
Insulin-Like Growth Factor 2 As a Possible Neuroprotective Agent and Memory Enhancer—Its Comparative Expression, Processing and Signaling in Mammalian CNS


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. IGF2 mRNA Expression in Different Species
2.1. Rodents (Mouse and Rat)
2.2. Artiodactyla (Sheep and Cows)
2.3. Humans and Other Primates
3. IGF2 Expression Regulation and Genomic Imprinting
4. IGF Family Protein Processing
4.1. Cleavage
4.2. Proteases Participating in the Processing of IGF-2
4.3. Glycosylation
5. IGF-Binding Proteins
- Inhibition of IGF activity by binding IGFs and reducing their access to receptors. For example, in human serum the concentration of IGF-2 is higher than that of insulin and, considering the fact that IGF-2 is able to activate insulin receptors, this would lead to hypoglycemia if IGF-2 was present in its unbound form [7]. But IGFs in blood are mostly confined to ternary protein complexes formed with IGFBP3/5 and ALS [37,118] and thus are unable to activate their receptors.
- Regulation of local IGF concentration: when proteases degrade IGFBPs, their affinity to IGFs decreases, leading to the release of IGFs and increase in their local concentration [37].
- Interaction with cell surface molecules and extracellular matrix [37].
- Interaction with other growth factors [127].
6. IGF-2 Receptors (IGF1R, IR, IGF2R)
6.1. IGF-1 Receptor
6.2. Insulin Receptor
6.3. IGF-2 Receptor
7. IGF-2 as a Memory Enhancer
8. IGF-2 as a Neuroprotective Agent
9. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bracko, O.; Singer, T.; Aigner, S.; Knobloch, M.; Winner, B.; Ray, J.; Clemenson, G.D., Jr.; Suh, H.; Couillard-Despres, S.; Aigner, L.; et al. Gene expression profiling of neural stem cells and their neuronal progeny reveals IGF2 as a regulator of adult hippocampal neurogenesis. J. Neurosci. 2012, 32, 3376–3387. [Google Scholar] [CrossRef] [Green Version]
- Martín-Montañez, E.; Millon, C.; Boraldi, F.; Garcia-Guirado, F.; Pedraza, C.; Lara, E.; Santin, L.J.; Pavia, J.; Garcia-Fernandez, M. IGF-II promotes neuroprotection and neuroplasticity recovery in a long-lasting model of oxidative damage induced by glucocorticoids. Redox Biol. 2017, 13, 69–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russo, V.C.; Gluckman, P.D.; Feldman, E.L.; Werther, G.A. The Insulin-Like Growth Factor System and Its Pleiotropic Functions in Brain. Endocr. Rev. 2005, 26, 916–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Yakar, S.; Liu, J.L.; Stannard, B.; Butler, A.; Accili, D.; Sauer, B.; LeRoith, D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc. Natl. Acad. Sci. USA 1999, 96, 7324–7329. [Google Scholar] [CrossRef] [Green Version]
- Le Roith, D.; Bondy, C.; Yakar, S.; Liu, J.-L.; Butler, A. The Somatomedin Hypothesis: 2001. Endocr. Rev. 2001, 22, 53–74. [Google Scholar] [CrossRef] [PubMed]
- van Doorn, J. Insulin-like growth factor-II and bioactive proteins containing a part of the E-domain of pro-insulin-like growth factor-II. Biofactors 2020, 46, 563–578. [Google Scholar] [CrossRef]
- Dupont, J.; LeRoith, D. Insulin and insulin-like growth factor I receptors: Similarities and differences in signal transduction. Horm. Res. 2001, 55 Suppl. 2, 22–26. [Google Scholar] [CrossRef]
- Dynkevich, Y.; Rother, K.I.; Whitford, I.; Qureshi, S.; Galiveeti, S.; Szulc, A.L.; Danoff, A.; Breen, T.L.; Kaviani, N.; Shanik, M.H.; et al. Tumors, IGF-2, and hypoglycemia: Insights from the clinic, the laboratory, and the historical archive. Endocr. Rev. 2013, 34, 798–826. [Google Scholar] [CrossRef] [Green Version]
- Bolin, D.R. Peptide Growth Factors and Their Receptors I and II.; Sporn, M.B., Roberts, A.B., Eds. Q. Rev. Biol. 1993, 68, 100–101. [Google Scholar] [CrossRef]
- Chen, D.Y.; Stern, S.A.; Garcia-Osta, A.; Saunier-Rebori, B.; Pollonini, G.; Bambah-Mukku, D.; Blitzer, R.D.; Alberini, C.M. A critical role for IGF-II in memory consolidation and enhancement. Nature 2011, 469, 491–497. [Google Scholar] [CrossRef] [PubMed]
- Stern, S.A.; Kohtz, A.S.; Pollonini, G.; Alberini, C.M. Enhancement of Memories by Systemic Administration of Insulin-Like Growth Factor II. Neuropsychopharmacology 2014, 39, 2179–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwamoto, T.; Ouchi, Y. Emerging evidence of insulin-like growth factor 2 as a memory enhancer: A unique animal model of cognitive dysfunction with impaired adult neurogenesis. Rev. Neurosci. 2014, 25, 559–574. [Google Scholar] [CrossRef] [PubMed]
- Bryzgalov, D.V.; Kuznetsova, I.L.; Rogaev, E.I. Enhancement of Declarative Memory: From Genetic Regulation to Non-invasive Stimulation. Biochemistry 2018, 83, 1124–1138. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; MacDonald, R.G.; Thinakaran, G.; Kar, S. Insulin-Like Growth Factor-II/Cation-Independent Mannose 6-Phosphate Receptor in Neurodegenerative Diseases. Mol. Neurobiol. 2017, 54, 2636–2658. [Google Scholar] [CrossRef] [Green Version]
- Pardo, M.; Cheng, Y.; Sitbon, Y.H.; Lowell, J.A.; Grieco, S.F.; Worthen, R.J.; Desse, S.; Barreda-Diaz, A. Insulin growth factor 2 (IGF2) as an emergent target in psychiatric and neurological disorders. Review. Neurosci. Res. 2019, 149, 1–13. [Google Scholar] [CrossRef]
- Yang, Y.-J.; Luo, T.; Zhao, Y.; Jiang, S.-Z.; Xiong, J.-W.; Zhan, J.-Q.; Yu, B.; Yan, K.; Wei, B. Altered insulin-like growth factor-2 signaling is associated with psychopathology and cognitive deficits in patients with schizophrenia. PLoS ONE 2020, 15, e0226688. [Google Scholar] [CrossRef] [Green Version]
- Akanji, A.O.; Ohaeri, J.U.; Al-Shammri, S.A.; Fatania, H.R. Associations of blood levels of insulin-like growth factor (IGF)-I, IGF-II and IGF binding protein (IGFBP)-3 in schizophrenic Arab subjects. Clin. Chem. Lab. Med. 2007, 45, 1229–1231. [Google Scholar] [CrossRef] [PubMed]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J. Alzheimers Dis. 2005, 7, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Pascual-Lucas, M.; Viana da Silva, S.; Di Scala, M.; Garcia-Barroso, C.; González-Aseguinolaza, G.; Mulle, C.; Alberini, C.M.; Cuadrado-Tejedor, M.; Garcia-Osta, A. Insulin-like growth factor 2 reverses memory and synaptic deficits in APP transgenic mice. EMBO Mol. Med. 2014, 6, 1246–1262. [Google Scholar] [CrossRef]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimers Dis. 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Agbemenyah, H.Y.; Agis-Balboa, R.C.; Burkhardt, S.; Delalle, I.; Fischer, A. Insulin growth factor binding protein 7 is a novel target to treat dementia. Neurobiol. Dis. 2014, 62, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Hertze, J.; Nägga, K.; Minthon, L.; Hansson, O. Changes in cerebrospinal fluid and blood plasma levels of IGF-II and its binding proteins in Alzheimer’s disease: An observational study. BMC Neurol. 2014, 14, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Åberg, D.; Johansson, P.; Isgaard, J.; Wallin, A.; Johansson, J.O.; Andreasson, U.; Blennow, K.; Zetterberg, H.; Åberg, N.D.; Svensson, J. Increased Cerebrospinal Fluid Level of Insulin-like Growth Factor-II in Male Patients with Alzheimer’s Disease. J. Alzheimers Dis. 2015, 48, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Heywood, W.E.; Galimberti, D.; Bliss, E.; Sirka, E.; Paterson, R.W.; Magdalinou, N.K.; Carecchio, M.; Reid, E.; Heslegrave, A.; Fenoglio, C.; et al. Identification of novel CSF biomarkers for neurodegeneration and their validation by a high-throughput multiplexed targeted proteomic assay. Mol. Neurodegener. 2015, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, Y.; Banno, Y.; Shimizu, Y.; Ando, S.; Hasegawa, H.; Adachi, K.; Iwamoto, T. Reduced adult hippocampal neurogenesis and working memory deficits in the Dgcr8-deficient mouse model of 22q11.2 deletion-associated schizophrenia can be rescued by IGF2. J. Neurosci. 2013, 33, 9408–9419. [Google Scholar] [CrossRef]
- Aros, S.; Mills, J.L.; Iñiguez, G.; Avila, A.; Conley, M.R.; Troendle, J.; Cox, C.; Cassorla, F. Effects of prenatal ethanol exposure on postnatal growth and the insulin-like growth factor axis. Horm. Res. Paediatr. 2011, 75, 166–173. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Kohtz, A.; Pollonini, G.; Riccio, A.; Alberini, C.M. Insulin Like Growth Factor 2 Expression in the Rat Brain Both in Basal Condition and following Learning Predominantly Derives from the Maternal Allele. PLoS ONE 2015, 10, e0141078. [Google Scholar] [CrossRef] [Green Version]
- Baral, K.; Rotwein, P. The insulin-like growth factor 2 gene in mammals: Organizational complexity within a conserved locus. PLoS ONE 2019, 14, e0219155. [Google Scholar] [CrossRef]
- Nielsen, F.C.; Nielsen, J.; Christiansen, J. A family of IGF-II mRNA binding proteins (IMP) involved in RNA trafficking. Scand. J. Clin. Lab. Invest. Suppl. 2001, 234, 93–99. [Google Scholar] [CrossRef]
- Cao, J.; Mu, Q.; Huang, H. The Roles of Insulin-Like Growth Factor 2 mRNA-Binding Protein 2 in Cancer and Cancer Stem Cells. Stem Cells Int. 2018, 2018, 4217259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Roth, A.; Yu, M.; Morris, R.; Bersani, F.; Rivera, M.N.; Lu, J.; Shioda, T.; Vasudevan, S.; Ramaswamy, S.; et al. The IGF2 intronic miR-483 selectively enhances transcription from IGF2 fetal promoters and enhances tumorigenesis. Genes Dev. 2013, 27, 2543–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rotwein, P. The complex genetics of human insulin-like growth factor 2 are not reflected in public databases. J. Biol. Chem. 2018, 293, 4324–4333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sun, J.; Yang, F. The role of long non-coding RNA H19 in breast cancer. Oncol. Lett. 2020, 19, 7–16. [Google Scholar] [CrossRef]
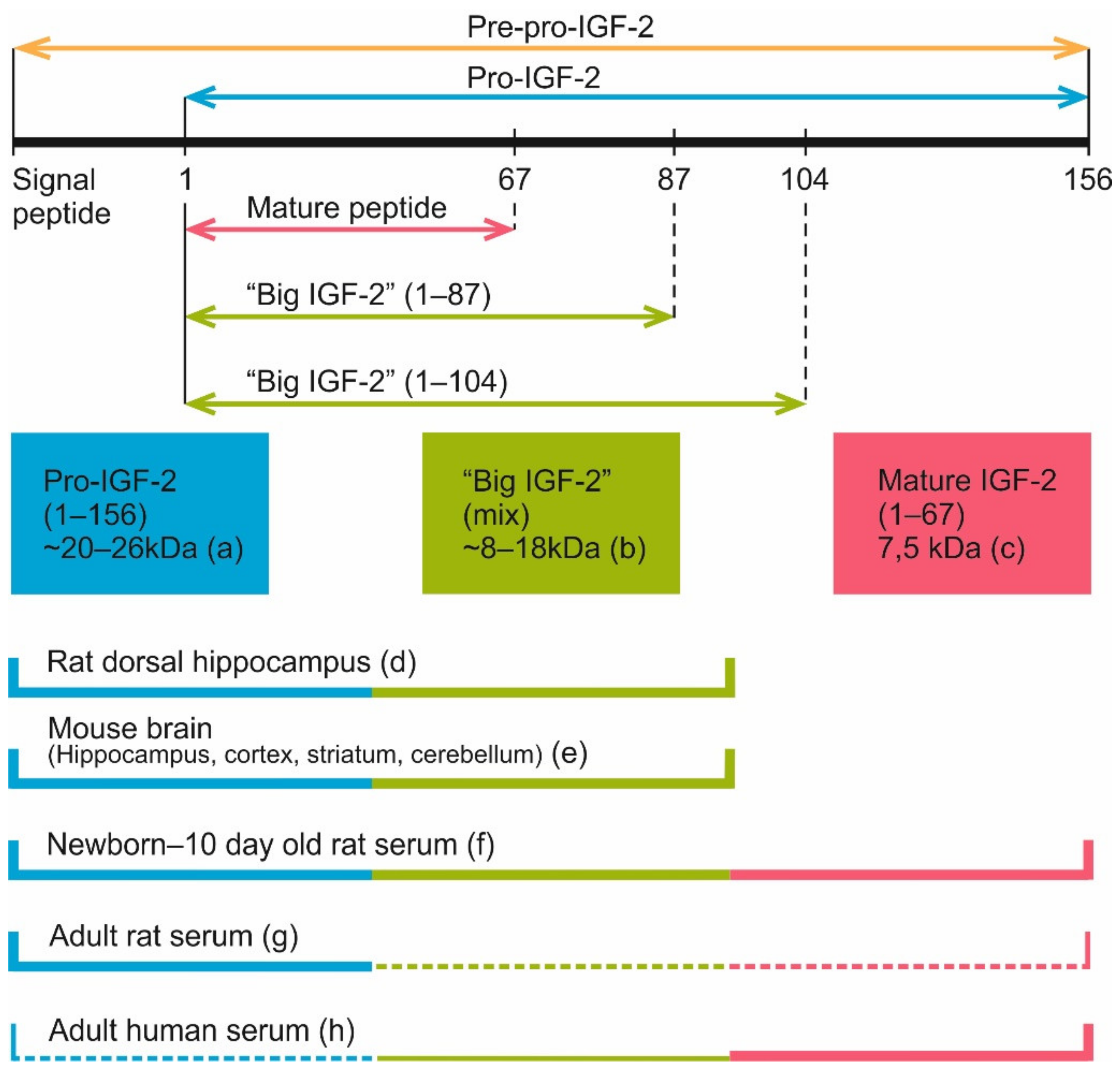
- Duguay, S.J.; Jin, Y.; Stein, J.; Duguay, A.N.; Gardner, P.; Steiner, D.F. Post-translational processing of the insulin-like growth factor-2 precursor. Analysis of O-glycosylation and endoproteolysis. J. Biol. Chem. 1998, 273, 18443–18451. [Google Scholar] [CrossRef] [Green Version]
- Boulle, N.; Gicquel, C.; Logié, A.; Christol, R.; Feige, J.J.; Le Bouc, Y. Fibroblast growth factor-2 inhibits the maturation of pro-insulin-like growth factor-II (Pro-IGF-II) and the expression of insulin-like growth factor binding protein-2 (IGFBP-2) in the human adrenocortical tumor cell line NCI-H295R. Endocrinology 2000, 141, 3127–3136. [Google Scholar] [CrossRef]
- Allard, J.B.; Duan, C. IGF-Binding Proteins: Why Do They Exist and Why Are There So Many? Front. Endocrinol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-S.; Rosenfeld, R.G.; Oh, Y. Biological roles of insulin-like growth factor binding proteins (IGFBPs). Exp. Mol. Med. 1997, 29, 85–96. [Google Scholar] [CrossRef]
- Rajaram, S.; Baylink, D.J.; Mohan, S. Insulin-like growth factor-binding proteins in serum and other biological fluids: Regulation and functions. Endocr. Rev. 1997, 18, 801–831. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Lee, Y.W.; Gao, Q.; Lee, Y.; Lee, H.E.; Ryu, J.H. Exogenous insulin-like growth factor 2 administration enhances memory consolidation and persistence in a time-dependent manner. Brain Res. 2015, 1622, 466–473. [Google Scholar] [CrossRef]
- Alberini, C.M.; Chen, D.Y. Memory enhancement: Consolidation, reconsolidation and insulin-like growth factor 2. Trends Neurosci. 2012, 35, 274–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Montañez, E.; Pavia, J.; Santin, L.J.; Boraldi, F.; Estivill-Torrus, G.; Aguirre, J.A.; Garcia-Fernandez, M. Involvement of IGF-II receptors in the antioxidant and neuroprotective effects of IGF-II on adult cortical neuronal cultures. Biochim. Biophys. Acta 2014, 1842, 1041–1051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahms, N.; Hancock, M.K. P-type lectins. Biochim. Biophys. Acta 2002, 1572, 317–340. [Google Scholar] [CrossRef]
- Werner, H.; LeRoith, D. Insulin and insulin-like growth factor receptors in the brain: Physiological and pathological aspects. Eur. Neuropsychopharmacol. 2014, 24, 1947–1953. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, T.; Chakraborty, C.; Gleeson, L.M.; Chidiac, P.; Lala, P.K. Stimulation of human extravillous trophoblast migration by IGF-II is mediated by IGF type 2 receptor involving inhibitory G protein(s) and phosphorylation of MAPK. J. Clin. Endocrinol. Metab. 2001, 86, 3665–3674. [Google Scholar] [CrossRef]
- Zhang, Q.; Tally, M.; Larsson, O.; Kennedy, R.T.; Huang, L.; Hall, K.; Berggren, P.O. Insulin-like growth factor II signaling through the insulin-like growth factor II/mannose-6-phosphate receptor promotes exocytosis in insulin-secreting cells. Proc. Natl. Acad. Sci. USA 1997, 94, 6232–6237. [Google Scholar] [CrossRef] [Green Version]
- El-Shewy, H.M.; Johnson, K.R.; Lee, M.H.; Jaffa, A.A.; Obeid, L.M.; Luttrell, L.M. Insulin-like growth factors mediate heterotrimeric G protein-dependent ERK1/2 activation by transactivating sphingosine 1-phosphate receptors. J. Biol. Chem. 2006, 281, 31399–31407. [Google Scholar] [CrossRef]
- Agis-Balboa, R.C.; Arcos-Diaz, D.; Wittnam, J.; Govindarajan, N.; Blom, K.; Burkhardt, S.; Haladyniak, U.; Agbemenyah, H.Y.; Zovoilis, A.; Salinas-Riester, G.; et al. A hippocampal insulin-growth factor 2 pathway regulates the extinction of fear memories. EMBO J. 2011, 30, 4071–4083. [Google Scholar] [CrossRef] [Green Version]
- García-Huerta, P.; Troncoso-Escudero, P.; Wu, D.; Thiruvalluvan, A.; Cisternas-Olmedo, M.; Henríquez, D.R.; Plate, L.; Chana-Cuevas, P.; Saquel, C.; Thielen, P.; et al. Insulin-like growth factor 2 (IGF2) protects against Huntington’s disease through the extracellular disposal of protein aggregates. Acta Neuropathol. 2020, 140, 737–764. [Google Scholar] [CrossRef]
- Ferrón, S.R.; Radford, E.J.; Domingo-Muelas, A.; Kleine, I.; Ramme, A.; Gray, D.; Sandovici, I.; Constancia, M.; Ward, A.; Menheniott, T.R.; et al. Differential genomic imprinting regulates paracrine and autocrine roles of IGF2 in mouse adult neurogenesis. Nat. Commun. 2015, 6, 8265. [Google Scholar] [CrossRef] [Green Version]
- Kasprzak, A.; Adamek, A. The insulin-like growth factor (IGF) signaling axis and hepatitis C virus-associated carcinogenesis (review). Int. J. Oncol. 2012, 41, 1919–1931. [Google Scholar] [CrossRef] [Green Version]
- Adamek, A.; Kasprzak, A. Insulin-Like Growth Factor (IGF) System in Liver Diseases. Int. J. Mol. Sci. 2018, 19, 1308. [Google Scholar] [CrossRef] [Green Version]
- Livingstone, C. IGF2 and cancer. Endocr. Relat. Cancer 2013, 20, R321–R339. [Google Scholar] [CrossRef] [Green Version]
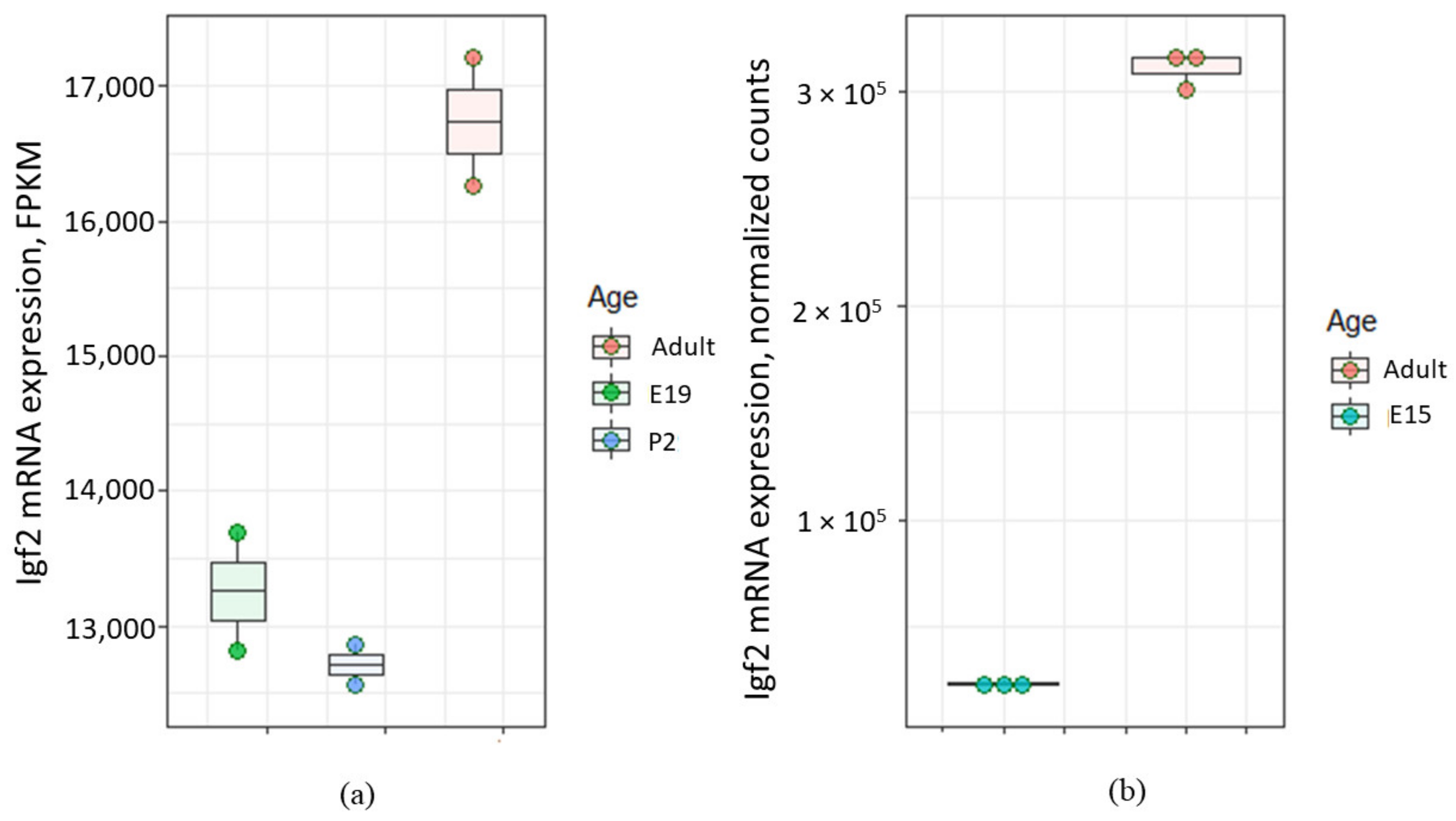
- Qiu, Q.; Jiang, J.Y.; Bell, M.; Tsang, B.K.; Gruslin, A. Activation of endoproteolytic processing of insulin-like growth factor-II in fetal, early postnatal, and pregnant rats and persistence of circulating levels in postnatal life. Endocrinology 2007, 148, 4803–4811. [Google Scholar] [CrossRef] [Green Version]
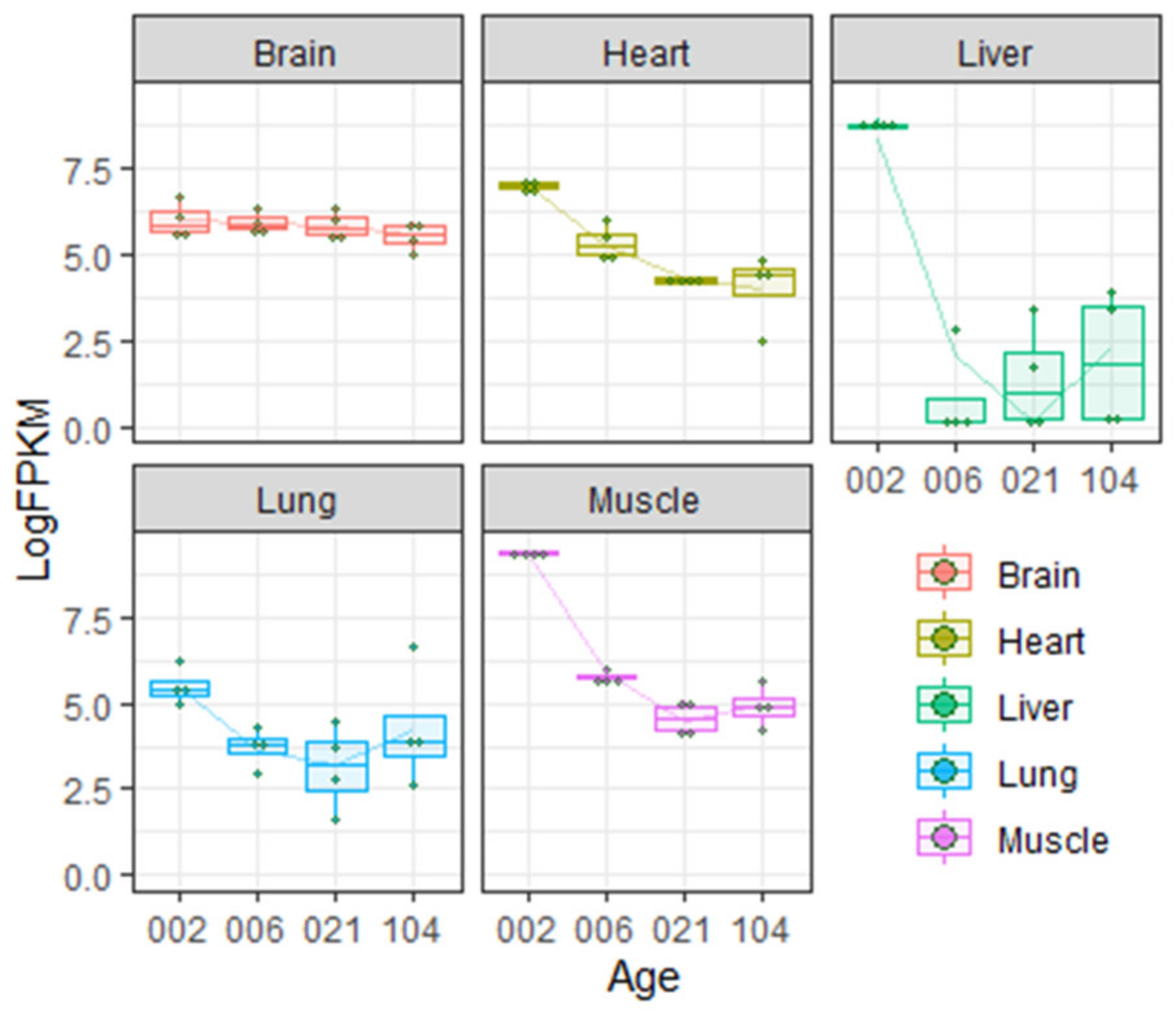
- Brown, A.L.; Graham, D.E.; Nissley, S.P.; Hill, D.J.; Strain, A.J.; Rechler, M.M. Developmental regulation of insulin-like growth factor II mRNA in different rat tissues. J. Biol. Chem. 1986, 261, 13144–13150. [Google Scholar] [CrossRef]
- Gray, A.; Tam, A.W.; Dull, T.J.; Hayflick, J.; Pintar, J.; Cavenee, W.K.; Koufos, A.; Ullrich, A. Tissue-specific and developmentally regulated transcription of the insulin-like growth factor 2 gene. DNA 1987, 6, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Gene Expression Omnibus. GEO accession number GSE53960. A rat RNA-Seq transcriptomic Bodymap across eleven organs and four developmental stages. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE53960 (accessed on 25 December 2020).
- Yu, Y.; Fuscoe, J.C.; Zhao, C.; Guo, C.; Jia, M.; Qing, T.; Bannon, D.I.; Lancashire, L.; Bao, W.; Du, T.; et al. A rat RNA-Seq transcriptomic BodyMap across 11 organs and 4 developmental stages. Nat. Commun. 2014, 5, 3230. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulou, F.; Herbert, J.; Soares, M.B.; Efstratiadis, A. Expression of the insulin-like growth factor II gene in the choroid plexus and the leptomeninges of the adult rat central nervous system. Proc. Natl. Acad. Sci. USA 1988, 85, 141–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynes, M.A.; Brooks, P.J.; Van Wyk, J.J.; Lund, P.K. Insulin-like growth factor II messenger ribonucleic acids are synthesized in the choroid plexus of the rat brain. Mol. Endocrinol. 1988, 2, 47–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehtinen, M.K.; Zappaterra, M.W.; Chen, X.; Yang, Y.J.; Hill, A.D.; Lun, M.; Maynard, T.; Gonzalez, D.; Kim, S.; Ye, P.; et al. The cerebrospinal fluid provides a proliferative niche for neural progenitor cells. Neuron 2011, 69, 893–905. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, A.N.; Schneider, J.S.; Qin, M.; Tyler, W.A.; Pintar, J.E.; Fraidenraich, D.; Wood, T.L.; Levison, S.W. IGF-II promotes stemness of neural restricted precursors. Stem Cells 2012, 30, 1265–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene Expression Omnibus. GEO accession number GSE44056. Gene expression data from lateral ventricle choroid plexuses of developing and adult rats. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE44056 (accessed on 25 December 2020).
- Kratzer, I.; Liddelow, S.A.; Saunders, N.R.; Dziegielewska, K.M.; Strazielle, N.; Ghersi-Egea, J.F. Developmental changes in the transcriptome of the rat choroid plexus in relation to neuroprotection. Fluids Barriers CNS 2013, 10, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gene Expression Omnibus. GEO accession number GSE44072. In Genome-Wide Analysis and Comparison of E15 and Adult Rat Lateral Ventricular Choroid Plexus Epithelial Cells. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE44072 (accessed on 25 December 2020).
- Liddelow, S.A.; Dziegielewska, K.M.; Ek, C.J.; Habgood, M.D.; Bauer, H.; Bauer, H.C.; Lindsay, H.; Wakefield, M.J.; Strazielle, N.; Kratzer, I.; et al. Mechanisms that determine the internal environment of the developing brain: A transcriptomic, functional and ultrastructural approach. PLoS ONE 2013, 8, e65629. [Google Scholar] [CrossRef] [Green Version]
- Terauchi, A.; Johnson-Venkatesh, E.M.; Bullock, B.; Lehtinen, M.K.; Umemori, H. Retrograde fibroblast growth factor 22 (FGF22) signaling regulates insulin-like growth factor 2 (IGF2) expression for activity-dependent synapse stabilization in the mammalian brain. eLife 2016, 5. [Google Scholar] [CrossRef]
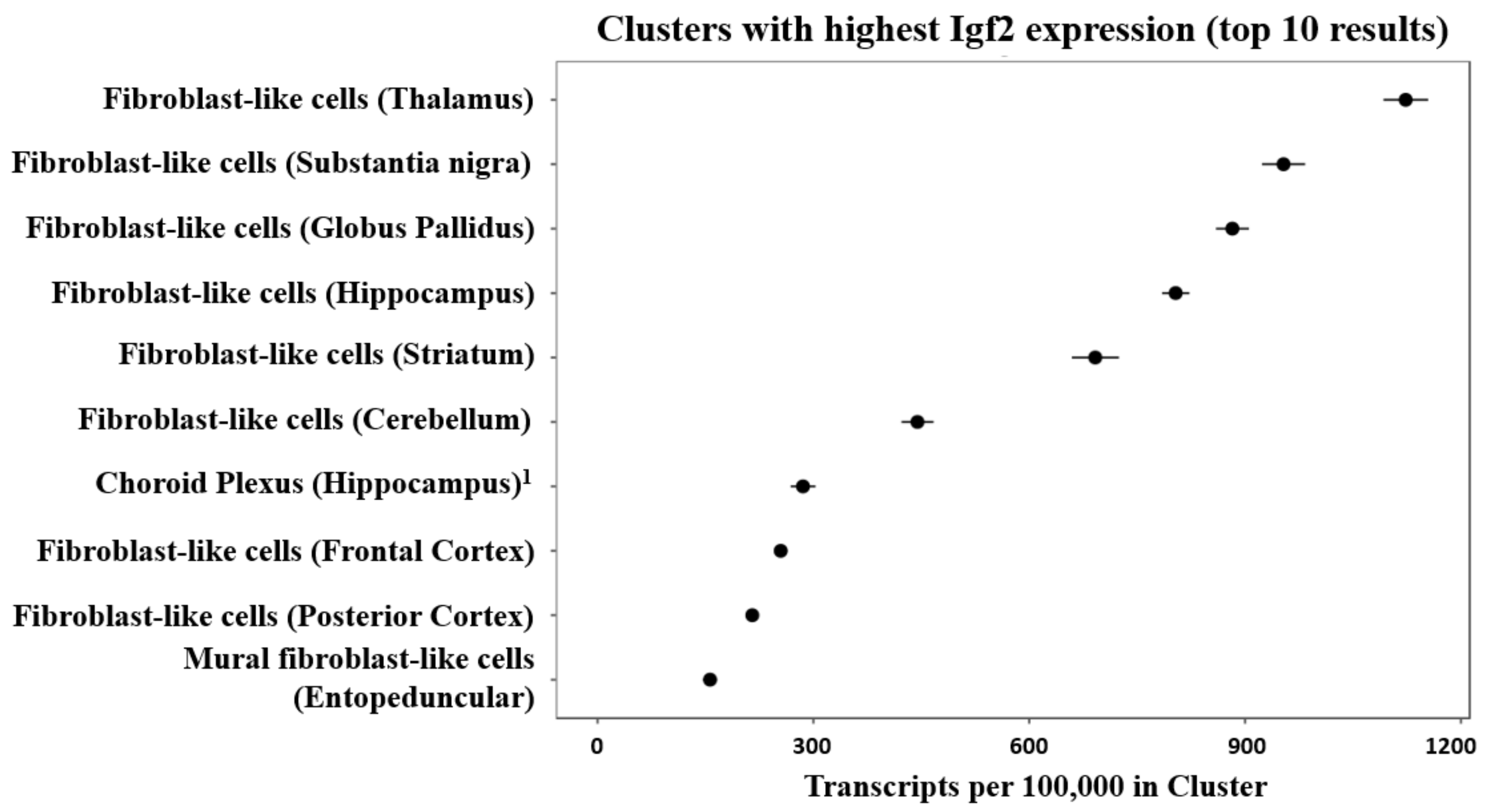
- DropViz. Exploring the Mouse Brain through Single Cell Expression Profiles. Igf2 Expression Levels by Cluster. Available online: http://dropviz.org/?_state_id_=73cd7e0d6071a9a9 (accessed on 25 December 2020).
- Saunders, A.; Macosko, E.Z.; Wysoker, A.; Goldman, M.; Krienen, F.M.; de Rivera, H.; Bien, E.; Baum, M.; Bortolin, L.; Wang, S.; et al. Molecular Diversity and Specializations among the Cells of the Adult Mouse Brain. Cell 2018, 174, 1015–1030. [Google Scholar] [CrossRef] [Green Version]
- Ghanipoor-Samami, M.; Javadmanesh, A.; Burns, B.M.; Thomsen, D.A.; Nattrass, G.S.; Estrella, C.A.S.; Kind, K.L.; Hiendleder, S. Atlas of tissue- and developmental stage specific gene expression for the bovine insulin-like growth factor (IGF) system. PLoS ONE 2018, 13, e0200466. [Google Scholar] [CrossRef]
- Delhanty, P.J.; Han, V.K. The expression of insulin-like growth factor (IGF)-binding protein-2 and IGF-II genes in the tissues of the developing ovine fetus. Endocrinology 1993, 132, 41–52. [Google Scholar] [CrossRef]
- Scott, J.; Cowell, J.; Robertson, M.E.; Priestley, L.M.; Wadey, R.; Hopkins, B.; Pritchard, J.; Bell, G.I.; Rall, L.B.; Graham, C.F.; et al. Insulin-like growth factor-II gene expression in Wilms’ tumour and embryonic tissues. Nature 1985, 317, 260–262. [Google Scholar] [CrossRef]
- Sandberg, A.C.; Engberg, C.; Lake, M.; von Holst, H.; Sara, V.R. The expression of insulin-like growth factor I and insulin-like growth factor II genes in the human fetal and adult brain and in glioma. Neurosci. Lett. 1988, 93, 114–119. [Google Scholar] [CrossRef]
- The Human Protein Atlas. IGF2 mRNA expression in the brain. Available online: https://www.proteinatlas.org/ENSG00000167244-IGF2/brain (accessed on 25 December 2020).
- Han, V.K.; Lund, P.K.; Lee, D.C.; D’Ercole, A.J. Expression of somatomedin/insulin-like growth factor messenger ribonucleic acids in the human fetus: Identification, characterization, and tissue distribution. J. Clin. Endocrinol. Metab. 1988, 66, 422–429. [Google Scholar] [CrossRef]
- HBT (Human Brain Transcriptome). IGF2 gene expression in neocortical areas. Available online: https://hbatlas.org/hbtd/images/nctxBrain/IGF2.pdf (accessed on 25 December 2020).
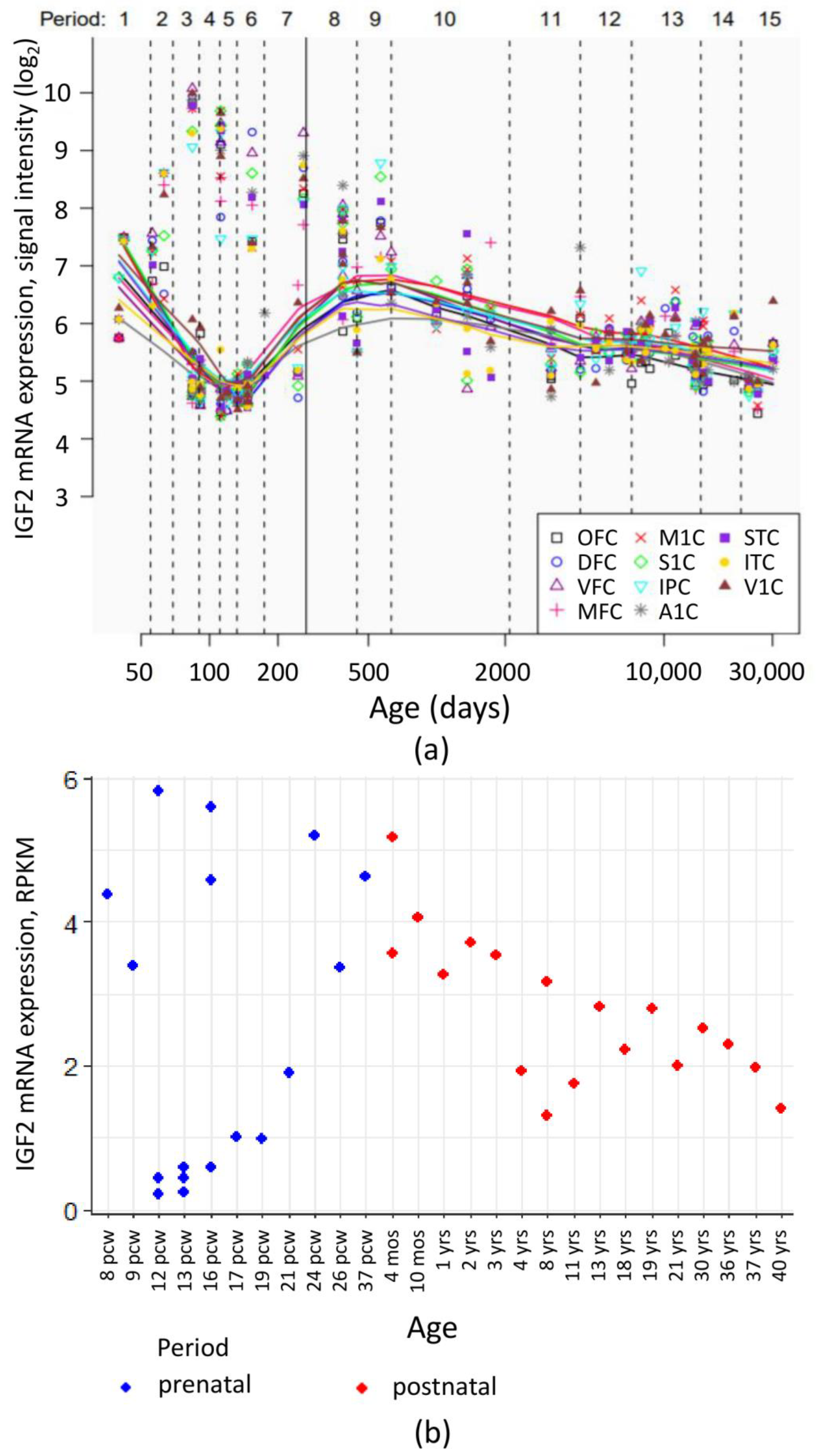
- Kang, H.J.; Kawasawa, Y.I.; Cheng, F.; Zhu, Y.; Xu, X.; Li, M.; Sousa, A.M.; Pletikos, M.; Meyer, K.A.; Sedmak, G.; et al. Spatio-temporal transcriptome of the human brain. Nature 2011, 478, 483–489. [Google Scholar] [CrossRef] [Green Version]
- BrainSpan Atlas of the Developing Human Brain. IGF2 gene level RPKM. Available online: http://www.brainspan.org/rnaseq/searches?exact_match=false&search_term=%22IGF2%22&search_type=gene (accessed on 25 December 2020).
- Miller, J.A.; Ding, S.-L.; Sunkin, S.M.; Smith, K.A.; Ng, L.; Szafer, A.; Ebbert, A.; Riley, Z.L.; Royall, J.J.; Aiona, K.; et al. Transcriptional landscape of the prenatal human brain. Nature 2014, 508, 199–206. [Google Scholar] [CrossRef]
- Haselbacher, G.K.; Schwab, M.E.; Pasi, A.; Humbel, R.E. Insulin-like growth factor II (IGF II) in human brain: Regional distribution of IGF II and of higher molecular mass forms. Proc. Natl. Acad. Sci. USA 1985, 82, 2153–2157. [Google Scholar] [CrossRef] [Green Version]
- McKelvie, P.A.; Rosen, K.M.; Kinney, H.C.; Villa-Komaroff, L. Insulin-like growth factor II expression in the developing human brain. J. Neuropathol. Exp. Neurol. 1992, 51, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.I.; Goldstein, O.; Han, V.K.M.; Tarantal, A.F. IGF-II and IGF Binding Protein (IGFBP-1, IGFBP-3) Gene Expression in Fetal Rhesus Monkey Tissues during the Second and Third Trimesters. Pediatr. Res. 2001, 49, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Suh, H.S.; Zhao, M.L.; Derico, L.; Choi, N.; Lee, S.C. Insulin-like growth factor 1 and 2 (IGF1, IGF2) expression in human microglia: Differential regulation by inflammatory mediators. J. Neuroinflamm. 2013, 10, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubo, S.; Ogino, S.; Fukushima, T.; Olson, P.R.; Kida, M.; Maruno, M.; Yoshimine, T.; Hyakawa, T. Immunohistochemical study of insulin-like growth factor II (IGF-II) and insulin-like growth factor binding protein-2 (IGFBP-2) in choroid plexus papilloma. Neurol. Res. 1999, 21, 339–344. [Google Scholar] [CrossRef]
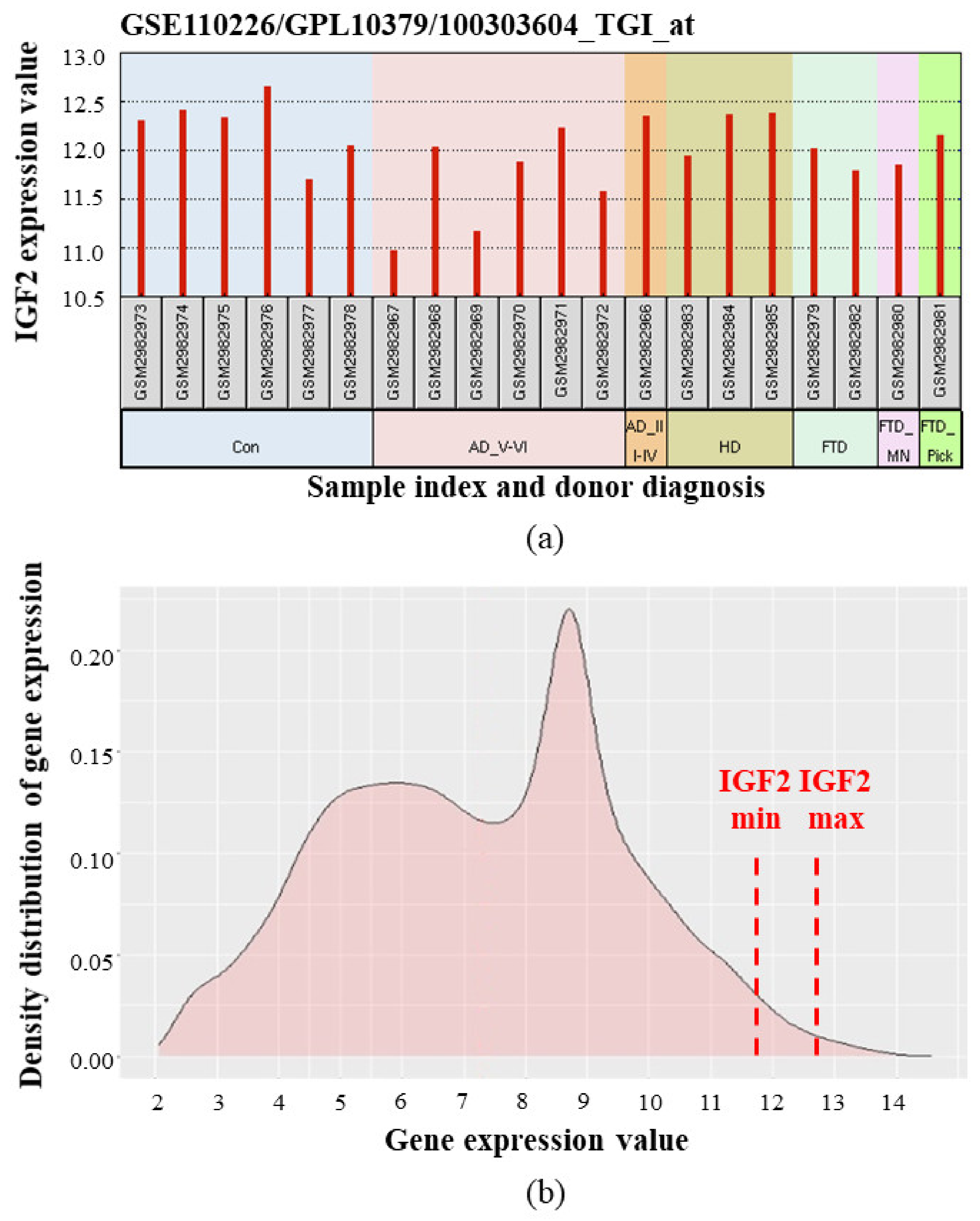
- Stopa, E.G.; Tanis, K.Q.; Miller, M.C.; Nikonova, E.V.; Podtelezhnikov, A.A.; Finney, E.M.; Stone, D.J.; Camargo, L.M.; Parker, L.; Verma, A.; et al. Comparative transcriptomics of choroid plexus in Alzheimer’s disease, frontotemporal dementia and Huntington’s disease: Implications for CSF homeostasis. Fluids Barriers CNS 2018, 15, 18. [Google Scholar] [CrossRef] [PubMed]
- Gene Expression Omnibus. GEO accession number GSE110226. Comparative Transcriptomics of Choroid Plexus in Alzheimer’s Disease, Huntington’s Disease and Frontotemporal Dementia: Implications for CSF Homeostasis and Dynamics. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE110226 (accessed on 25 December 2020).
- GEO2R interactive web tool -instructions for use. Available online: https://www.ncbi.nlm.nih.gov/geo/info/geo2r.html (accessed on 4 December 2020).
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—update. Nucleic Acids Res. 2013, 41, D991–D995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.-M.; Yang, D.; Liu, Z.-F.; Hu, S.-Z.; Yan, S.-H.; He, X.-W. Density distribution of gene expression profiles and evaluation of using maximal information coefficient to identify differentially expressed genes. PLoS ONE 2019, 14, e0219551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Mistry, J.; Nicar, M.J.; Khosravi, M.J.; Diamandis, A.; van Doorn, J.; Juul, A. Insulin-like growth factors (IGF-I, free IGF-I and IGF-II) and insulin-like growth factor binding proteins (IGFBP-2, IGFBP-3, IGFBP-6, and ALS) in blood circulation. J. Clin. Lab. Anal. 1999, 13, 166–172. [Google Scholar] [CrossRef]
- Zapf, J.; Walter, H.; Froesch, E.R. Radioimmunological determination of insulinlike growth factors I and II in normal subjects and in patients with growth disorders and extrapancreatic tumor hypoglycemia. J. Clin. Invest. 1981, 68, 1321–1330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodenburg, R.J.; Holthuizen, P.E.; Sussenbach, J.S. A functional Sp1 binding site is essential for the activity of the adult liver-specific human insulin-like growth factor II promoter. Mol. Endocrinol. 1997, 11, 237–250. [Google Scholar] [CrossRef]
- Rotwein, P. Structure, evolution, expression and regulation of insulin-like growth factors I and II. Growth Factors 1991, 5, 3–18. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.A.; Ferguson-Smith, A.C. Mechanisms regulating imprinted genes in clusters. Curr. Opin. Cell Biol. 2007, 19, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Vu, T.H.; Hoffman, A.R. Promoter-specific imprinting of the human insulin-like growth factor-II gene. Nature 1994, 371, 714–717. [Google Scholar] [CrossRef]
- Baran, Y.; Subramaniam, M.; Biton, A.; Tukiainen, T.; Tsang, E.K.; Rivas, M.A.; Pirinen, M.; Gutierrez-Arcelus, M.; Smith, K.S.; Kukurba, K.R.; et al. The landscape of genomic imprinting across diverse adult human tissues. Genome Res. 2015, 25, 927–936. [Google Scholar] [CrossRef] [Green Version]
- Ordyan, N.E.; Malysheva, O.V.; Akulova, V.K.; Pivina, S.G.; Kholova, G.I. The Capability to Learn and Expression of the Insulin-Like Growth Factor II Gene in the Brain of Male Rats Whose Fathers Were Subjected to Stress Factors in the “Stress–Restress” Paradigm. Neurochem. J. 2020, 14, 191–196. [Google Scholar] [CrossRef]
- Schmeisser, M.J.; Baumann, B.; Johannsen, S.; Vindedal, G.F.; Jensen, V.; Hvalby, Ø.C.; Sprengel, R.; Seither, J.; Maqbool, A.; Magnutzki, A.; et al. IκB kinase/nuclear factor κB-dependent insulin-like growth factor 2 (Igf2) expression regulates synapse formation and spine maturation via Igf2 receptor signaling. J. Neurosci. 2012, 32, 5688–5703. [Google Scholar] [CrossRef]
- Dellapolla, A.; Kloehn, I.; Pancholi, H.; Callif, B.; Wertz, D.; Rohr, K.E.; Hurley, M.M.; Baker, K.M.; Hattar, S.; Gilmartin, M.R.; et al. Long days enhance recognition memory and increase insulin-like growth factor 2 in the hippocampus. Sci. Rep. 2017, 7, 3925. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Gao, Y.; Shi, H.S.; Song, L.; Wang, J.C.; Shao, J.; Geng, X.H.; Xue, G.; Li, J.L.; Hou, Y.N. Overexpression of SIRT6 in the hippocampal CA1 impairs the formation of long-term contextual fear memory. Sci. Rep. 2016, 6, 18982. [Google Scholar] [CrossRef] [Green Version]
- Pardo, M.; Cheng, Y.; Velmeshev, D.; Magistri, M.; Eldar-Finkelman, H.; Martinez, A.; Faghihi, M.A.; Jope, R.S.; Beurel, E. Intranasal siRNA administration reveals IGF2 deficiency contributes to impaired cognition in Fragile X syndrome mice. JCI Insight 2017, 2, e91782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sárvári, M.; Kalló, I.; Hrabovszky, E.; Solymosi, N.; Rodolosse, A.; Liposits, Z. Long-Term Estrogen Receptor Beta Agonist Treatment Modifies the Hippocampal Transcriptome in Middle-Aged Ovariectomized Rats. Front. Cell. Neurosci. 2016, 10, 149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sárvári, M.; Kalló, I.; Hrabovszky, E.; Solymosi, N.; Tóth, K.; Likó, I.; Molnár, B.; Tihanyi, K.; Liposits, Z. Estradiol replacement alters expression of genes related to neurotransmission and immune surveillance in the frontal cortex of middle-aged, ovariectomized rats. Endocrinology 2010, 151, 3847–3862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahmoradi, A.; Radyushkin, K.; Rossner, M.J. Enhanced memory consolidation in mice lacking the circadian modulators Sharp1 and -2 caused by elevated Igf2 signaling in the cortex. Proc. Natl. Acad. Sci. USA 2015, 112, E3582–E3589. [Google Scholar] [CrossRef] [Green Version]
- Poggini, S.; Golia, M.T.; Alboni, S.; Milior, G.; Sciarria, L.P.; Viglione, A.; Matte Bon, G.; Brunello, N.; Puglisi-Allegra, S.; Limatola, C.; et al. Combined Fluoxetine and Metformin Treatment Potentiates Antidepressant Efficacy Increasing IGF2 Expression in the Dorsal Hippocampus. Neural Plast. 2019, 2019, 4651031. [Google Scholar] [CrossRef] [PubMed]
- Romanus, J.A.; Yang, Y.W.; Adams, S.O.; Sofair, A.N.; Tseng, L.Y.; Nissley, S.P.; Rechler, M.M. Synthesis of insulin-like growth factor II (IGF-II) in fetal rat tissues: Translation of IGF-II ribonucleic acid and processing of pre-pro-IGF-II. Endocrinology 1988, 122, 709–716. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.W.; Romanus, J.A.; Liu, T.Y.; Nissley, S.P.; Rechler, M.M. Biosynthesis of rat insulin-like growth factor II. I. Immunochemical demonstration of a approximately 20-kilodalton biosynthetic precursor of rat insulin-like growth factor II in metabolically labeled BRL-3A rat liver cells. J. Biol. Chem. 1985, 260, 2570–2577. [Google Scholar] [CrossRef]
- Steinmetz, A.B.; Johnson, S.A.; Iannitelli, D.E.; Pollonini, G.; Alberini, C.M. Insulin-like growth factor 2 rescues aging-related memory loss in rats. Neurobiol. Aging 2016, 44, 9–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, S.; Ariyasu, H.; Uraki, S.; Takeshima, K.; Morita, S.; Inaba, H.; Iwakura, H.; Doi, A.; Ohashi, T.; Kawago, M.; et al. Imbalanced Expression of IGF2 and PCSK4 Is Associated With Overproduction of Big IGF2 in SFT With NICTH: A Pilot Study. J. Clin. Endocrinol. Metab. 2018, 103, 2728–2734. [Google Scholar] [CrossRef]
- Qiu, Q.; Yan, X.; Bell, M.; Di, J.; Tsang, B.K.; Gruslin, A. Mature IGF-II prevents the formation of "big" IGF-II/IGFBP-2 complex in the human circulation. Growth Horm. IGF Res. 2010, 20, 110–117. [Google Scholar] [CrossRef]
- Steinmetz, A.B.; Stern, S.A.; Kohtz, A.S.; Descalzi, G.; Alberini, C.M. Insulin-Like Growth Factor II Targets the mTOR Pathway to Reverse Autism-Like Phenotypes in Mice. J. Neurosci. 2018, 38, 1015–1029. [Google Scholar] [CrossRef] [PubMed]
- Marks, A.G.; Carroll, J.M.; Purnell, J.Q.; Roberts, C.T., Jr. Plasma distribution and signaling activities of IGF-II precursors. Endocrinology 2011, 152, 922–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Weiss, M.A.; Arunagiri, A.; Yong, J.; Rege, N.; Sun, J.; Haataja, L.; Kaufman, R.J.; Arvan, P. Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes. Metab. 2018, 20, 28–50. [Google Scholar] [CrossRef]
- Weiss, M.; Steiner, D.F.; Philipson, L.H. Insulin Biosynthesis, Secretion, Structure, and Structure-Activity Relationships. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, J., Kaltsas, G., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Haselbacher, G.; Humbel, R. Evidence for two species of insulin-like growth factor II (IGF II and "big" IGF II) in human spinal fluid. Endocrinology 1982, 110, 1822–1824. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Q.; Basak, A.; Mbikay, M.; Tsang, B.K.; Gruslin, A. Role of pro-IGF-II processing by proprotein convertase 4 in human placental development. Proc. Natl. Acad. Sci. USA 2005, 102, 11047–11052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenall, S.A.; Bentley, J.D.; Pearce, L.A.; Scoble, J.A.; Sparrow, L.G.; Bartone, N.A.; Xiao, X.; Baxter, R.C.; Cosgrove, L.J.; Adams, T.E. Biochemical characterization of individual human glycosylated pro-insulin-like growth factor (IGF)-II and big-IGF-II isoforms associated with cancer. J. Biol. Chem. 2013, 288, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Khosravi, M.J.; Diamandi, A.; Mistry, J.; Krishna, R.G.; Khare, A. Acid-Labile Subunit of Human Insulin-Like Growth Factor-Binding Protein Complex: Measurement, Molecular, and Clinical Evaluation. J. Clin. Endocrinol. Metab. 1997, 82, 3944–3951. [Google Scholar] [CrossRef]
- Holly, J.M.P.; Biernacka, K.; Perks, C.M. The Neglected Insulin: IGF-II, a Metabolic Regulator with Implications for Diabetes, Obesity, and Cancer. Cells 2019, 8, 1207. [Google Scholar] [CrossRef] [Green Version]
- Frystyk, J.; Skjaerbaek, C.; Dinesen, B.; Orskov, H. Free insulin-like growth factors (IGF-I and IGF-II) in human serum. FEBS Lett. 1994, 348, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Hwa, V.; Oh, Y.; Rosenfeld, R.G. The insulin-like growth factor-binding protein (IGFBP) superfamily. Endocr. Rev. 1999, 20, 761–787. [Google Scholar] [CrossRef]
- Lewitt, M.S.; Boyd, G.W. The Role of Insulin-Like Growth Factors and Insulin-Like Growth Factor-Binding Proteins in the Nervous System. Biochem. Insight. 2019, 12, 1178626419842176. [Google Scholar] [CrossRef] [Green Version]
- Daza, D.O.; Sundström, G.; Bergqvist, C.A.; Duan, C.; Larhammar, D. Evolution of the insulin-like growth factor binding protein (IGFBP) family. Endocrinology 2011, 152, 2278–2289. [Google Scholar] [CrossRef] [Green Version]
- Forbes, B.E.; McCarthy, P.; Norton, R.S. Insulin-like growth factor binding proteins: A structural perspective. Front. Endocrinol. 2012, 3, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guler, H.P.; Zapf, J.; Schmid, C.; Froesch, E.R. Insulin-like growth factors I and II in healthy man. Estimations of half-lives and production rates. Acta Endocrinol. 1989, 121, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Goto, Y.I.; Itoh, M. Insulin-Like Growth Factor Binding Protein-3 Deficiency Leads to Behavior Impairment with Monoaminergic and Synaptic Dysfunction. Am. J. Pathol. 2017, 187, 390–400. [Google Scholar] [CrossRef] [Green Version]
- Wang, E.A.; Chen, W.-Y.; Wong, C.-H. Multiple Growth Factor Targeting by Engineered Insulin-like Growth Factor Binding Protein-3 Augments EGF Receptor Tyrosine Kinase Inhibitor Efficacy. Sci. Rep. 2020, 10, 2735. [Google Scholar] [CrossRef]
- Yamada, P.M.; Lee, K.W. Perspectives in mammalian IGFBP-3 biology: Local vs. systemic action. Am. J. Physiol. Cell Physiol. 2009, 296, C954–C976. [Google Scholar] [CrossRef] [Green Version]
- Baxter, R.C. Nuclear actions of insulin-like growth factor binding protein-3. Gene 2015, 569, 7–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikonen, M.; Liu, B.; Hashimoto, Y.; Ma, L.; Lee, K.W.; Niikura, T.; Nishimoto, I.; Cohen, P. Interaction between the Alzheimer’s survival peptide humanin and insulin-like growth factor-binding protein 3 regulates cell survival and apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 13042–13047. [Google Scholar] [CrossRef] [Green Version]
- Bonham, L.W.; Geier, E.G.; Steele, N.Z.R.; Holland, D.; Miller, B.L.; Dale, A.M.; Desikan, R.S.; Yokoyama, J.S. Insulin-Like Growth Factor Binding Protein 2 Is Associated With Biomarkers of Alzheimer’s Disease Pathology and Shows Differential Expression in Transgenic Mice. Front. Neurosci. 2018, 12, 476. [Google Scholar] [CrossRef]
- McGrath, E.R.; Himali, J.J.; Levy, D.; Conner, S.C.; DeCarli, C.S.; Pase, M.P.; Courchesne, P.; Satizabal, C.L.; Vasan, R.S.; Beiser, A.S.; et al. Circulating IGFBP-2: A novel biomarker for incident dementia. Ann. Clin. Transl. Neurol. 2019, 6, 1659–1670. [Google Scholar] [CrossRef] [Green Version]
- Khan, S. IGFBP-2 Signaling in the Brain: From Brain Development to Higher Order Brain Functions. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Lu, X.; Huang, Q.; Tang, J.; Weng, J.; Yang, Z.; Lv, M.; Xu, X.; Xia, F.; Zhang, M.; et al. IGFBP2 Plays an Essential Role in Cognitive Development during Early Life. Adv. Sci. 2019, 6, 1901152. [Google Scholar] [CrossRef] [Green Version]
- Boisclair, Y.R.; Rhoads, R.P.; Ueki, I.; Wang, J.; Ooi, G.T. The acid-labile subunit (ALS) of the 150 kDa IGF-binding protein complex: An important but forgotten component of the circulating IGF system. J. Endocrinol. 2001, 170, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bond, J.J.; Meka, S.; Baxter, R.C. Binding characteristics of pro-insulin-like growth factor-II from cancer patients: Binary and ternary complex formation with IGF binding proteins-1 to -6. J. Endocrinol. 2000, 165, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Veggel, K.M.; Huits, R.M.; Donker, G.H.; Lentjes, E.G.; van Doorn, J. Column chromatographic characterization of complex formation of pro-IGF-II isoforms with acid labile subunit and IGF-binding proteins associated with non-islet cell tumour induced hypoglycaemia. Growth Horm. IGF Res. 2014, 24, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Ide, S.; Takashima, S.; Kudo, S.; Nomura, Y.; Segawa, M.; Kubota, T.; Mori, H.; Tanaka, S.; Horie, H.; et al. Methyl CpG-binding protein 2 (a mutation of which causes Rett syndrome) directly regulates insulin-like growth factor binding protein 3 in mouse and human brains. J. Neuropathol. Exp. Neurol. 2007, 66, 117–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scala, M.; Mojarrad, M.; Riazuddin, S.; Brigatti, K.W.; Ammous, Z.; Cohen, J.S.; Hosny, H.; Usmani, M.A.; Shahzad, M.; Riazuddin, S.; et al. RSRC1 loss-of-function variants cause mild to moderate autosomal recessive intellectual disability. Brain 2020, 143, e31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez, Y.; Menascu, S.; Cohen, I.; Kadir, R.; Basha, O.; Shorer, Z.; Romi, H.; Meiri, G.; Rabinski, T.; Ofir, R.; et al. RSRC1 mutation affects intellect and behaviour through aberrant splicing and transcription, downregulating IGFBP3. Brain 2018, 141, 961–970. [Google Scholar] [CrossRef]
- Honda, M.; Eriksson, K.S.; Zhang, S.; Tanaka, S.; Lin, L.; Salehi, A.; Hesla, P.E.; Maehlen, J.; Gaus, S.E.; Yanagisawa, M.; et al. IGFBP3 Colocalizes with and Regulates Hypocretin (Orexin). PLoS ONE 2009, 4, e4254. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, H.S.G.; Dempsey, R.J. IGFBP-3 Inhibits the Proliferation of Neural Progenitor Cells. Neurochem. Res. 2011, 36, 406–411. [Google Scholar] [CrossRef] [PubMed]
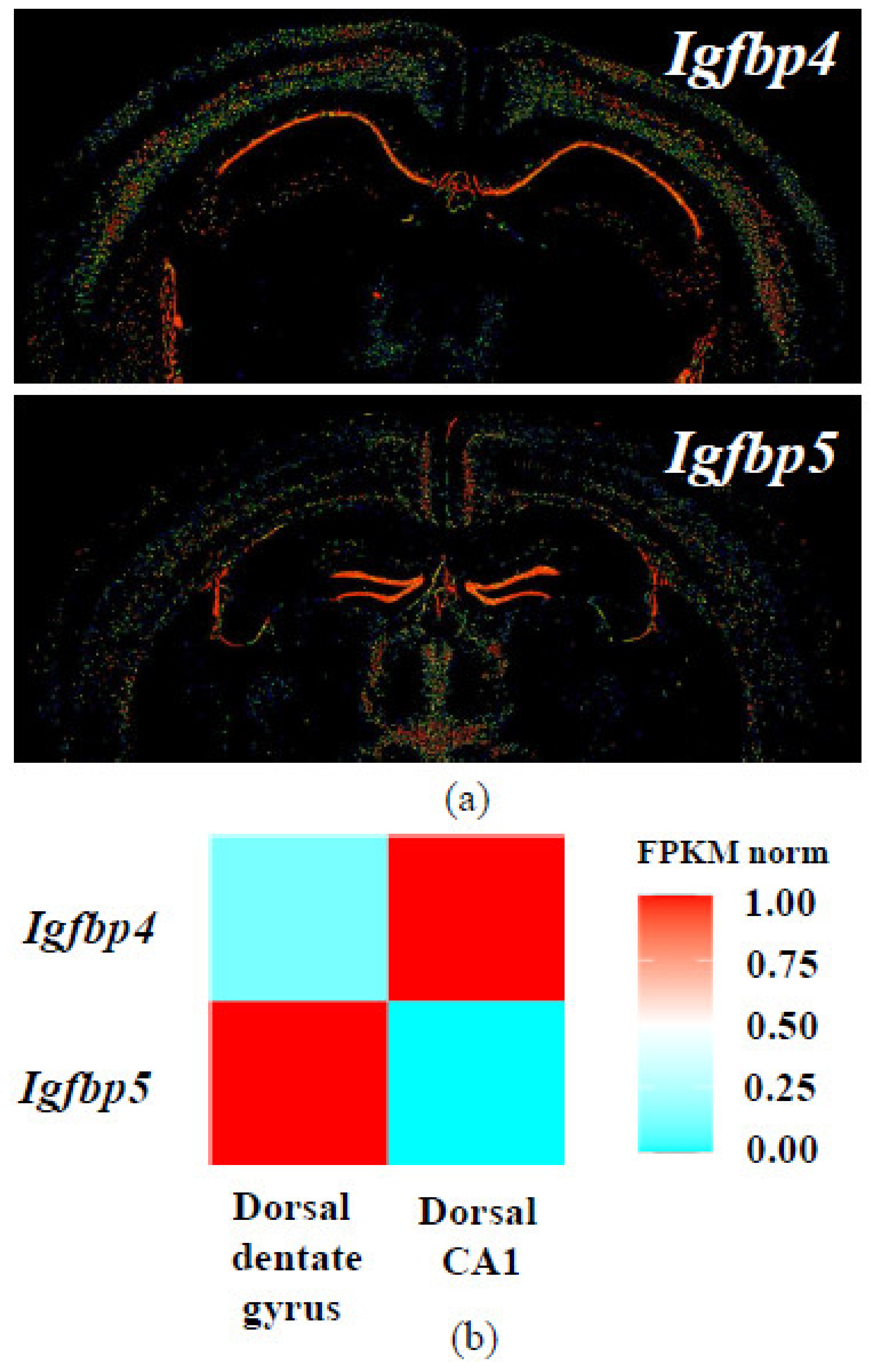
- Jiang, X.; Zhao, J.; Ju, L.; Liu, Y.; Wang, B.; Zou, X.; Xu, C. Temporal expression patterns of insulin-like growth factor binding protein-4 in the embryonic and postnatal rat brain. BMC Neurosci. 2013, 14, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen Mouse Brain Atlas. Expression pattern of Igfbp4, coronal sections. Available online: https://mouse.brain-map.org/experiment/show/71924311 (accessed on 25 December 2020).
- Allen Mouse Brain Atlas. Expression pattern of Igfbp5, coronal sections. Available online: https://mouse.brain-map.org/experiment/show/73592530 (accessed on 25 December 2020).
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature 2007, 445, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Hippocampus RNA-seq atlas. Available online: https://hipposeq.janelia.org/full/t1/27C4508E-0E80-11EB-B29E-9FD29DC49958/t2/1F321C52-0E81-11EB-9976-9ED29DC49958/ (accessed on 25 December 2020).
- Cembrowski, M.S.; Wang, L.; Sugino, K.; Shields, B.C.; Spruston, N. Hipposeq: A comprehensive RNA-seq database of gene expression in hippocampal principal neurons. eLife 2016, 5, e14997. [Google Scholar] [CrossRef]
- Sandberg Nordqvist, A.C.; von Holst, H.; Holmin, S.; Sara, V.R.; Bellander, B.M.; Schalling, M. Increase of insulin-like growth factor (IGF)-1, IGF binding protein-2 and −4 mRNAs following cerebral contusion. Mol. Brain Res. 1996, 38, 285–293. [Google Scholar] [CrossRef]
- Mostafavi, S.; Gaiteri, C.; Sullivan, S.E.; White, C.C.; Tasaki, S.; Xu, J.; Taga, M.; Klein, H.-U.; Patrick, E.; Komashko, V.; et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 2018, 21, 811–819. [Google Scholar] [CrossRef]
- Yu, L.; Petyuk, V.A.; Gaiteri, C.; Mostafavi, S.; Young-Pearse, T.; Shah, R.C.; Buchman, A.S.; Schneider, J.A.; Piehowski, P.D.; Sontag, R.L.; et al. Targeted brain proteomics uncover multiple pathways to Alzheimer’s dementia. Ann. Neurol. 2018, 84, 78–88. [Google Scholar] [CrossRef]
- Buchman, A.S.; Yu, L.; Petyuk, V.A.; Gaiteri, C.; Tasaki, S.; Blizinsky, K.D.; Schneider, J.A.; De Jager, P.L.; Bennett, D.A. Cognition may link cortical IGFBP5 levels with motor function in older adults. PLoS ONE 2019, 14, e0220968. [Google Scholar] [CrossRef]
- Siddle, K. Molecular basis of signaling specificity of insulin and IGF receptors: Neglected corners and recent advances. Front. Endocrinol. 2012, 3, 34. [Google Scholar] [CrossRef] [Green Version]
- Kavran, J.M.; McCabe, J.M.; Byrne, P.O.; Connacher, M.K.; Wang, Z.; Ramek, A.; Sarabipour, S.; Shan, Y.; Shaw, D.E.; Hristova, K.; et al. How IGF-1 activates its receptor. eLife 2014, 3, e03772. [Google Scholar] [CrossRef]
- Nadimpalli, S.K.; Amancha, P.K. Evolution of mannose 6-phosphate receptors (MPR300 and 46): Lysosomal enzyme sorting proteins. Curr. Protein Pept. Sci. 2010, 11, 68–90. [Google Scholar] [CrossRef] [PubMed]
- El-Shewy, H.M.; Luttrell, L.M. Insulin-like growth factor-2/mannose-6 phosphate receptors. Vitam. Horm. 2009, 80, 667–697. [Google Scholar] [CrossRef] [PubMed]
- Frasca, F.; Pandini, G.; Scalia, P.; Sciacca, L.; Mineo, R.; Costantino, A.; Goldfine, I.D.; Belfiore, A.; Vigneri, R. Insulin receptor isoform A, a newly recognized, high-affinity insulin-like growth factor II receptor in fetal and cancer cells. Mol. Cell. Biol. 1999, 19, 3278–3288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, M.; Nørgaard-Pedersen, D.; Brandt, J.; Pettersson, I.; Slaaby, R. IGF1 and IGF2 specificities to the two insulin receptor isoforms are determined by insulin receptor amino acid 718. PLoS ONE 2017, 12, e0178885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, T.E.; Epa, V.C.; Garrett, T.P.; Ward, C.W. Structure and function of the type 1 insulin-like growth factor receptor. Cell. Mol. Life Sci. 2000, 57, 1050–1093. [Google Scholar] [CrossRef]
- Nissley, S.P.; Rechler, M.M. Somatomedin/insulin-like growth factor tissue receptors. Clin. Endocrinol. Metab. 1984, 13, 43–67. [Google Scholar] [CrossRef]
- Kar, S.; Seto, D.; Doré, S.; Hanisch, U.; Quirion, R. Insulin-like growth factors-I and -II differentially regulate endogenous acetylcholine release from the rat hippocampal formation. Proc. Natl. Acad. Sci. USA 1997, 94, 14054–14059. [Google Scholar] [CrossRef] [Green Version]
- Olson, L.J.; Castonguay, A.C.; Lasanajak, Y.; Peterson, F.C.; Cummings, R.D.; Smith, D.F.; Dahms, N.M. Identification of a fourth mannose 6-phosphate binding site in the cation-independent mannose 6-phosphate receptor. Glycobiology 2015, 25, 591–606. [Google Scholar] [CrossRef]
- Harris, L.K.; Westwood, M. Biology and significance of signalling pathways activated by IGF-II. Growth Factors 2012, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Morrione, A.; Valentinis, B.; Xu, S.-q.; Yumet, G.; Louvi, A.; Efstratiadis, A.; Baserga, R. Insulin-like growth factor II stimulates cell proliferation through the insulin receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 3777–3782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.-W.; Pandey, K.; Katzman, A.C.; Alberini, C.M. A role for CIM6P/IGF2 receptor in memory consolidation and enhancement. eLife 2020, 9, e54781. [Google Scholar] [CrossRef]
- Fernandez, A.M.; Torres-Alemán, I. The many faces of insulin-like peptide signalling in the brain. Nat. Rev. Neurosci. 2012, 13, 225–239. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar] [CrossRef] [Green Version]
- Kappeler, L.; De Magalhaes Filho, C.; Dupont, J.; Leneuve, P.; Cervera, P.; Périn, L.; Loudes, C.; Blaise, A.; Klein, R.; Epelbaum, J.; et al. Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol. 2008, 6, e254. [Google Scholar] [CrossRef]
- Bondy, C.A.; Werner, H.; Roberts, C.T., Jr.; LeRoith, D. Cellular pattern of insulin-like growth factor-I (IGF-I) and type I IGF receptor gene expression in early organogenesis: Comparison with IGF-II gene expression. Mol. Endocrinol. 1990, 4, 1386–1398. [Google Scholar] [CrossRef] [PubMed]
- Sinor, A.D.; Lillien, L. Akt-1 expression level regulates CNS precursors. J. Neurosci. 2004, 24, 8531–8541. [Google Scholar] [CrossRef]
- Kulik, G.; Weber, M.J. Akt-dependent and -independent survival signaling pathways utilized by insulin-like growth factor I. Mol. Cell. Biol. 1998, 18, 6711–6718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ness, J.K.; Scaduto, R.C., Jr.; Wood, T.L. IGF-I prevents glutamate-mediated bax translocation and cytochrome C release in O4+ oligodendrocyte progenitors. Glia 2004, 46, 183–194. [Google Scholar] [CrossRef]
- Han, J.; Wang, B.; Xiao, Z.; Gao, Y.; Zhao, Y.; Zhang, J.; Chen, B.; Wang, X.; Dai, J. Mammalian target of rapamycin (mTOR) is involved in the neuronal differentiation of neural progenitors induced by insulin. Mol. Cell. Neurosci. 2008, 39, 118–124. [Google Scholar] [CrossRef]
- Samuels, I.S.; Karlo, J.C.; Faruzzi, A.N.; Pickering, K.; Herrup, K.; Sweatt, J.D.; Saitta, S.C.; Landreth, G.E. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J. Neurosci. 2008, 28, 6983–6995. [Google Scholar] [CrossRef] [PubMed]
- O’Kusky, J.; Ye, P. Neurodevelopmental effects of insulin-like growth factor signaling. Front. Neuroendocrinol. 2012, 33, 230–251. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Chen, R.; Wu, L.; Chen, Q.; Hu, A.; Zhang, T.; Wang, Z.; Zhu, X. The regulatory mechanism of neurogenesis by IGF-1 in adult mice. Mol. Neurobiol. 2015, 51, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Lichtenwalner, R.J.; Forbes, M.E.; Bennett, S.A.; Lynch, C.D.; Sonntag, W.E.; Riddle, D.R. Intracerebroventricular infusion of insulin-like growth factor-I ameliorates the age-related decline in hippocampal neurogenesis. Neuroscience 2001, 107, 603–613. [Google Scholar] [CrossRef]
- Gazit, N.; Vertkin, I.; Shapira, I.; Helm, M.; Slomowitz, E.; Sheiba, M.; Mor, Y.; Rizzoli, S.; Slutsky, I. IGF-1 Receptor Differentially Regulates Spontaneous and Evoked Transmission via Mitochondria at Hippocampal Synapses. Neuron 2016, 89, 583–597. [Google Scholar] [CrossRef] [Green Version]
- Xing, C.; Yin, Y.; Chang, R.; Gong, X.; He, X.; Xie, Z. Effects of insulin-like growth factor 1 on synaptic excitability in cultured rat hippocampal neurons. Exp. Neurol. 2007, 205, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Logan, S.; Pharaoh, G.A.; Marlin, M.C.; Masser, D.R.; Matsuzaki, S.; Wronowski, B.; Yeganeh, A.; Parks, E.E.; Premkumar, P.; Farley, J.A.; et al. Insulin-like growth factor receptor signaling regulates working memory, mitochondrial metabolism, and amyloid-β uptake in astrocytes. Mol. Metab. 2018, 9, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Garzón, E.; Fernandez, A.M.; Perez-Alvarez, A.; Genis, L.; Bascuñana, P.; Fernandez de la Rosa, R.; Delgado, M.; Angel Pozo, M.; Moreno, E.; McCormick, P.J.; et al. The insulin-like growth factor I receptor regulates glucose transport by astrocytes. Glia 2016, 64, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Vaynman, S.; Akhavan, M.; Ying, Z.; Gomez-Pinilla, F. Insulin-like growth factor I interfaces with brain-derived neurotrophic factor-mediated synaptic plasticity to modulate aspects of exercise-induced cognitive function. Neuroscience 2006, 140, 823–833. [Google Scholar] [CrossRef]
- Soto, M.; Cai, W.; Konishi, M.; Kahn, C.R. Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc. Natl. Acad. Sci. USA 2019, 116, 6379–6384. [Google Scholar] [CrossRef] [Green Version]
- Nishijima, T.; Piriz, J.; Duflot, S.; Fernandez, A.M.; Gaitan, G.; Gomez-Pinedo, U.; Verdugo, J.M.; Leroy, F.; Soya, H.; Nuñez, A.; et al. Neuronal activity drives localized blood-brain-barrier transport of serum insulin-like growth factor-I into the CNS. Neuron 2010, 67, 834–846. [Google Scholar] [CrossRef] [Green Version]
- Voss, M.W.; Erickson, K.I.; Prakash, R.S.; Chaddock, L.; Kim, J.S.; Alves, H.; Szabo, A.; Phillips, S.M.; Wójcicki, T.R.; Mailey, E.L.; et al. Neurobiological markers of exercise-related brain plasticity in older adults. Brain Behav. Immun. 2013, 28, 90–99. [Google Scholar] [CrossRef] [Green Version]
- Stern, S.A.; Chen, D.Y.; Alberini, C.M. The effect of insulin and insulin-like growth factors on hippocampus- and amygdala-dependent long-term memory formation. Learn. Mem. 2014, 21, 556–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Géloën, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Azpurua, J.; Yang, J.-N.; Van Meter, M.; Liu, Z.; Kim, J.; Lobo Ladd, A.A.B.; Coppi, A.A.; Gorbunova, V.; Seluanov, A. IGF1R levels in the brain negatively correlate with longevity in 16 rodent species. Aging 2013, 5, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.; Dillin, A. The insulin paradox: Aging, proteotoxicity and neurodegeneration. Nat. Rev. Neurosci. 2008, 9, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banks, W.A.; Owen, J.B.; Erickson, M.A. Insulin in the brain: There and back again. Pharmacol. Ther. 2012, 136, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Brüning, J.C.; Gautam, D.; Burks, D.J.; Gillette, J.; Schubert, M.; Orban, P.C.; Klein, R.; Krone, W.; Müller-Wieland, D.; Kahn, C.R. Role of brain insulin receptor in control of body weight and reproduction. Science 2000, 289, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Serrano, R.; Villar, M.; Martínez, C.; Carrascosa, J.M.; Gallardo, N.; Andrés, A. Differential gene expression of insulin receptor isoforms A and B and insulin receptor substrates 1, 2 and 3 in rat tissues: Modulation by aging and differentiation in rat adipose tissue. J. Mol. Endocrinol. 2005, 34, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Garwood, C.J.; Ratcliffe, L.E.; Morgan, S.V.; Simpson, J.E.; Owens, H.; Vazquez-Villaseñor, I.; Heath, P.R.; Romero, I.A.; Ince, P.G.; Wharton, S.B. Insulin and IGF1 signalling pathways in human astrocytes in vitro and in vivo; characterisation, subcellular localisation and modulation of the receptors. Mol. Brain 2015, 8, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, B.; Rank, L.; Metcalf, J.; Desplats, P. Identification of Insulin Receptor Splice Variant B in Neurons by in situ Detection in Human Brain Samples. Sci. Rep. 2018, 8, 4070. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.W.; Mudd, L.; Boyd, F.T., Jr.; Fields, M.; Raizada, M.K. Insulin is released from rat brain neuronal cells in culture. J. Neurochem. 1986, 47, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.M.; Hernandez-Garzón, E.; Perez-Domper, P.; Perez-Alvarez, A.; Mederos, S.; Matsui, T.; Santi, A.; Trueba-Saiz, A.; García-Guerra, L.; Pose-Utrilla, J.; et al. Insulin Regulates Astrocytic Glucose Handling Through Cooperation With IGF-I. Diabetes 2017, 66, 64–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Cáceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heidenreich, K.A.; Brandenburg, D. Oligosaccharide heterogeneity of insulin receptors. Comparison of N-linked glycosylation of insulin receptors in adipocytes and brain. Endocrinology 1986, 118, 1835–1842. [Google Scholar] [CrossRef]
- Dridi, L.; Seyrantepe, V.; Fougerat, A.; Pan, X.; Bonneil, E.; Thibault, P.; Moreau, A.; Mitchell, G.A.; Heveker, N.; Cairo, C.W.; et al. Positive regulation of insulin signaling by neuraminidase 1. Diabetes 2013, 62, 2338–2346. [Google Scholar] [CrossRef] [Green Version]
- Grillo, C.A.; Piroli, G.G.; Kaigler, K.F.; Wilson, S.P.; Wilson, M.A.; Reagan, L.P. Downregulation of hypothalamic insulin receptor expression elicits depressive-like behaviors in rats. Behav. Brain Res. 2011, 222, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Grillo, C.A.; Piroli, G.G.; Lawrence, R.C.; Wrighten, S.A.; Green, A.J.; Wilson, S.P.; Sakai, R.R.; Kelly, S.J.; Wilson, M.A.; Mott, D.D.; et al. Hippocampal Insulin Resistance Impairs Spatial Learning and Synaptic Plasticity. Diabetes 2015, 64, 3927–3936. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Chen, H.; Xu, H.; Moore, E.; Meiri, N.; Quon, M.J.; Alkon, D.L. Brain insulin receptors and spatial memory. Correlated changes in gene expression, tyrosine phosphorylation, and signaling molecules in the hippocampus of water maze trained rats. J. Biol. Chem. 1999, 274, 34893–34902. [Google Scholar] [CrossRef] [Green Version]
- Dou, J.T.; Chen, M.; Dufour, F.; Alkon, D.L.; Zhao, W.Q. Insulin receptor signaling in long-term memory consolidation following spatial learning. Learn. Mem. 2005, 12, 646–655. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, A.N.; Chidambaram, S.; Forbes, B.E.; Wood, T.L.; Levison, S.W. Insulin-like growth factor-II (IGF-II) and IGF-II analogs with enhanced insulin receptor-a binding affinity promote neural stem cell expansion. J. Biol. Chem. 2014, 289, 4626–4633. [Google Scholar] [CrossRef] [Green Version]
- Pandini, G.; Conte, E.; Medico, E.; Sciacca, L.; Vigneri, R.; Belfiore, A. IGF-II binding to insulin receptor isoform A induces a partially different gene expression profile from insulin binding. Ann. NY Acad. Sci. 2004, 1028, 450–456. [Google Scholar] [CrossRef]
- Sacco, A.; Morcavallo, A.; Pandini, G.; Vigneri, R.; Belfiore, A. Differential signaling activation by insulin and insulin-like growth factors I and II upon binding to insulin receptor isoform A. Endocrinology 2009, 150, 3594–3602. [Google Scholar] [CrossRef] [Green Version]
- Scalia, P.; Giordano, A.; Martini, C.; Williams, S.J. Isoform- and Paralog-Switching in IR-Signaling: When Diabetes Opens the Gates to Cancer. Biomolecules 2020, 10, 1617. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Fung, M.R.; Barlow, D.P.; Wagner, E.F. Regulation of embryonic growth and lysosomal targeting by the imprinted Igf2/Mpr gene. Nature 1994, 372, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, T.; Eggenschwiler, J.; Fisher, P.; D’Ercole, A.J.; Davenport, M.L.; Efstratiadis, A. Mouse mutants lacking the type 2 IGF receptor (IGF2R) are rescued from perinatal lethality in Igf2 and Igf1r null backgrounds. Dev. Biol. 1996, 177, 517–535. [Google Scholar] [CrossRef] [Green Version]
- Nolan, C.M.; Kyle, J.W.; Watanabe, H.; Sly, W.S. Binding of insulin-like growth factor II (IGF-II) by human cation-independent mannose 6-phosphate receptor/IGF-II receptor expressed in receptor-deficient mouse L cells. Cell Regul. 1990, 1, 197–213. [Google Scholar] [CrossRef] [Green Version]
- Sleat, D.E.; Lackland, H.; Wang, Y.; Sohar, I.; Xiao, G.; Li, H.; Lobel, P. The human brain mannose 6-phosphate glycoproteome: A complex mixture composed of multiple isoforms of many soluble lysosomal proteins. Proteomics 2005, 5, 1520–1532. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Dahms, N.M.; Kornfeld, S. Mannose 6-phosphate receptors: New twists in the tale. Nat. Rev. Mol. Cell Biol. 2003, 4, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Seaman, M.N.J. Retromer and the cation-independent mannose 6-phosphate receptor-Time for a trial separation? Traffic 2018, 19, 150–152. [Google Scholar] [CrossRef]
- Puertollano, R.; Aguilar, R.C.; Gorshkova, I.; Crouch, R.J.; Bonifacino, J.S. Sorting of mannose 6-phosphate receptors mediated by the GGAs. Science 2001, 292, 1712–1716. [Google Scholar] [CrossRef]
- Kucera, A.; Borg Distefano, M.; Berg-Larsen, A.; Skjeldal, F.; Repnik, U.; Bakke, O.; Progida, C. Spatiotemporal Resolution of Rab9 and CI-MPR Dynamics in the Endocytic Pathway. Traffic 2016, 17, 211–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Gabin, A.G.; Yin, X.; Si, Q.; Larocca, J.N. Transport of mannose-6-phosphate receptors from the trans-Golgi network to endosomes requires Rab31. Exp. Cell Res. 2009, 315, 2215–2230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonetti, B.; Danson, C.M.; Heesom, K.J.; Cullen, P.J. Sequence-dependent cargo recognition by SNX-BARs mediates retromer-independent transport of CI-MPR. J. Cell Biol. 2017, 216, 3695–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leksa, V.; Ilková, A.; Vičíková, K.; Stockinger, H. Unravelling novel functions of the endosomal transporter mannose 6-phosphate/insulin-like growth factor receptor (CD222) in health and disease: An emerging regulator of the immune system. Immunol. Lett. 2017, 190, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, P.; Kornfeld, S. The cytoplasmic tail of the cation-independent mannose 6-phosphate receptor contains four binding sites for AP-1. Arch. Biochem. Biophys. 2004, 426, 225–230. [Google Scholar] [CrossRef]
- Blanchard, F.; Duplomb, L.; Raher, S.; Vusio, P.; Hoflack, B.; Jacques, Y.; Godard, A. Mannose 6-Phosphate/Insulin-like growth factor II receptor mediates internalization and degradation of leukemia inhibitory factor but not signal transduction. J. Biol. Chem. 1999, 274, 24685–24693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheel, G.; Herzog, V. Mannose 6-phosphate receptor in porcine thyroid follicle cells. Localization and possible implications for the intracellular transport of thyroglobulin. Eur. J. Cell Biol. 1989, 49, 140–148. [Google Scholar]
- Van Kesteren, C.A.; Danser, A.H.; Derkx, F.H.; Dekkers, D.H.; Lamers, J.M.; Saxena, P.R.; Schalekamp, M.A. Mannose 6-phosphate receptor-mediated internalization and activation of prorenin by cardiac cells. Hypertension 1997, 30, 1389–1396. [Google Scholar] [CrossRef]
- Lee, S.J.; Nathans, D. Proliferin secreted by cultured cells binds to mannose 6-phosphate receptors. J. Biol. Chem. 1988, 263, 3521–3527. [Google Scholar] [CrossRef]
- Groskopf, J.C.; Syu, L.J.; Saltiel, A.R.; Linzer, D.I. Proliferin induces endothelial cell chemotaxis through a G protein-coupled, mitogen-activated protein kinase-dependent pathway. Endocrinology 1997, 138, 2835–2840. [Google Scholar] [CrossRef]
- Braulke, T.; Tippmer, S.; Neher, E.; von Figura, K. Regulation of the mannose 6-phosphate/IGF II receptor expression at the cell surface by mannose 6-phosphate, insulin like growth factors and epidermal growth factor. EMBO J. 1989, 8, 681–686. [Google Scholar] [CrossRef]
- York, S.J.; Arneson, L.S.; Gregory, W.T.; Dahms, N.M.; Kornfeld, S. The rate of internalization of the mannose 6-phosphate/insulin-like growth factor II receptor is enhanced by multivalent ligand binding. J. Biol. Chem. 1999, 274, 1164–1171. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Murayama, Y.; Okamoto, T.; Yokota, T.; Ikezu, T.; Takahashi, S.; Giambarella, U.; Ogata, E.; Nishimoto, I. Conversion of G-protein specificity of insulin-like growth factor II/mannose 6-phosphate receptor by exchanging of a short region with beta-adrenergic receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 11772–11776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikezu, T.; Okamoto, T.; Giambarella, U.; Yokota, T.; Nishimoto, I. In vivo coupling of insulin-like growth factor II/mannose 6-phosphate receptor to heteromeric G proteins. Distinct roles of cytoplasmic domains and signal sequestration by the receptor. J. Biol. Chem. 1995, 270, 29224–29228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murayama, Y.; Okamoto, T.; Ogata, E.; Asano, T.; Iiri, T.; Katada, T.; Ui, M.; Grubb, J.H.; Sly, W.S.; Nishimoto, I. Distinctive regulation of the functional linkage between the human cation-independent mannose 6-phosphate receptor and GTP-binding proteins by insulin-like growth factor II and mannose 6-phosphate. J. Biol. Chem. 1990, 265, 17456–17462. [Google Scholar] [CrossRef]
- Chu, C.H.; Tzang, B.S.; Chen, L.M.; Liu, C.J.; Tsai, F.J.; Tsai, C.H.; Lin, J.A.; Kuo, W.W.; Bau, D.T.; Yao, C.H.; et al. Activation of insulin-like growth factor II receptor induces mitochondrial-dependent apoptosis through G(alpha)q and downstream calcineurin signaling in myocardial cells. Endocrinology 2009, 150, 2723–2731. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Tosh, D.N.; Zhang, S.; McMillen, I.C.; Duffield, J.A.; Brooks, D.A.; Morrison, J.L. IGF-2R-Gαq signaling and cardiac hypertrophy in the low-birth-weight lamb. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 308, R627–R635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.C.; Botting, K.J.; Padhee, M.; Zhang, S.; McMillen, I.C.; Suter, C.M.; Brooks, D.A.; Morrison, J.L. Early origins of heart disease: Low birth weight and the role of the insulin-like growth factor system in cardiac hypertrophy. Clin. Exp. Pharmacol. Physiol. 2012, 39, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Brooks, D.A.; Botting, K.J.; Morrison, J.L. IGF-2R-mediated signaling results in hypertrophy of cultured cardiomyocytes from fetal sheep. Biol. Reprod. 2012, 86, 183. [Google Scholar] [CrossRef]
- Wang, K.C.; Brooks, D.A.; Thornburg, K.L.; Morrison, J.L. Activation of IGF-2R stimulates cardiomyocyte hypertrophy in the late gestation sheep fetus. J. Physiol. 2012, 590, 5425–5437. [Google Scholar] [CrossRef]
- Körner, C.; Nürnberg, B.; Uhde, M.; Braulke, T. Mannose 6-phosphate/insulin-like growth factor II receptor fails to interact with G-proteins. Analysis of mutant cytoplasmic receptor domains. J. Biol. Chem. 1995, 270, 287–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konishi, Y.; Fushimi, S.; Shirabe, T. Immunohistochemical distribution of cation-dependent mannose 6-phosphate receptors in the mouse central nervous system: Comparison with that of cation-independent mannose 6-phophate receptors. Neurosci. Lett. 2005, 378, 7–12. [Google Scholar] [CrossRef]
- Couce, M.E.; Weatherington, A.J.; McGinty, J.F. Expression of insulin-like growth factor-II (IGF-II) and IGF-II/mannose-6-phosphate receptor in the rat hippocampus: An in situ hybridization and immunocytochemical study. Endocrinology 1992, 131, 1636–1642. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.; Kar, S. Insulin-like growth factor-II/mannose-6-phosphate receptor: Widespread distribution in neurons of the central nervous system including those expressing cholinergic phenotype. J. Comp. Neurol. 2003, 458, 113–127. [Google Scholar] [CrossRef]
- Fushimi, S.; Shirabe, T. Expression of insulin-like growth factors in remyelination following ethidium bromide-induced demyelination in the mouse spinal cord. Neuropathology 2004, 24, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.S.; Cosenza-Nashat, M.; Choi, N.; Zhao, M.L.; Li, J.F.; Pollard, J.W.; Jirtle, R.L.; Goldstein, H.; Lee, S.C. Insulin-like growth factor 2 receptor is an IFNgamma-inducible microglial protein that facilitates intracellular HIV replication: Implications for HIV-induced neurocognitive disorders. Am. J. Pathol. 2010, 177, 2446–2458. [Google Scholar] [CrossRef]
- Cheng, X.-T.; Xie, Y.-X.; Zhou, B.; Huang, N.; Farfel-Becker, T.; Sheng, Z.-H. Characterization of LAMP1-labeled nondegradative lysosomal and endocytic compartments in neurons. J. Cell Biol. 2018, 217, 3127–3139. [Google Scholar] [CrossRef] [Green Version]
- Sahu, A.; Dube, M.G.; Phelps, C.P.; Sninsky, C.A.; Kalra, P.S.; Kalra, S.P. Insulin and insulin-like growth factor II suppress neuropeptide Y release from the nerve terminals in the paraventricular nucleus: A putative hypothalamic site for energy homeostasis. Endocrinology 1995, 136, 5718–5724. [Google Scholar] [CrossRef]
- Hawkes, C.; Jhamandas, J.H.; Harris, K.H.; Fu, W.; MacDonald, R.G.; Kar, S. Single transmembrane domain insulin-like growth factor-II/mannose-6-phosphate receptor regulates central cholinergic function by activating a G-protein-sensitive, protein kinase C-dependent pathway. J. Neurosci. 2006, 26, 585–596. [Google Scholar] [CrossRef]
- Amritraj, A.; Rauw, G.; Baker, G.B.; Kar, S. Leu27 insulin-like growth factor-II, an insulin-like growth factor-II analog, attenuates depolarization-evoked GABA release from adult rat hippocampal and cortical slices. Neuroscience 2010, 170, 722–730. [Google Scholar] [CrossRef]
- Amritraj, A.; Posse de Chaves, E.I.; Hawkes, C.; Macdonald, R.G.; Kar, S. Single-transmembrane domain IGF-II/M6P receptor: Potential interaction with G protein and its association with cholesterol-rich membrane domains. Endocrinology 2012, 153, 4784–4798. [Google Scholar] [CrossRef] [Green Version]
- Cruz, E.; Descalzi, G.; Steinmetz, A.; Scharfman, H.E.; Katzman, A.; Alberini, C.M. CIM6P/IGF-2 Receptor Ligands Reverse Deficits in Angelman Syndrome Model Mice. Autism Res. 2020. [Google Scholar] [CrossRef]
- Napoli, I.; Blusztajn, J.K.; Mellott, T.J. Prenatal choline supplementation in rats increases the expression of IGF2 and its receptor IGF2R and enhances IGF2-induced acetylcholine release in hippocampus and frontal cortex. Brain Res. 2008, 1237, 124–135. [Google Scholar] [CrossRef]
- Konishi, Y.; Takahashi, K.; Chui, D.H.; Rosenfeld, R.G.; Himeno, M.; Tabira, T. Insulin-like growth factor II promotes in vitro cholinergic development of mouse septal neurons: Comparison with the effects of insulin-like growth factor I. Brain Res. 1994, 649, 53–61. [Google Scholar] [CrossRef]
- Hawkes, C.; Kabogo, D.; Amritraj, A.; Kar, S. Up-regulation of cation-independent mannose 6-phosphate receptor and endosomal-lysosomal markers in surviving neurons after 192-IgG-saporin administrations into the adult rat brain. Am. J. Pathol. 2006, 169, 1140–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, R.A.; Cataldo, A.M. Lysosomal system pathways: Genes to neurodegeneration in Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 277–289. [Google Scholar] [CrossRef]
- Matrone, C.; Dzamko, N.; Madsen, P.; Nyegaard, M.; Pohlmann, R.; Søndergaard, R.V.; Lassen, L.B.; Andresen, T.L.; Halliday, G.M.; Jensen, P.H.; et al. Mannose 6-Phosphate Receptor Is Reduced in -Synuclein Overexpressing Models of Parkinsons Disease. PLoS ONE 2016, 11, e0160501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amritraj, A.; Hawkes, C.; Phinney, A.L.; Mount, H.T.; Scott, C.D.; Westaway, D.; Kar, S. Altered levels and distribution of IGF-II/M6P receptor and lysosomal enzymes in mutant APP and APP + PS1 transgenic mouse brains. Neurobiol. Aging 2009, 30, 54–70. [Google Scholar] [CrossRef]
- van Rahden, V.A.; Brand, K.; Najm, J.; Heeren, J.; Pfeffer, S.R.; Braulke, T.; Kutsche, K. The 5-phosphatase OCRL mediates retrograde transport of the mannose 6-phosphate receptor by regulating a Rac1-cofilin signalling module. Hum. Mol. Genet. 2012, 21, 5019–5038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tammineni, P.; Jeong, Y.Y.; Feng, T.; Aikal, D.; Cai, Q. Impaired axonal retrograde trafficking of the retromer complex augments lysosomal deficits in Alzheimer’s disease neurons. Hum. Mol. Genet. 2017, 26, 4352–4366. [Google Scholar] [CrossRef] [PubMed]
- Mellott, T.J.; Pender, S.M.; Burke, R.M.; Langley, E.A.; Blusztajn, J.K. IGF2 ameliorates amyloidosis, increases cholinergic marker expression and raises BMP9 and neurotrophin levels in the hippocampus of the APPswePS1dE9 Alzheimer’s disease model mice. PLoS ONE 2014, 9, e94287. [Google Scholar] [CrossRef] [PubMed]
- Dobryakova, Y.V.; Volobueva, M.N.; Manolova, A.O.; Medvedeva, T.M.; Kvichansky, A.A.; Gulyaeva, N.V.; Markevich, V.A.; Stepanichev, M.Y.; Bolshakov, A.P. Cholinergic Deficit Induced by Central Administration of 192IgG-Saporin Is Associated With Activation of Microglia and Cell Loss in the Dorsal Hippocampus of Rats. Front. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Wiley, R.G.; Berbos, T.G.; Deckwerth, T.L.; Johnson, E.M.; Lappi, D.A. Destruction of the cholinergic basal forebrain using immunotoxin to rat NGF receptor: Modeling the cholinergic degeneration of Alzheimer’s disease. J. Neurol. Sci. 1995, 128, 157–166. [Google Scholar] [CrossRef]
- Wang, Y.; Buggia-Prévot, V.; Zavorka, M.E.; Bleackley, R.C.; MacDonald, R.G.; Thinakaran, G.; Kar, S. Overexpression of the Insulin-Like Growth Factor II Receptor Increases β-Amyloid Production and Affects Cell Viability. Mol. Cell. Biol. 2015, 35, 2368–2384. [Google Scholar] [CrossRef] [Green Version]
- Villaseñor, R.; Kalaidzidis, Y.; Zerial, M. Signal processing by the endosomal system. Curr. Opin. Cell Biol. 2016, 39, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Bingol, B.; Sheng, M. Deconstruction for reconstruction: The role of proteolysis in neural plasticity and disease. Neuron 2011, 69, 22–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Monreal, M.; Brown, T.C.; Royo, M.; Esteban, J.A. The balance between receptor recycling and trafficking toward lysosomes determines synaptic strength during long-term depression. J. Neurosci. 2012, 32, 13200–13205. [Google Scholar] [CrossRef] [Green Version]
- Goo, M.S.; Sancho, L.; Slepak, N.; Boassa, D.; Deerinck, T.J.; Ellisman, M.H.; Bloodgood, B.L.; Patrick, G.N. Activity-dependent trafficking of lysosomes in dendrites and dendritic spines. J. Cell Biol. 2017, 216, 2499–2513. [Google Scholar] [CrossRef] [Green Version]
- Padamsey, Z.; McGuinness, L.; Bardo, S.J.; Reinhart, M.; Tong, R.; Hedegaard, A.; Hart, M.L.; Emptage, N.J. Activity-Dependent Exocytosis of Lysosomes Regulates the Structural Plasticity of Dendritic Spines. Neuron 2017, 93, 132–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sleat, D.E.; Sohar, I.; Lackland, H.; Majercak, J.; Lobel, P. Rat brain contains high levels of mannose-6-phosphorylated glycoproteins including lysosomal enzymes and palmitoyl-protein thioesterase, an enzyme implicated in infantile neuronal lipofuscinosis. J. Biol. Chem. 1996, 271, 19191–19198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Bartholdson, S.J.; Couch, A.C.M.; Yusa, K.; Wright, G.J. Genome-scale identification of cellular pathways required for cell surface recognition. Genome Res. 2018, 28, 1372–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cioni, J.M.; Lin, J.Q.; Holtermann, A.V.; Koppers, M.; Jakobs, M.A.H.; Azizi, A.; Turner-Bridger, B.; Shigeoka, T.; Franze, K.; Harris, W.A.; et al. Late Endosomes Act as mRNA Translation Platforms and Sustain Mitochondria in Axons. Cell 2019, 176, 56–72. [Google Scholar] [CrossRef] [Green Version]
- Tsien, R.Y. Very long-term memories may be stored in the pattern of holes in the perineuronal net. Proc. Natl. Acad. Sci. USA 2013, 110, 12456–12461. [Google Scholar] [CrossRef] [Green Version]
- Ramos-DeSimone, N.; Hahn-Dantona, E.; Sipley, J.; Nagase, H.; French, D.L.; Quigley, J.P. Activation of matrix metalloproteinase-9 (MMP-9) via a converging plasmin/stromelysin-1 cascade enhances tumor cell invasion. J. Biol. Chem. 1999, 274, 13066–13076. [Google Scholar] [CrossRef] [Green Version]
- Michaluk, P.; Kaczmarek, L. Matrix metalloproteinase-9 in glutamate-dependent adult brain function and dysfunction. Cell Death Differ. 2007, 14, 1255–1258. [Google Scholar] [CrossRef]
- Davis, G.E.; Pintar Allen, K.A.; Salazar, R.; Maxwell, S.A. Matrix metalloproteinase-1 and -9 activation by plasmin regulates a novel endothelial cell-mediated mechanism of collagen gel contraction and capillary tube regression in three-dimensional collagen matrices. J. Cell Sci. 2001, 114, 917–930. [Google Scholar]
- Kostoulas, G.; Lang, A.; Nagase, H.; Baici, A. Stimulation of angiogenesis through cathepsin B inactivation of the tissue inhibitors of matrix metalloproteinases. FEBS Lett. 1999, 455, 286–290. [Google Scholar] [CrossRef] [Green Version]
- Murphy, N.; Lynch, M.A. Activation of the P2X₇ receptor induces migration of glial cells by inducing cathepsin B degradation of tissue inhibitor of metalloproteinase 1. J. Neurochem. 2012, 123, 761–770. [Google Scholar] [CrossRef]
- Godár, S.; Horejsi, V.; Weidle, U.H.; Binder, B.R.; Hansmann, C.; Stockinger, H. M6P/IGFII-receptor complexes urokinase receptor and plasminogen for activation of transforming growth factor-beta1. Eur. J. Immunol. 1999, 29, 1004–1013. [Google Scholar] [CrossRef]
- Mort, J.S. Chapter 406—Cathepsin, B. In Handbook of Proteolytic Enzymes, 3rd ed.; Rawlings, N.D., Salvesen, G., Eds.; Academic Press: London, UK, 2013; pp. 1784–1791. [Google Scholar] [CrossRef]
- Chang, M.H.; Kuo, W.W.; Chen, R.J.; Lu, M.C.; Tsai, F.J.; Kuo, W.H.; Chen, L.Y.; Wu, W.J.; Huang, C.Y.; Chu, C.H. IGF-II/mannose 6-phosphate receptor activation induces metalloproteinase-9 matrix activity and increases plasminogen activator expression in H9c2 cardiomyoblast cells. J. Mol. Endocrinol. 2008, 41, 65–74. [Google Scholar] [CrossRef] [Green Version]
- Beilharz, E.J.; Bassett, N.S.; Sirimanne, E.S.; Williams, C.E.; Gluckman, P.D. Insulin-like growth factor II is induced during wound repair following hypoxic-ischemic injury in the developing rat brain. Brain Res. Mol. Brain Res. 1995, 29, 81–91. [Google Scholar] [CrossRef]
- Gluckman, P.; Klempt, N.; Guan, J.; Mallard, C.; Sirimanne, E.; Dragunow, M.; Klempt, M.; Singh, K.; Williams, C.; Nikolics, K. A role for IGF-1 in the rescue of CNS neurons following hypoxic-ischemic injury. Biochem. Biophys. Res. Commun. 1992, 182, 593–599. [Google Scholar] [CrossRef]
- Gustafson, K.; Hagberg, H.; Bengtsson, B.A.; Brantsing, C.; Isgaard, J. Possible protective role of growth hormone in hypoxia-ischemia in neonatal rats. Pediatr. Res. 1999, 45, 318–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Bennet, L.; Gluckman, P.D.; Gunn, A.J. Insulin-like growth factor-1 and post-ischemic brain injury. Prog. Neurobiol. 2003, 70, 443–462. [Google Scholar] [CrossRef]
- Cheng, B.; Mattson, M.P. IGF-I and IGF-II protect cultured hippocampal and septal neurons against calcium-mediated hypoglycemic damage. J. Neurosci. 1992, 12, 1558–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattson, M.P.; Zhang, Y.; Bose, S. Growth factors prevent mitochondrial dysfunction, loss of calcium homeostasis, and cell injury, but not ATP depletion in hippocampal neurons deprived of glucose. Exp. Neurol. 1993, 121, 1–13. [Google Scholar] [CrossRef]
- García-Fernández, M.; Delgado, G.; Puche, J.E.; González-Barón, S.; Castilla Cortázar, I. Low doses of insulin-like growth factor I improve insulin resistance, lipid metabolism, and oxidative damage in aging rats. Endocrinology 2008, 149, 2433–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castilla-Cortázar, I.; García-Fernández, M.; Delgado, G.; Puche, J.E.; Sierra, I.; Barhoum, R.; González-Barón, S. Hepatoprotection and neuroprotection induced by low doses of IGF-II in aging rats. J. Transl. Med. 2011, 9, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, K.B.; Loddick, S.A.; Naeve, G.S.; Vana, A.M.; Verge, G.M.; Foster, A.C. Neuroprotective effects of insulin-like growth factor-binding protein ligand inhibitors in vitro and in vivo. J. Cereb. Blood Flow Metab. 2003, 23, 1160–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, J.; Williams, C.E.; Skinner, S.J.; Mallard, E.C.; Gluckman, P.D. The effects of insulin-like growth factor (IGF)-1, IGF-2, and des-IGF-1 on neuronal loss after hypoxic-ischemic brain injury in adult rats: Evidence for a role for IGF binding proteins. Endocrinology 1996, 137, 893–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vafaee, F.; Zarifkar, A.; Emamghoreishi, M.; Namavar, M.R.; Shirzad, S.; Ghazavi, H.; Mahdavizadeh, V. Insulin-Like Growth Factor 2 (IGF-2) Regulates Neuronal Density and IGF-2 Distribution Following Hippocampal Intracerebral Hemorrhage. J. Stroke Cerebrovasc. Dis. 2020, 29, 105128. [Google Scholar] [CrossRef] [PubMed]
- Cotman, C.W.; Berchtold, N.C.; Christie, L.A. Exercise builds brain health: Key roles of growth factor cascades and inflammation. Trends Neurosci. 2007, 30, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Trejo, J.L.; Carro, E.; Torres-Aleman, I. Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J. Neurosci. 2001, 21, 1628–1634. [Google Scholar] [CrossRef] [Green Version]
- Trejo, J.L.; Llorens-Martín, M.V.; Torres-Alemán, I. The effects of exercise on spatial learning and anxiety-like behavior are mediated by an IGF-I-dependent mechanism related to hippocampal neurogenesis. Mol. Cell. Neurosci. 2008, 37, 402–411. [Google Scholar] [CrossRef]
- Carro, E.; Trejo, J.L.; Busiguina, S.; Torres-Aleman, I. Circulating insulin-like growth factor I mediates the protective effects of physical exercise against brain insults of different etiology and anatomy. J. Neurosci. 2001, 21, 5678–5684. [Google Scholar] [CrossRef] [Green Version]
- Carro, E.; Nuñez, A.; Busiguina, S.; Torres-Aleman, I. Circulating insulin-like growth factor I mediates effects of exercise on the brain. J. Neurosci. 2000, 20, 2926–2933. [Google Scholar] [CrossRef]
- Allodi, I.; Comley, L.; Nichterwitz, S.; Nizzardo, M.; Simone, C.; Benitez, J.A.; Cao, M.; Corti, S.; Hedlund, E. Differential neuronal vulnerability identifies IGF-2 as a protective factor in ALS. Sci. Rep. 2016, 6, 25960. [Google Scholar] [CrossRef] [Green Version]
- Kaspar, B.K.; Lladó, J.; Sherkat, N.; Rothstein, J.D.; Gage, F.H. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science 2003, 301, 839–842. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, A.C.; Briquez, P.S.; Hubbell, J.A.; Cochran, J.R. Engineering growth factors for regenerative medicine applications. Acta Biomater. 2016, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Jiang, W. From One-Cell to Tissue: Reprogramming, Cell Differentiation and Tissue Engineering. Bioscience 2015, 65, 468–475. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.T.; Bendriem, R.M.; Wu, W.W.; Shen, R.F. 3D brain Organoids derived from pluripotent stem cells: Promising experimental models for brain development and neurodegenerative disorders. J. Biomed. Sci. 2017, 24, 59. [Google Scholar] [CrossRef] [PubMed]
- Pravtcheva, D.D.; Wise, T.L. Metastasizing mammary carcinomas in H19 enhancers-Igf2 transgenic mice. J. Exp. Zool. 1998, 281, 43–57. [Google Scholar] [CrossRef]
- Goldblatt, E.M.; Lee, W.-H. From bench to bedside: The growing use of translational research in cancer medicine. Am. J. Transl. Res. 2010, 2, 1–18. [Google Scholar]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Camirand, A.; Pollak, M. Co-targeting IGF-1R and c-kit: Synergistic inhibition of proliferation and induction of apoptosis in H 209 small cell lung cancer cells. Br. J. Cancer 2004, 90, 1825–1829. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Su, K.; Zhang, Y.; Zhang, W.; Zhao, Q.; Chu, D.; Guo, R. IR-A/IGF-1R-mediated signals promote epithelial-mesenchymal transition of endometrial carcinoma cells by activating PI3K/AKT and ERK pathways. Cancer Biol. Ther. 2019, 20, 295–306. [Google Scholar] [CrossRef]
- Jiang, J.; Ren, H.-Y.; Geng, G.-J.; Mi, Y.-J.; Liu, Y.; Li, N.; Yang, S.-Y.; Shen, D.-Y. Oncogenic activity of insulin in the development of non-small cell lung carcinoma. Oncol. Lett. 2018, 15, 447–452. [Google Scholar] [CrossRef]
- Yamada, N.; Matsuda, R.; Morishige, N.; Yanai, R.; Chikama, T.I.; Nishida, T.; Ishimitsu, T.; Kamiya, A. Open clinical study of eye-drops containing tetrapeptides derived from substance P and insulin-like growth factor-1 for treatment of persistent corneal epithelial defects associated with neurotrophic keratopathy. Br. J. Ophthalmol. 2008, 92, 896–900. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Identifier: NCT01207908. Safety and Efficacy Study of IGF-1 in Duchenne Muscular Dystrophy. Available online: https://www.clinicaltrials.gov/ct2/show/results/NCT01207908?term=igf-1&draw=2&rank=7 (accessed on 25 December 2020).
- ClinicalTrials.gov. Identifier: NCT01207908. A Pilot Treatment Study of Insulin-Like Growth Factor-1 (IGF-1) in Autism Spectrum Disorder. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01970345?term=igf-1&draw=2&rank=5 (accessed on 25 December 2020).
- Wise, T.L.; Pravtcheva, D.D. Delayed onset of Igf2-induced mammary tumors in Igf2r transgenic mice. Cancer Res. 2006, 66, 1327–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, J.; Burns, J.L.; Foulstone, E.J.; Pignatelli, M.; Zaina, S.; Hassan, A.B. Soluble IGF2 receptor rescues Apc(Min/+) intestinal adenoma progression induced by Igf2 loss of imprinting. Cancer Res. 2006, 66, 1940–1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, J.M.; Lian, W.S.; Qiu, M.K.; Dai, Y.X.; Dong, Q.; Shen, J.; Dong, P.; Wang, X.F.; Liu, Y.B.; Quan, Z.W.; et al. Knockdown of IGF2R suppresses proliferation and induces apoptosis in hemangioma cells in vitro and in vivo. Int. J. Oncol. 2014, 45, 1241–1249. [Google Scholar] [CrossRef] [Green Version]
- Thorne, R.G.; Emory, C.R.; Ala, T.A.; Frey, W.H., II. Quantitative analysis of the olfactory pathway for drug delivery to the brain. Brain Res. 1995, 692, 278–282. [Google Scholar] [CrossRef]
- Born, J.; Lange, T.; Kern, W.; McGregor, G.P.; Bickel, U.; Fehm, H.L. Sniffing neuropeptides: A transnasal approach to the human brain. Nat. Neurosci. 2002, 5, 514–516. [Google Scholar] [CrossRef]
- Illum, L. Transport of drugs from the nasal cavity to the central nervous system. Eur. J. Pharm. Sci. 2000, 11, 1–18. [Google Scholar] [CrossRef]
- Gomez, D.; Martinez, J.A.; Hanson, L.R.; Frey, W.H., 2nd; Toth, C.C. Intranasal treatment of neurodegenerative diseases and stroke. Front. Biosci. 2012, 4, 74–89. [Google Scholar] [CrossRef]
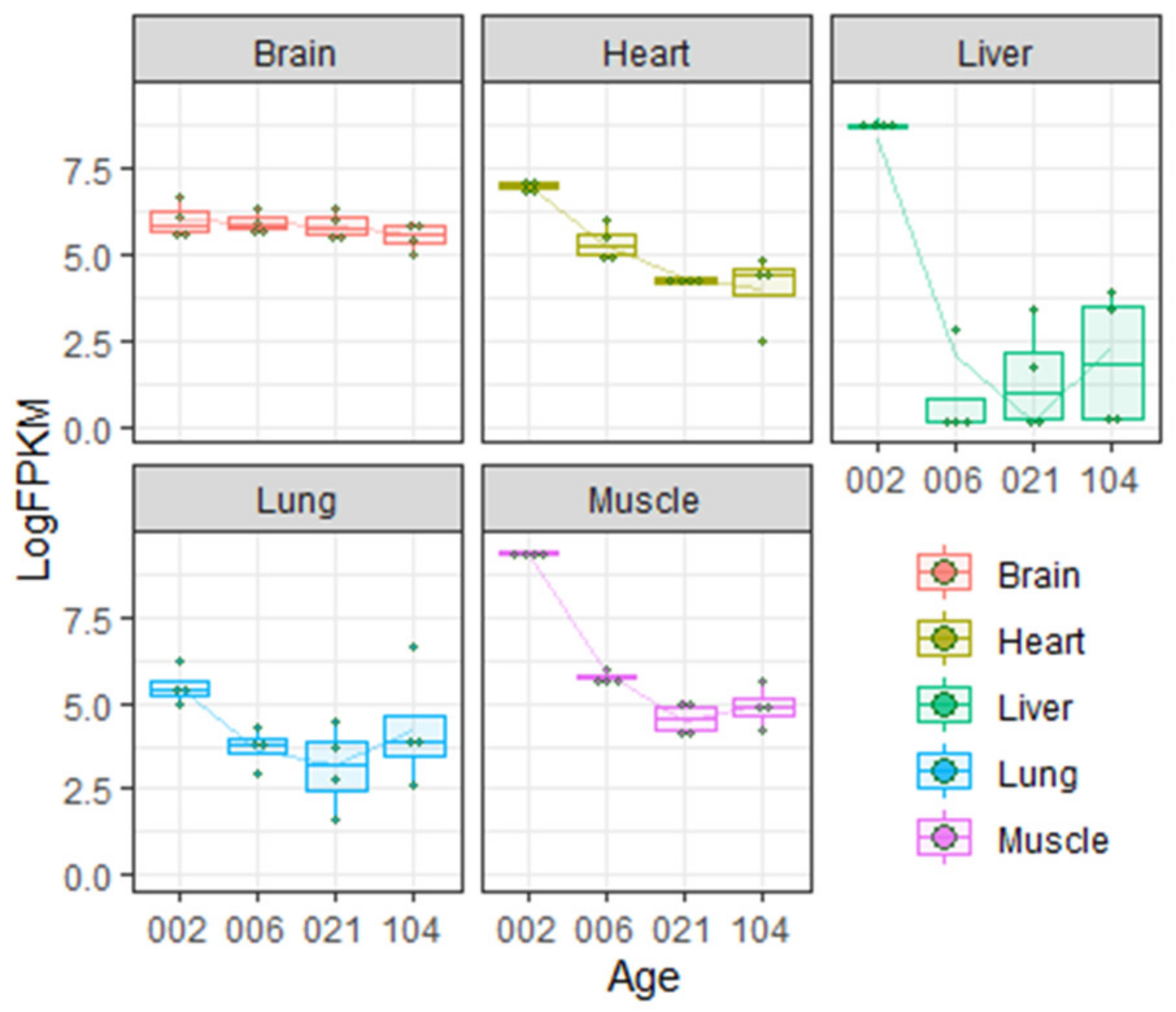



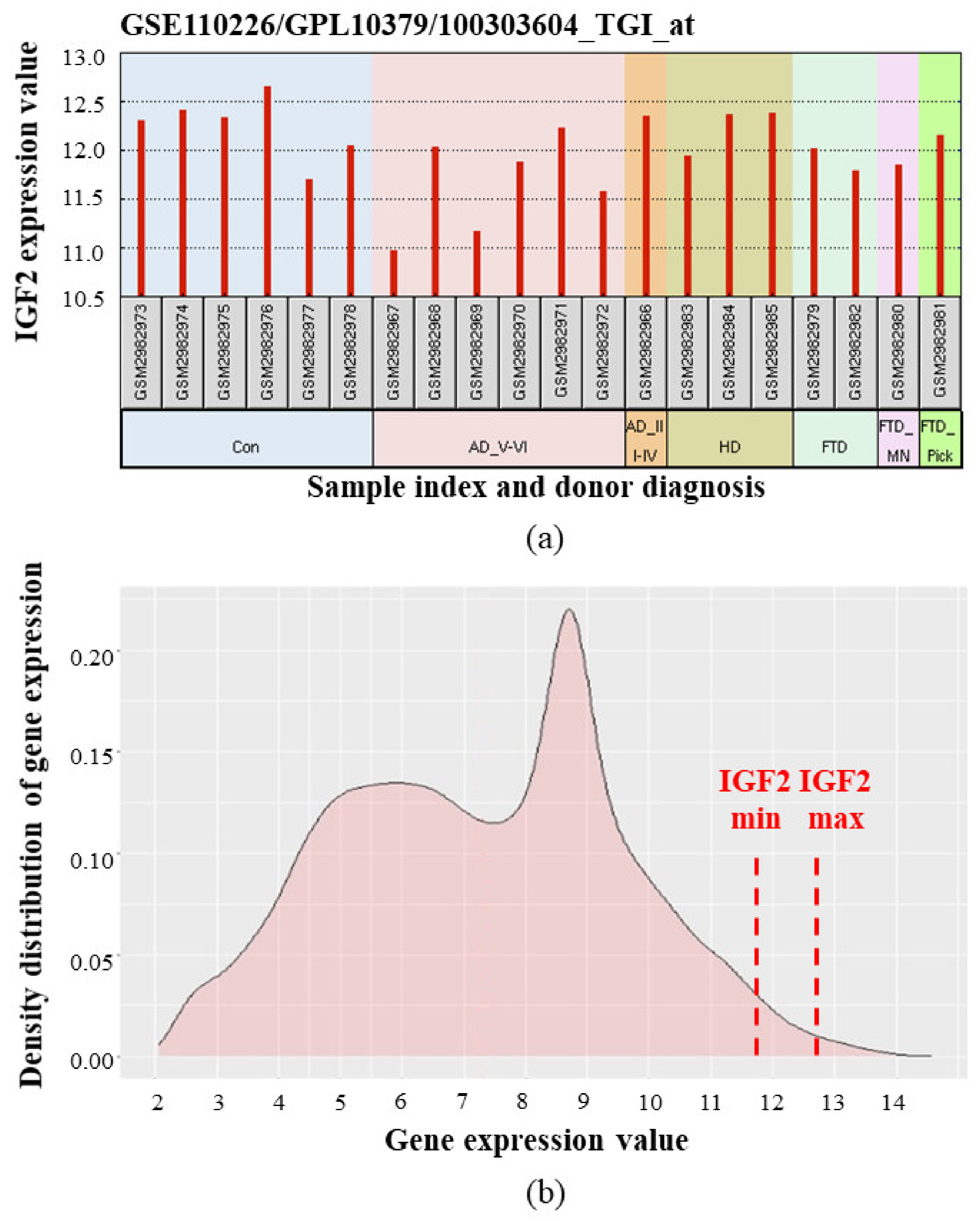
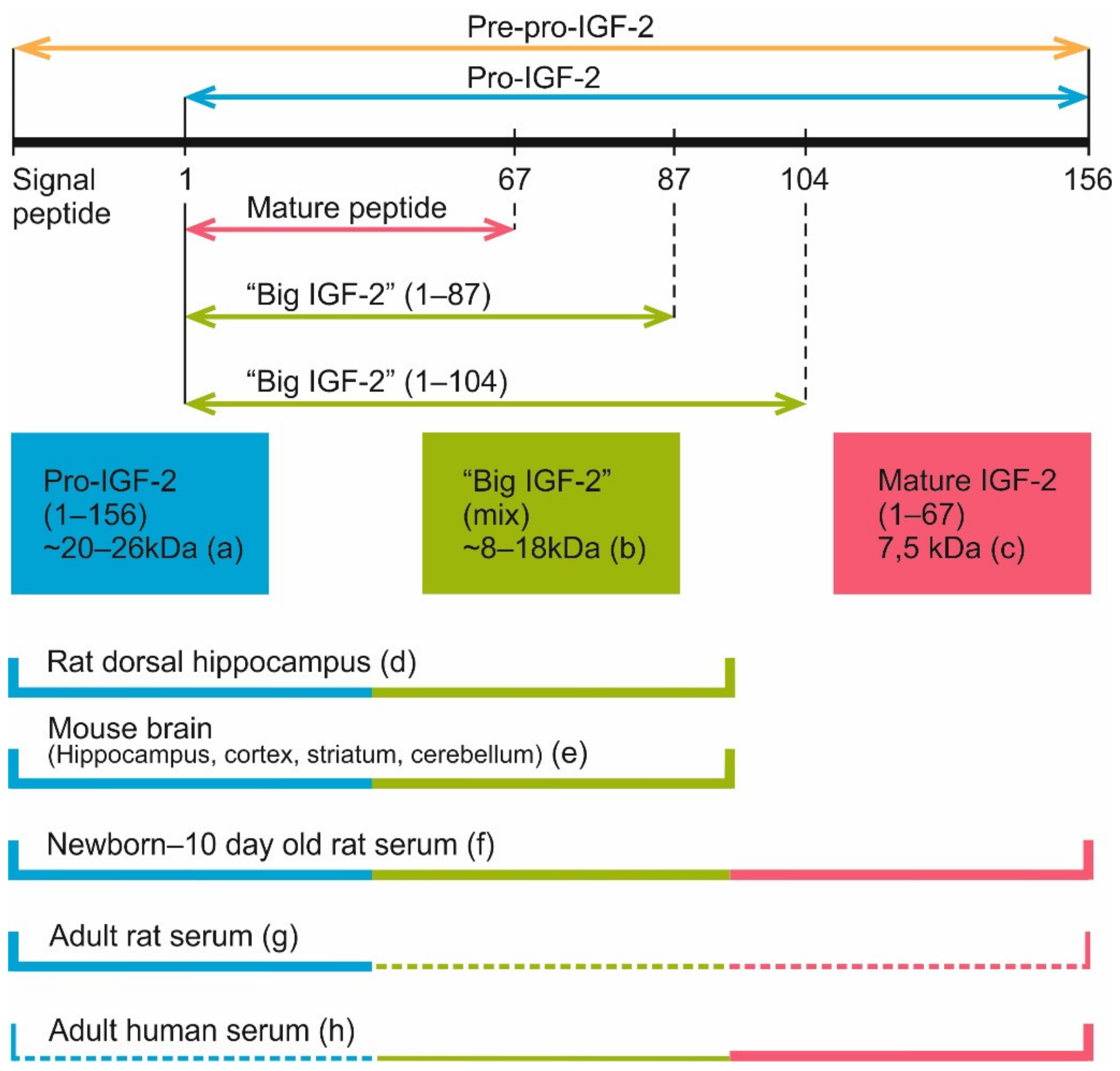
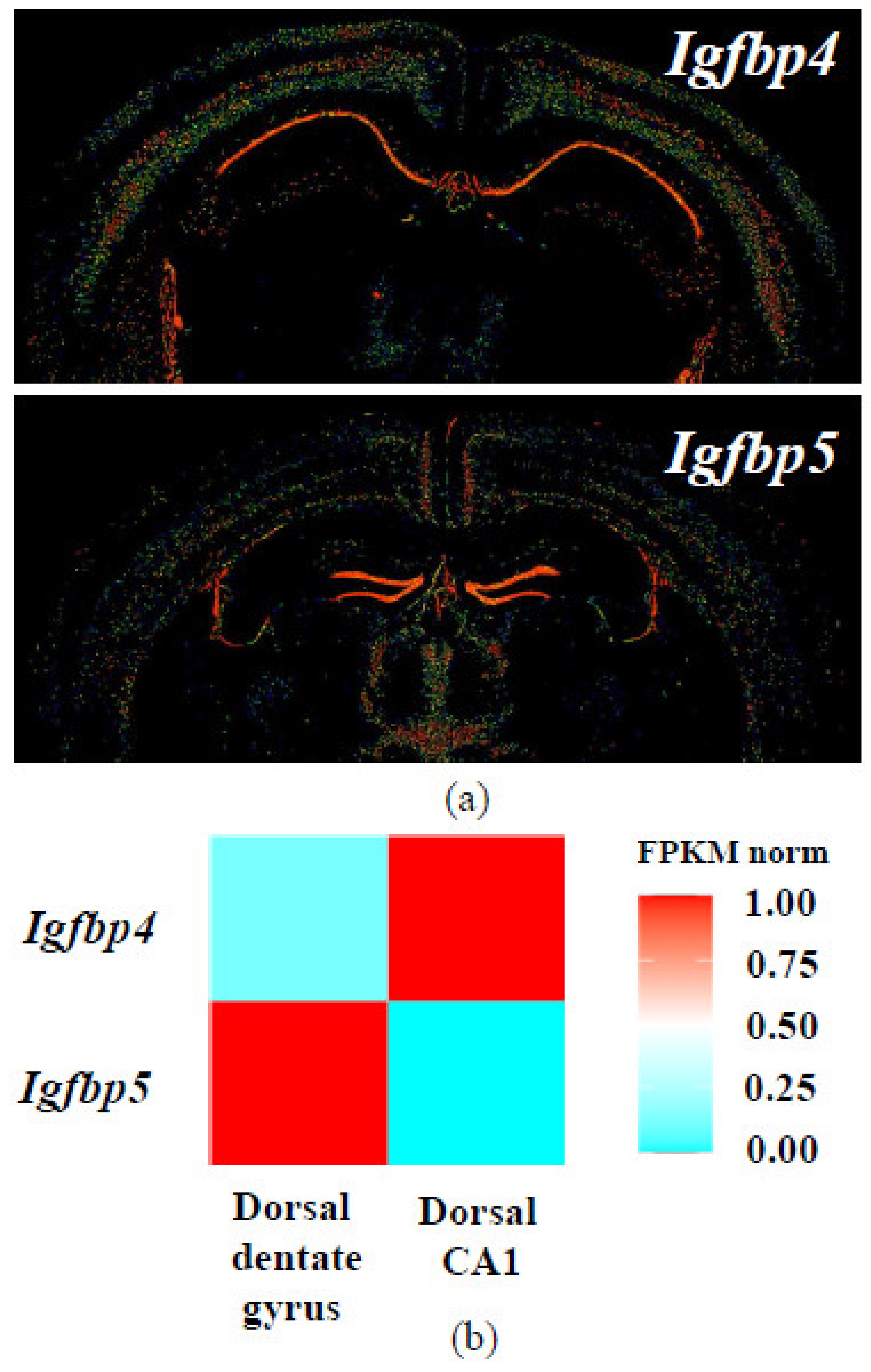
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beletskiy, A.; Chesnokova, E.; Bal, N. Insulin-Like Growth Factor 2 As a Possible Neuroprotective Agent and Memory Enhancer—Its Comparative Expression, Processing and Signaling in Mammalian CNS. Int. J. Mol. Sci. 2021, 22, 1849. https://doi.org/10.3390/ijms22041849
Beletskiy A, Chesnokova E, Bal N. Insulin-Like Growth Factor 2 As a Possible Neuroprotective Agent and Memory Enhancer—Its Comparative Expression, Processing and Signaling in Mammalian CNS. International Journal of Molecular Sciences. 2021; 22(4):1849. https://doi.org/10.3390/ijms22041849
Chicago/Turabian StyleBeletskiy, Alexander, Ekaterina Chesnokova, and Natalia Bal. 2021. "Insulin-Like Growth Factor 2 As a Possible Neuroprotective Agent and Memory Enhancer—Its Comparative Expression, Processing and Signaling in Mammalian CNS" International Journal of Molecular Sciences 22, no. 4: 1849. https://doi.org/10.3390/ijms22041849
APA StyleBeletskiy, A., Chesnokova, E., & Bal, N. (2021). Insulin-Like Growth Factor 2 As a Possible Neuroprotective Agent and Memory Enhancer—Its Comparative Expression, Processing and Signaling in Mammalian CNS. International Journal of Molecular Sciences, 22(4), 1849. https://doi.org/10.3390/ijms22041849