by HPLC-MS. Ions with [M-H]
− masses 161 (for 7-hydroxycoumarin) (see
Figure S8 in the Supplementary Materials) and 163 (for 7-hydroxy-3,4-dihydrocoumarin) were not detected showing a complete conversion of substrate and its reduced form, respectively. Moreover, the ions with [M-H]
− mass 181 (for 3-(2,4-dihydroxyphenyl)propionic acid) and with [M+H]
+ mass 212 (for 6-(2-carboxyethyl)-4-oxo-1,4-dihydropyridine-2-carboxylic acid) were observable, showing the accumulation of a reduced and hydrolyzed form of the substrate that was further converted to (2
E,4
E)-2,4-dihydroxy-6-oxonona-2,4-dienedioic acid by HcdA and HcdB enzymes. Some part of the produced (2
E,4
E)-2,4-dihydroxy-6-oxonona-2,4-dienedioic acid reacted with ammonium anions that were in bioconversion broth resulting in a formation of picolinic acid (
Figure 4) as described previously [
18].
These results were compared to the bioconversion of 7-hydroxycoumarin by E. coli cells containing hcdD, hcdE, hcdF and hcdG genes only, which showed the accumulation of only one reaction product with ion [M-H]− mass of 181 Da. This led to the conclusion that hypothetical HcdD, HcdE, HcdF and HcdG proteins might be used in the early stages of 7-hydroxycoumarin metabolism by P. mandelii 7HK4 bacteria, producing 3-(2,4-dihydroxyphenyl)propionic acid, which later could be as a substrate for proteins encoded by the hcdABC gene cluster.
A BLAST analysis of
hcdE and
hcdG sequences revealed that these genes encode the putative zinc-dependent alcohol dehydrogenase (
Figure 5) and NAD(P)H-dependent FMN reductase proteins, respectively. However, two other HcdD and HcdF proteins belong to less characterized groups of proteins, with similarities to
Bacillus chorismate mutase-like (BCM-like) or cupin-like protein families, respectively.
2.5. Isolation and Identification of 7-Hydroxycoumarin Bioconversion Product
Previously, it was shown that
Pseudomonas sp. 30–1 and
Aspergillus niger ATCC 11394 utilized a putative zinc-binding and NADH-dependent oxidoreductases to reduce coumarin generating dihydrocoumarin [
3,
21]. Therefore, we decided to further analyze HcdE protein that has the most resemblance to alcohol dehydrogenases (
Figure 5).
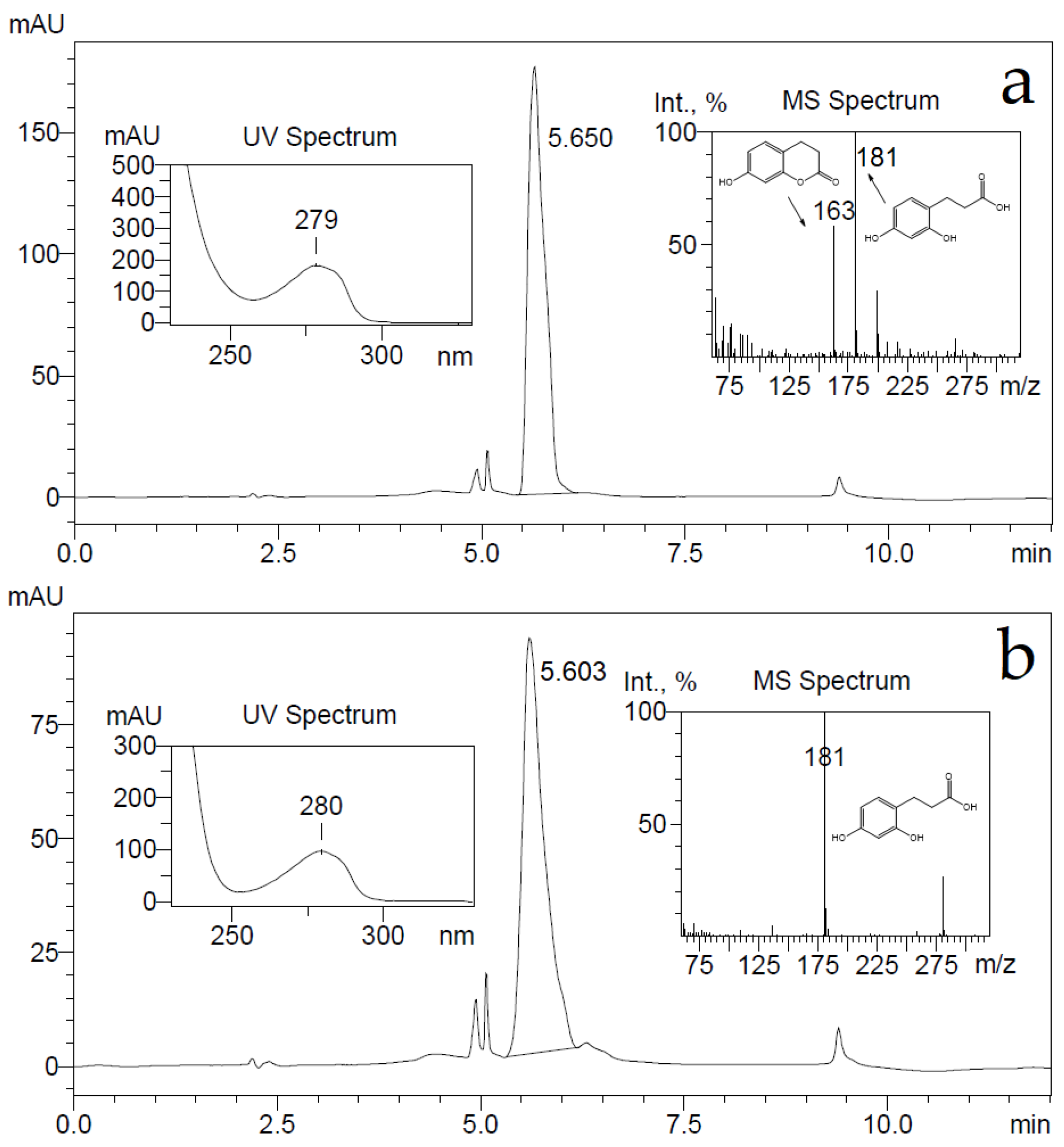
E. coli BL21 cells harboring the pHP7 plasmid were used for the conversion of 7-hydroxycoumarin as described in Materials and Methods. The consumption of the substrate was monitored by UV−VIS absorption spectroscopy and HPLC-MS. The UV absorption maximum changed from 340 to 270 nm, also, after conversion, a HPLC-MS analysis showed the accumulation of reaction products with ion [M-H]
− masses of 163 and 181 Da, and some substrate retained with ion [M-H]
− mass of 161 Da (
Figure 6). The detected ion [M-H]
− masses of 181 and 161 Da implied that the bioconversion reaction was incomplete, probably due to an inhibition by the substrate, and the presumed reaction product 7-hydroxy-3,4-dihydrocoumarin might been hydrolyzed by forming 3-(2,4-dihydroxyphenyl)propionic acid.
A conversion product was purified, yielding 3-(2,4-dihydroxyphenyl)propionic acid only as it was proven by HPLC-MS,
1H and
13C NMR analysis. To check whether the supposed reaction product—7-hydroxy-3,4-dihydrocoumarin—could be transformed through hydrolysis during the conversion reaction, 7-hydroxy-3,4-dihydrocoumarin was chemically synthesized as described in Materials and Methods, and the possible changes of it were investigated imitating the bioconversion conditions. Small amounts (a final concentration of 2 mM) of either 7-hydroxy-3,4-dihydrocoumarin, or 3-(2,4-dihydroxyphenyl)propionic acid were dissolved in 50 mM potassium phosphate buffer (pH 7.2) and incubated for several hours. Later, these compounds were extracted from aqueous solution with either dichloromethane, or ethyl acetate, then the solvent was removed and precipitates were dissolved in acetonitrile. Analysis of these samples by HPLC-MS showed that dichloromethane could extract 7-hydroxy-3,4-dihydrocoumarin only, on the other hand an extraction by ethyl acetate worked equally well for both analytes. Both ion [M-H]¯ masses of 163 and 181 Da were detected in the 7-hydroxy-3,4-dihydrocoumarin samples after an extraction by ethyl acetate from the aqueous solution (
Figure 7).
Further, the elution time of a compound from the 7-hydroxy-3,4-dihydrocoumarin sample was the same as one from the 3-(2,4-dihydroxyphenyl)propionic acid sample. A prolonged incubation (24 h) of 7-hydroxy-3,4-dihydrocoumarin in aqueous buffer solutions led to the disappearance of the [M-H]− ion mass of 163 Da in HPLC-MS analysis. This experiment confirmed that 7-hydroxy-3,4-dihydrocoumarin is prone to hydrolysis during incubation in aqueous solution and extraction by organic solvents.
Based on all experiments, it was concluded that 7-hydroxycoumarin was reduced by the HcdE enzyme resulting in the formation of 7-hydroxy-3,4-dihydrocoumarin. However, due to a strong inhibition of the HcdE enzyme by its substrate, the conversion rates were low enough that a prolonged conversion affected the reaction product 7-hydroxy-3,4-dihydrocoumarin, which underwent hydrolysis in aqueous solutions.
2.6. Characterization of HcdE Protein
The
hcdE gene was expressed and the recombinant C-terminally His
6-tagged protein was produced in
E. coli BL21 cells. A soluble and colorless HcdE protein was purified by affinity chromatography and analyzed by SDS-PAGE. The enzyme migrated as a ~40 kDa protein band (
Figure S2). The specificity of the enzyme was investigated for several cofactors, however the HcdE was able to utilize NADPH only. The addition of FAD or FMN to the reaction mixtures showed no changes in either NADH or NADPH oxidation. The optimum reaction conditions for the HcdE activity were found to be a low ionic strength phosphate-citrate buffer of pH 7.0 at room temperature (
Figures S3 and S4).
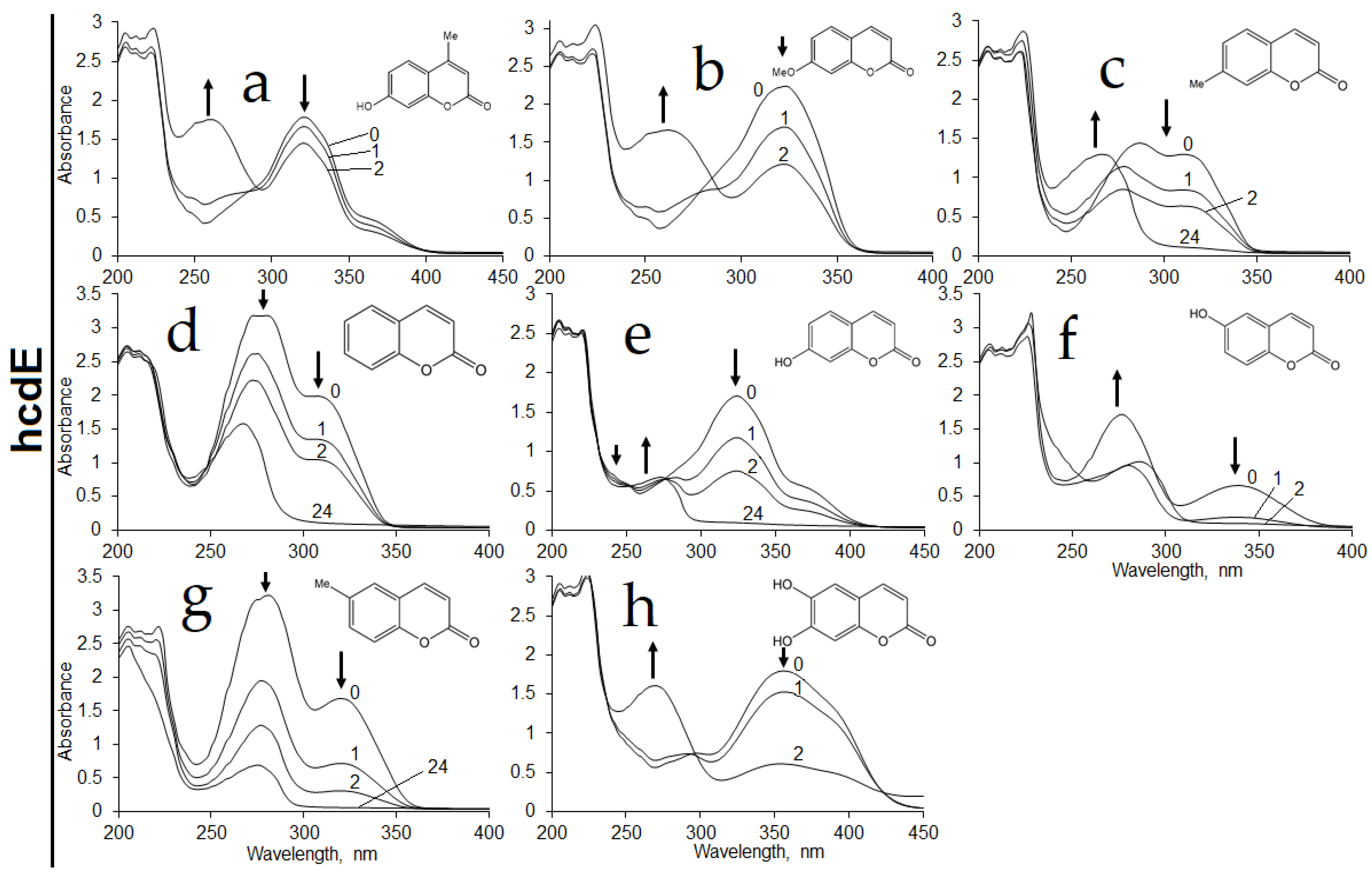
Further, the activity of the HcdE enzyme was assayed in the presence of NADPH cofactor against various coumarin substrates. The highest enzymatic activity was recorded in the presence of 7-hydroxycoumarin. Rates lower by 1.6-, 2-, 3.4- and 17-fold were observed when 6,7-dihydroxycoumarin, 6-hydroxycoumarin, 6-methylcoumarin and coumarin were used as the substrates, respectively. The HcdE was not active towards 7-methylcoumarin, 7-methoxycoumarin, 7-ethoxycoumarin, 4-methyl-7-hydroxycoumarin, 3-hydroxycoumarin, 4-hydroxycoumarin,
trans-cinnamic,
trans-2,4-dihydroxycinnamic,
o-coumaric,
m-coumaric,
p-coumaric and caffeic acids, cinnamyl alcohol, 7-hydroxyquinoline-(1
H)-2-one, 3,4-dihydroquinoline-(1
H)-2-one, 2-hydroxyquinoline, 3,4-dihydro-7-hydroxyquinoline-(1
H)-2-one and indole (
Table S1). Some data of in vitro experiments contradicted the results obtained by using the whole
E. coli BL21 cells harboring the pHP7 plasmid. Hence, those cells showed some activities towards 7-methylcoumarin, 7-methoxycoumarin and even 4-methyl-7-hydroxycoumarin (
Figure 8).
It was proposed that a strong inhibition of HcdE took place in vitro, when such effect was bypassed within E. coli BL21 cells by limiting substrate transportation through the cell membrane, thus maintaining a low concentration of substrate within the cells.
The initial velocities were measured for HcdE protein by varying both NADPH and 7-hydroxycoumarin concentrations to determine the kinetic properties of the HcdE enzyme. The estimation of the kinetic parameters was performed by fitting the data to the Michaelis−Menten expression of the bimolecular reaction rate (Equation (1)):
where [NADPH] was the concentration of NADPH; [7HK] was the concentration of 7-hydroxycoumarin;
v was the observed rate;
kbim was the bimolecular rate constant of 7-hydroxycoumarin reduction, expressed as
(where
);
kNADPH was the rate constant of NADPH oxidation;
was the
Km for NADPH at saturating 7-hydroxycoumarin levels (where
); and
Ki was the inhibition constant of 7-hydroxycoumarin. The action of 7-hydroxycoumarin reduction and NADPH oxidation was represented by the proposed reaction schemes (Equations (2)–(4)).
The reductive half-reaction sequence was modeled as shown in Equation (2), where E0 was an oxidized form of HcdE, E1 was the hcdEox-NADPH charge-transfer complex, and E2 was a reduced form of HcdE bound to NADP+. The oxidative half-reaction sequence was modeled as shown in Equation (3), where 7HK was 7-hydroxycoumarin, E3 was hcdEred-7HK charge-transfer complex and bound NADP+, and 7HKH2 was 7-hydroxy-3,4-dihydrocoumarin.
It was also observed that 7-hydroxycoumarin acted as a strong inhibitor of the HcdE enzyme at higher concentrations (see
Figure S5). We proposed that inhibition occurred when 7-hydroxycoumarin bound to the hcdE
ox-NADPH charge-transfer complex (E
1) rather than the oxidized HcdE enzyme form (E
0) (Equation (4)). Therefore, 7-hydroxycoumarin was likely a pseudo-competitive inhibitor characterized by the inhibition constant
Ki (expressed as
) that was equal to 22 mM for 7-hydroxycoumarin. The determined
kbim value of reduction of 7-hydroxycoumarin by HcdE was equal to 1490 mM
−1s
−1. Additionally, a rate constant (
kcat) of 27 s
−1 and a
of 20 μM have been determined for NADPH.
3. Discussion
Thus far, only a few studies have been published discussing the reduction of coumarins in microorganisms [
3,
21]. To date, it was known that coumarin could be reduced at the double bond in the α-pyrone ring by coumarin reductase and NADH as the first step resulting in dihydrocoumarin, followed by hydrolysis between the oxygen and carbonyl carbon atoms of the ring [
3,
21]. On the other hand, the initial attack on coumarin could also be hydrolysis to
o-coumaric acid, only then followed by reduction to melilotic acid [
4]. Some enzymes having coumarin reducing activity have been biochemically characterized [
3,
17], however scarce genetic data or enzyme mechanisms have been presented. In the present study, we discovered a 7-hydroxycoumarin-inducible alcohol dehydrogenase HcdE responsible for reduction of the α-pyrone ring of 7-hydroxycoumarin in
P. mandelii 7HK4 bacteria.
The HcdE protein is expressed in P. mandelii 7HK4 cells when grown with 7-hydroxycoumarin as the sole source of carbon and energy. HcdE catalyzes the NADPH-dependent reduction of several coumarin derivatives including coumarin itself, 6-methylcoumarin, 6-hydroxycoumarin, 7-hydroxycoumarin and 6,7-dihydroxycoumarin. HcdE appears to have a high specificity for coumarin derivatives with unsubstituted and non-hydrolyzed lactone moiety, thus other structurally related substrates, such as quinolin-2(1H)-one or 7-hydroxyquinolin-2(1H)-one are not used by the HcdE enzyme. There is also one discrepancy between conversions with whole cells and pure enzyme in this research. Whole cells of E. coli BL21 harboring hcdE gene are able to reduce low levels of 7-methylcoumarin, 7-methoxycoumarin and 4-methyl-7-hydroxycoumarin, while the purified HcdE enzyme alone is not. Although the results are not conclusive, we propose that the whole cells limit substrate transportation through the cell membrane maintaining a noninhibitive concentration of substrate within cells. We showed that the HcdE dehydrogenase is inhibited by its substrate 7-hydroxycoumarin at higher concentrations with the inhibition constant Ki that is equal to 22 mM for 7-hydroxycoumarin. The inhibition occurs when 7-hydroxycoumarin binds to the HcdEox-NADPH charge-transfer complex (E1), thus reducing the NADPH oxidation rate.
HcdE dehydrogenase is, to our best knowledge, the first ene-reductase of the MDR family involved in the in vivo degradation of coumarin compounds. Only one functionally related enzyme, such as XenA reductase from
Pseudomonas putida 86 is known [
17]. XenA reductase has well-described primary and secondary structures, yet it belongs to another family of ERs—OYEs, catalyzing FMN and NAD(P)H dependent reduction of coumarin and 8-hydroxycoumarin. The determined
kbim value of reduction of 7-hydroxycoumarin by HcdE dehydrogenase was equal to 1490 mM
−1s
−1. Additionally, a rate constant (
kcat) of 27 s
−1 has been determined for NADPH oxidation by hcdE dehydrogenase, which is almost 10-fold lower compared to the rate constant of 215 s
−1 for NADPH oxidation by XenA reductase from
Pseudomonas putida 86 [
10,
22]. The difference in NADPH oxidation between two functionally similar hcdE and XenA reductases could be due to observable competitive inhibition of HcdE enzyme by its substrate while XenA reductase is not inhibited.
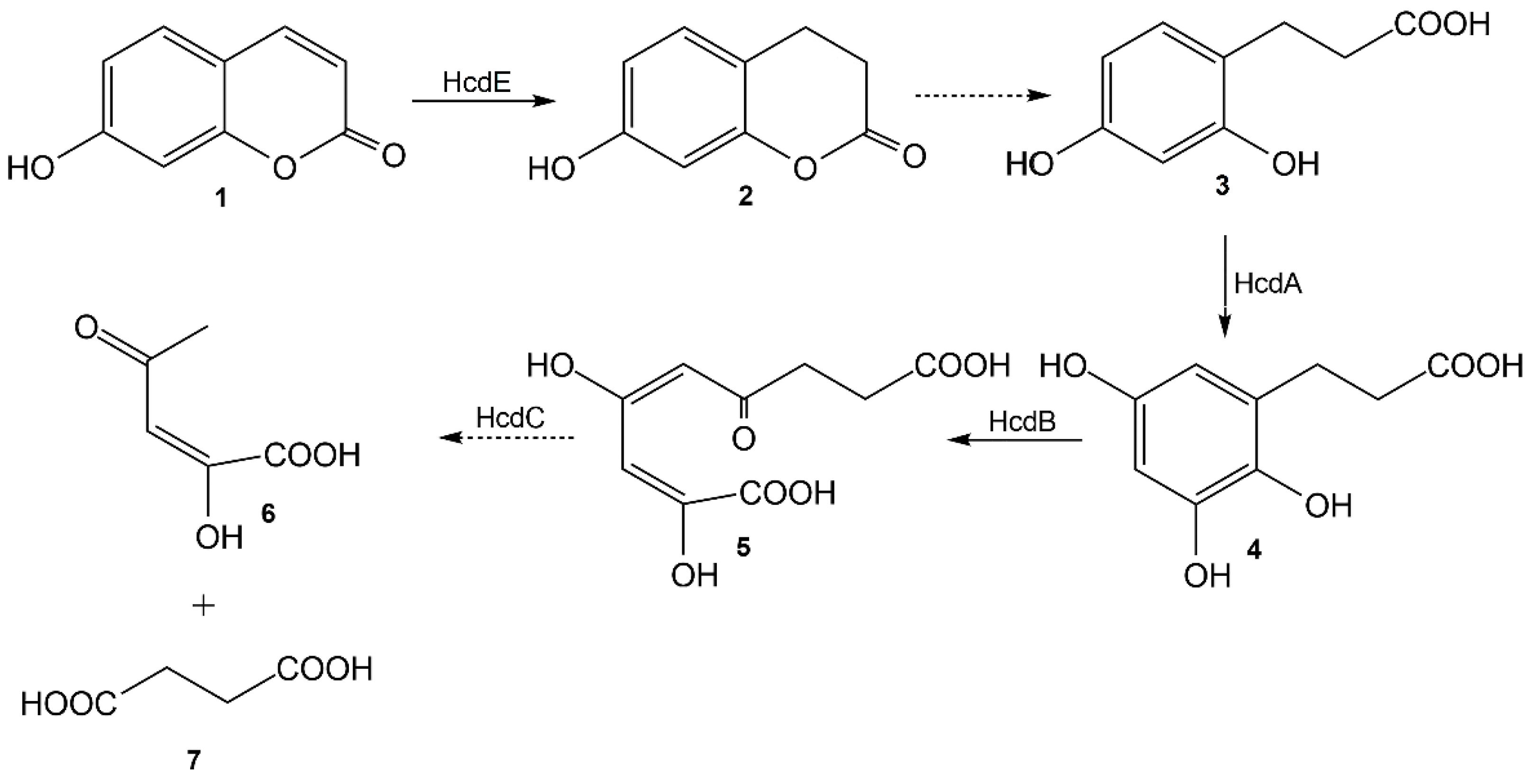
The bioconversion of 7-hydroxycoumarin was completed in vivo by the HcdE protein, and the reduced product was confirmed by HPLC-MS and NMR analysis as well as by chemical synthesis to be 7-hydroxy-3,4-dihydrocoumarin. However due to a strong inhibition of the HcdE enzyme by its substrate the conversion rates were low enough that the prolonged conversion time affected the reaction product 7-hydroxy-3,4-dihydrocoumarin. The latter compound is a lactone (a cyclic ester) that is much more reactive than its acyclic analogs. Additionally, it has a phenyl ring attached and the fact that it is a phenolic ester, 7-hydroxy-3,4-dihydrocoumarin is significantly more reactive against hydroxyl anions. Hence, a hydrolysis in aqueous solutions takes place, resulting in the formation of 3-(2,4-dihydroxyphenyl)propionic acid. Therefore, this work in combination with the previously published data [
18] allow an elucidation of a pathway of catabolism of 7-hydroxycoumarin in
Pseudomonas sp. 7HK4 bacteria (
Figure 9).
During the analysis of the
P. mandelii 7HK4 genome sequences adjacent to the
hcdE gene it was also expected to detect genes encoding hydrolase-like enzymes that could be responsible for the hydrolysis of 7-hydroxy-3,4-dihydrocoumarin. Transcriptional analysis revealed that like
hcdE, the
hcdD gene was also induced in
P. mandelii 7HK4 cells pregrown with 7-hydroxycoumarin. In addition,
hcdD and
hcdE together with
hcdF and
hcdG genes are arranged on the same DNA strand, and are separated by short intergenic regions, suggesting that these genes are organized into an operon. The HcdD protein is similar to
Bacillus chorismate mutase-like (BCM-like) family proteins, and is in agreement with the first representatives of Pfam family (formerly DUF1185) [
23]. We propose that this protein can stimulate a 7-hydroxy-3,4-dihydrocoumarin formation similarly to the homologous protein TgnF, which stimulates TgnE-dependent succinic acid semialdehyde oxidation in
Acinetobacter baylyi ADP1 bacteria [
24]. However, all attempts to observe such an effect in the case of HcdD have been unsuccessful. Further, HcdF is a cupin-like protein, which can be classified as a member of the RmlC-like cupins superfamily. It comprises families with members displaying diverse functions ranging from enzymatic activities like dioxygenases, hydrolases and epimerases to nonenzymatic functions [
25,
26,
27]. We believe that HcdF might act as a 7-hydroxy-3,4-dihydrocoumarin hydrolase or as an enzyme catalyzing removal of a methoxy group from the 7-
O-methylated 7-hydroxycoumarin, however further investigation is needed to elucidate its exact function in
P. mandelii 7HK4 cells. The last protein encoded by the
hcdDEFG gene cluster—HcdG—belongs to the NAD(P)H-dependent FMN reductase superfamily, members of which are widely involved in the electron-transfer reactions required for the metabolism of various compounds in bacteria [
28,
29,
30].
Finally, an in silico analysis of the P. mandelii 7HK4 genome provided evidence that 7HK4 cells harbor also other ER enzymes capable of utilizing various coumarin derivatives. Two putative xenobiotic reductases XenA38 and XenA45 from 7HK4 strain were produced in E. coli bacteria, and their activities have been determined in vivo. Coumarin, 6-hydroxycoumarin, 6-methylcoumarin, 7-hydroxycoumarin, 7-methylcoumarin and 6,7-dihydroxycoumarin are reduced by the XenA38 and XenA45 reductases. However, an analysis of mRNA synthesis shows that the production of XenA38 and XenA45 proteins are not induced by any coumarin in P. mandelii 7HK4 cells, although XenA38 enzyme has higher expression levels in 7HK4 cells grown in the absence of coumarins. An elucidation of a role of such enzymes as XenA38 with the constitutive expression and a broad substrate specificity needs additional studies.
In summary, in addition to previously reported
hcdABC gene cluster that is responsible for the degradation of 3-(2,4-dihydroxyphenyl)propionic acid [
18] here we report the fundamentally new insights into a 7-hydroxycoumarin catabolic pathway in
Pseudomonas mandelii 7HK4 bacteria. A new gene encoding an enzyme responsible for the reduction of 7-hydroxycoumarin has been isolated and identified. Our results show that the reduction of 7-hydroxycoumarin in
Pseudomonas mandelii 7HK4 involves a similar approach such as in the previously characterized coumarin catabolic routes in
Pseudomonas sp. 30–1 and
Aspergillus niger ATCC 11394 [
3,
21]. However, it has been shown that
Pseudomonas mandelii 7HK4 bacteria employ a unique NADPH-dependent alcohol dehydrogenase HcdE for the reduction of the C-3/C-4 double bond of 7-hydroxycoumarin lactone moiety forming 7-hydroxy-3,4-dihydrocoumarin. The HcdE dehydrogenase described in this paper has a significantly distant sequence homology (identity of 14.1%, only) from the previously characterized enzymes implicated in the degradation of structurally similar substrate, for example, such as 8-hydroxycoumarin in
Pseudomonas putida 86 [
17].
Thus, the HcdE provides not only a fundamentally new insight into the degradation of hydroxycoumarins by soil microorganisms, but also has a potential in alkene hydrogenation that is one of the most widely used industrial reactions producing fine chemicals, pharmaceuticals and agrochemical intermediates [
9,
31,
32]. Further studies are required to explore various possibilities of biocatalytic and chemo-enzymatic approaches using the HcdE dehydrogenase.
4. Materials and Methods
4.1. Bioconversions with Whole Cells
E. coli BL21 (DE3) bacteria containing recombinant genes were grown aerobically in 20 mL of LB medium at 30 °C overnight. High density bacterial culture was centrifuged and re-suspended in 30 mL of minimal C-750501 medium [
33,
34], in which the synthesis of proteins was induced with 1 mM of IPTG after 1.5 h incubation at 20 °C. Incubation at 20 °C was continued for another 24–48 h.
E. coli cells were sedimented by centrifugation (3220×
g, 15 min). The collected cells were washed twice with 15 mL of 0.9% NaCl solution. For whole cell conversion experiments, cells from 20 mL of culture were resuspended in 1 mL of 50 mM potassium phosphate buffer (pH 7.2). All small-scale bioconversions with whole cells were made in 50 mM potassium phosphate buffer, pH 7.2, which contained 0.5–1 mM of the substrate. The reaction mixtures were kept in thermoblock at 30 °C and 500 rpm. Bioconversion mixtures were centrifuged for 2 min at 10,000×
g and 100 µL of the supernatant were analyzed by absorption measurements in the UV−VIS range (200–450 nm). Measurements were repeated to record the changes in the absorption intensity over time. All measurements were performed with PowerWave XS microplate reader (BioTek Instruments, Inc., Highland Park, IL, USA).
4.2. Quantitative RT-PCR
P. mandelii 7HK4 was cultivated overnight in minimal medium containing 0.05% of glucose as the sole carbon source. Then cells were sedimented by centrifugation (3220×
g, 10 min) and resuspended in 50 mM potassium phosphate buffer, pH 7.2.
P. mandelii 7HK4 cells were supplemented with 1 mM of various coumarin derivatives and incubated for additional 3 h at 30 °C with shaking. Total RNA was isolated using a RiboPure Bacteria RNA Purification Kit (Thermo Fisher Scientific, Vilnius, Lithuania). cDNA synthesis was performed using a High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Vilnius, Lithuania) with 340 ng input of total RNA per sample. Quantitative-PCR (qPCR) amplification was performed using a Fast SYBR Green Master Mix (Thermo Fisher Scientific) on 7500 Fast Real-time PCR system (Thermo Fisher Scientific, Vilnius, Lithuania). qPCR was conducted in 20 µL of reaction mixture containing 10 µL of Fast SYBR Green Master Mix, 500 nM of each primer (
Table S3), and 2 µL of the cDNA sample. For quantitative analysis, fluorescence data were recorded after the annealing step. All experiments were carried out in triplicate. To verify the absence of DNA in the total RNA samples, the qPCR was performed directly for RNA samples. The threshold cycle (CT) (threshold value, 5% of amplification curve plateau) values were obtained using 7500 Fast Real-time PCR Software v2.0 (Thermo Fisher Scientific, Vilnius, Lithuania).
4.3. Protein Purification
Proteins were purified with Äkta purifier 900 chromatography systems (GE Healthcare, Uppsala, Sweden). Cell-free extracts were applied to Ni
2+ Chelating HiTrap HP column (GE Healthcare, Uppsala, Sweden) equilibrated with 50 mM potassium phosphate buffer, pH 7.0, at 1.0 mL/min. The column was washed with at least 3 volumes of the same buffer. Then the bound proteins were eluted with 0.5 M imidazole in 50 mM potassium phosphate buffer, pH 7.0. The fractions containing the purified enzyme applied to HiTrap Desalting column with Sephadex G-25 resin (GE Healthcare, Uppsala, Sweden) and eluted with 50 mM potassium phosphate buffer, pH 7.0. Proteins were analyzed by SDS-PAGE according Laemmli [
35]. Protein concentration was determined by Lowry method [
36].
4.4. Enzyme Assay
Reducing activity of HcdE protein was measured spectrophotometrically by monitoring absorption changes of bimolecular reaction at 340 nm wavelength due to the oxidation of NADPH and reduction of various coumarin derivatives (ε340 = 5580 M−1cm−1 (for 7-hydroxycoumarin), ε340 = 4780 M−1cm−1 (for 6-hydroxycoumarin), ε340 = 4440 M−1cm−1 (for 6-methylcoumarin), ε340 = 8500 M−1cm−1 (for 6,7-dihydroxycoumarin), ε340 = 3690 M−1cm−1 (for coumarin)) after the addition of the enzyme. The activity measurements were made with cell-free extracts or the purified protein. The standard measurements of the enzyme activity were carried out at room temperature in 0.8 mL of reaction mixture, containing 50 mM potassium phosphate buffer (pH 7.0), 160 µM NADPH, and 60 µM aromatic substrate.
Kinetic characterization of HcdE protein was performed by monitoring absorption changes of reaction mixture at 365 nm wavelength due to the oxidation of NADPH only (ε365 = 3500 M−1cm−1), after the addition of the enzyme. The measurements of the enzyme activity were carried out at room temperature in 0.8 mL of reaction mixture, containing 50 mM potassium phosphate buffer (pH 7.0), 5–200 µM NADPH, 5–150 µM 7-hydroxycoumarin, and 99 nM HcdE protein. Each measurement was performed three times. Kinetic data were analyzed using Wolfram Mathematica software (Wolfram Research, Inc., Oxfordshire, UK).
4.5. In Vivo Bioconversion of 7-Hydroxycoumarin and Purification of Bioconversion Products
E. coli BL21 (DE3) cells, containing pHP7 plasmid, were grown in 6 × 200 mL of LB medium at 30 °C and 180 rpm overnight. High density bacterial cultures were supplied with 0.5 mM of IPTG for induction of protein synthesis, and incubated at 20 °C and 180 rpm. After 48 h of induction cells were centrifuged and resuspended in respective volumes of 50 mM potassium phosphate buffer (pH 7.2). Then, 7-hydroxycoumarin was added to the final concentration of 0.3 mM. Bioconversion mixture was incubated at 30 °C with shaking overnight. Cells were removed by centrifugation for 40 min at 3220× g and the supernatants were used for product purification.
Bioconversion product was purified as described in [
37] with minor changes. The biotransformation broth was treated with concentrated HCl aq. in order to bring the pH between 4 and 5. The solution was saturated with NaCl and extracted with CH
2Cl
2 (4 × 100 mL). The combined organic phases were dried (Na
2SO
4) and concentrated under reduced pressure resulting in 7-hydroxycoumarin residues (MS (ESI+):
m/
z 161 [M-H]
−). Ethyl acetate (100 mL) was then added to the aqueous solution and the mixture was filtered on a celite pad. The phases were separated and the aqueous phase was extracted with further solvent. The combined organic phases were dried (Na
2SO
4) and concentrated under reduced pressure. The obtained pale-yellow precipitates consisted of 3-(2,4-dihydroxyphenyl)propionic acid. MS (ESI+):
m/
z 181 [M-H]
−;
1H NMR (DMSO-
d6, 400 MHz): δ 11.97 (s, 1H), 9.14 (s, 1H), 8.96 (s, 1H), 6.80 (d,
J = 8.2 Hz, 1H), 6.26 (d,
J = 2.4 Hz, 1H), 6.11 (dd,
J = 8.1, 2.4 Hz, 1H), 2.63 (dd,
J = 8.6, 6.9 Hz, 2H), 2.40 (dd,
J = 8.5, 6.9 Hz, 2H);
13C NMR (DMSO-
d6, 100 MHz): δ 174.73, 157.00, 156.23, 130.41, 117.79, 106.33, 102.84, 34.63, 25.37. Given NMR spectra complies with known spectra of 3-(2,4-dihydroxyphenyl)propionic acid standard (see section NMR spectra of 3-(2,4-dihydroxyphenyl)propionic acid standard in
Supplementary Material). The yield of 3-(2,4-dihydroxyphenyl)propionic acid from 59 mg of starting material was 50 mg (75.7% of the theoretical yield).
4.6. Organic Synthesis of 7-Hydroxy-3,4-Dihydrocoumarin
The analytical sample of 7-hydroxy-3,4-dihydrocoumarin was synthesized in good yield by thermal cyclization of 3-(2,4-dihydroxyphenyl)propionic acid [
38]. In short, 3-(2,4-dihydroxyphenyl)propionic acid (182 mg, 1 mmol) was heated in an oven at 135 °C for 3 h. Resulting brown colored melt was cooled to room temperature and dissolved in a hot toluene. The solution was treated with silica, filtered and the solvent was removed under vacuum. The residue then was crystallized from a small amount of toluene. The product was filtered and vacuum dried; Yield: 112 mg (68%). MS (ESI+):
m/
z 163 [M-H]
− (see
Figure S6).
1H NMR (400 MHz, DMSO-
d6): δ 9.61 (s, 1H), 7.05 (d,
J = 8.2 Hz, 1H), 6.52 (dd,
J = 8.2, 2.4 Hz, 1H), 6.43 (d,
J = 2.4 Hz, 1H), 2.86–2.82 (m, 2H), 2.74–2.70 (m, 2H).
13C NMR (100 MHz, DMSO-
d6): δ 168.5, 157.1, 152.3, 128.7, 113.2, 111.2, 103.2, 29.0, 22.1.
4.7. Other Analytical Methods
For the HPLC-MS analysis, most of the analytes were dissolved in acetonitrile, otherwise the aqueous samples were mixed with an equal part of acetonitrile and centrifuged before the analysis. High-performance liquid chromatography and mass spectrometry (HPLC-MS) were carried out using the system consisting of the CBM-20 control unit, two LC-2020AD pumps, SIL-30AC auto sampler and CTO-20AC column thermostat, using the SPD-M20A detector and LCMS-2020 mass spectrometer with ESI source (Shimadzu, Japan).
Chromatographic fractionation was conducted using YMC-Pack Pro C18 column, 150 × 3 mm (YMC) at 40 °C, with water and acetonitrile gradient from 5% to 95%.
Mass spectra were recorded from m/z 10 up to 500 m/z at 350 °C and ±4500 V using N2. Mass spectrometry analysis was carried out using both the positive and negative ionization modes. The data were analyzed using LabSolutions LC/MS software (Shimadzu, Japan).
1H NMR and 13C NMR spectra were recorded in DMSO-d6 on Avance III 400 NMR spectrometer, at 400 MHz for 1H and 100 MHz for 13C, chemical shifts are reported in ppm relative to solvent resonance signal as an internal standard (1H NMR: δ (DMSO-d6) = 2.50 ppm; 13C NMR: δ (DMSO-d6) = 39.52 ppm).
4.8. Accession Numbers
Accession numbers for the sequences of HcdE, XenA38 and XenA45 genes are MW310254, MW310255, MW310256, respectively. The whole fragment of
P. mandelii 7HK4 genome containing
hcd genes (
Figure 3c) could be found by the accession number MW310253.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








