
The PKR/P38/RIPK1 Signaling Pathway as a Therapeutic Target in Alzheimer’s Disease



Abstract
:1. Introduction
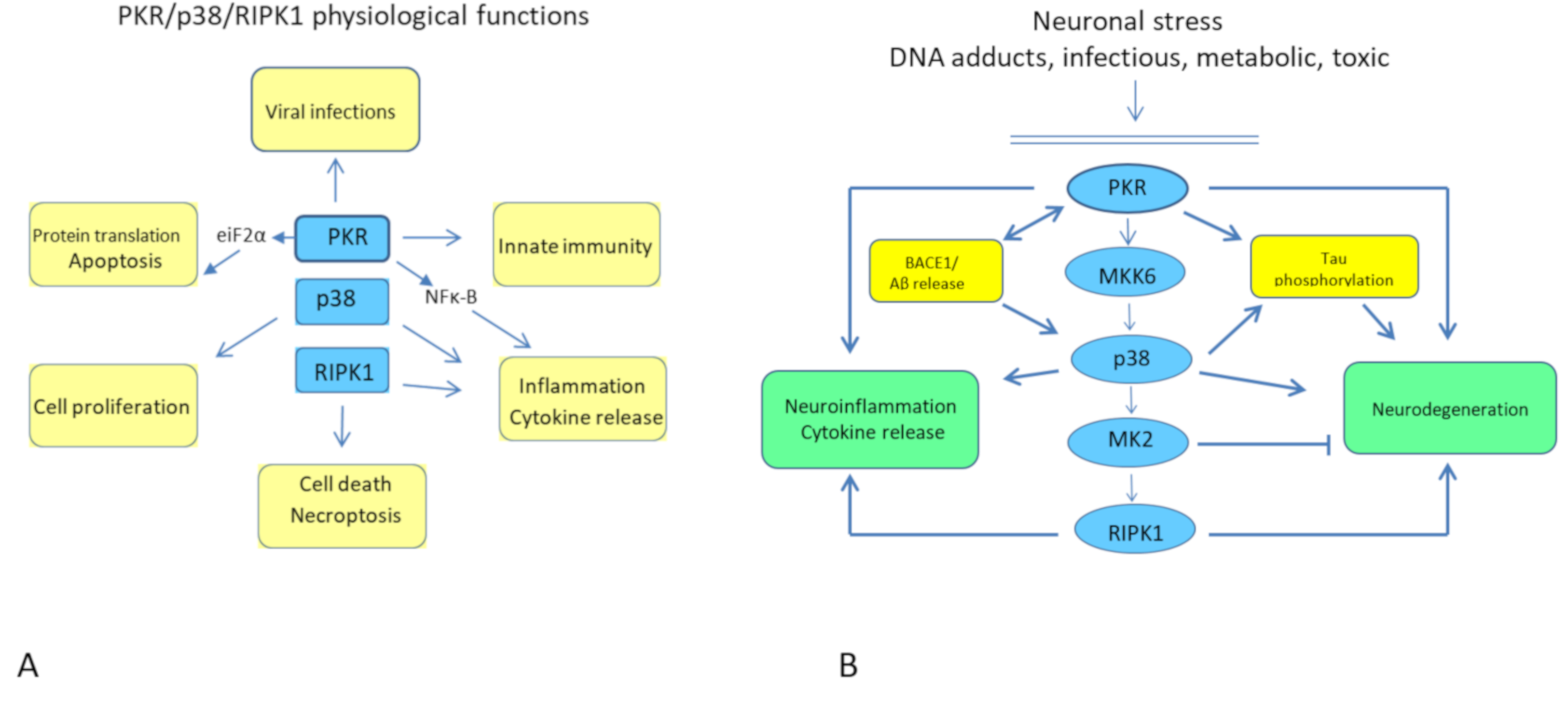
2. The PKR/p38/RIPK1 Pathway
The Physiological Pathway
3. PKR and AD
4. MKK6 and AD
5. P38 and AD
6. MK2 and AD
7. RIPK1 and AD
8. Conclusions
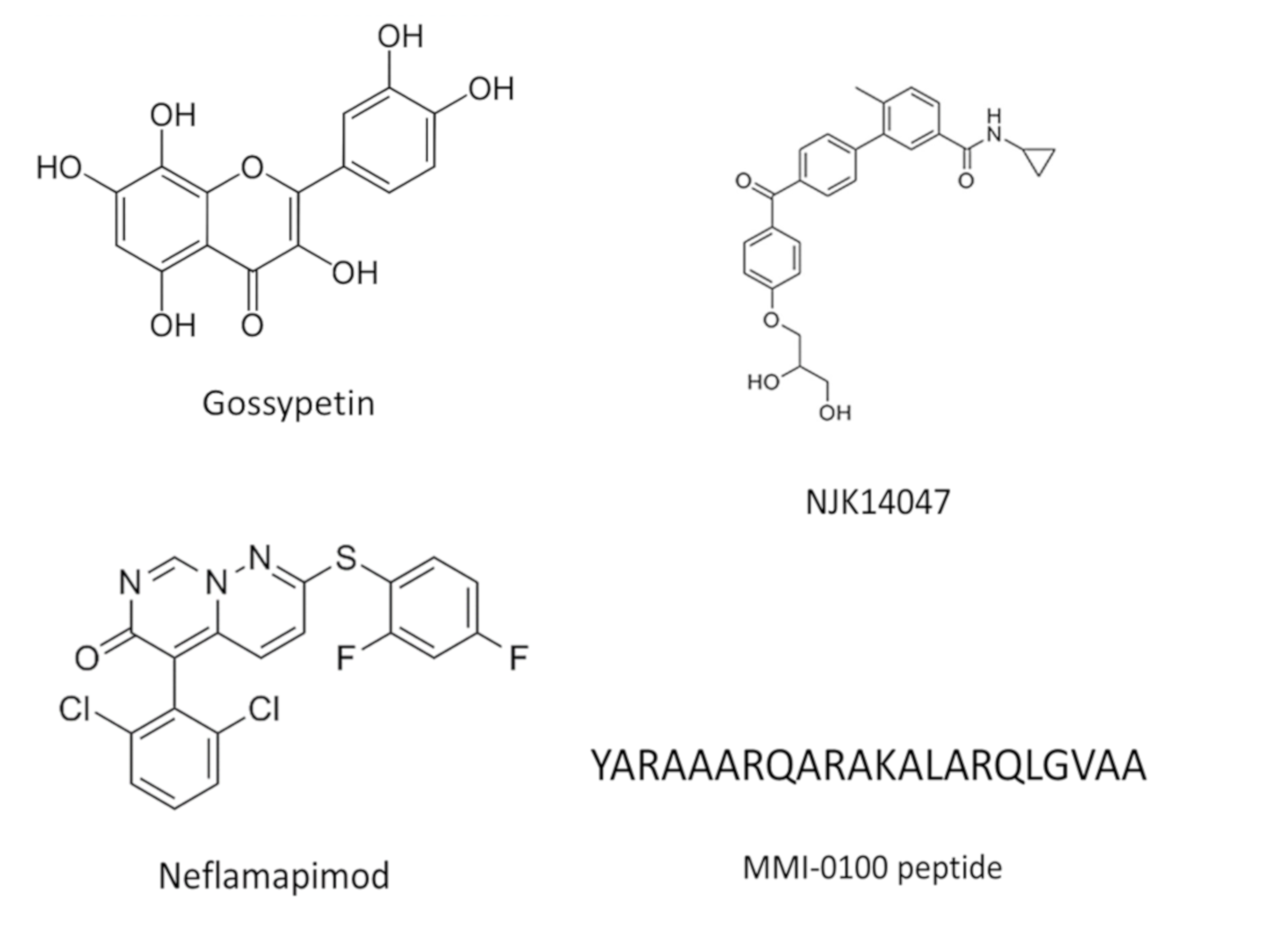
Targeting Several Kinases in AD Patients
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
List of Abbreviations
References
- Scheltens, P.; Blennow, K.; Breteler, M.M.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; Van der Flier, W.M. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Duyckaerts, C.; Delatour, B.; Potier, M.C. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009, 118, 5–36. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Van der Kant, R.; Goldstein, L.S.B.; Ossenkoppele, R. Amyloid-beta-independent regulators of tau pathology in Alzheimer disease. Nat. Rev. Neurosci. 2020, 21, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Zetterberg, H.; Bendlin, B.B. Biomarkers for Alzheimer’s disease-Preparing for a new era of disease-modifying therapies. Mol. Psychiatry 2020, 26, 296–308. [Google Scholar] [CrossRef]
- Ramusino, M.C.; Garibotto, V.; Bacchin, R.; Altomare, D.; Dodich, A.; Assal, F.; Mendes, A.; Costa, A.; Tinazzi, M.; Morbelli, S.D.; et al. Incremental value of amyloid-PET versus CSF in the diagnosis of Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Aquilani, R.; Costa, A.; Maestri, R.; Cotta Ramusino, M.; Pierobon, A.; Dossena, M.; Solerte, S.B.; Condino, A.M.; Torlaschi, V.; Bini, P.; et al. Mini Nutritional Assessment May Identify a Dual Pattern of Perturbed Plasma Amino Acids in Patients with Alzheimer’s Disease: A Window to Metabolic and Physical Rehabilitation? Nutrients 2020, 12, 1845. [Google Scholar] [CrossRef]
- Doody, R.S.; Thomas, R.G.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; Raman, R.; Sun, X.; Aisen, P.S.; et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N. Engl. J. Med. 2014, 370, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Lovestone, S.; Boada, M.; Dubois, B.; Hull, M.; Rinne, J.O.; Huppertz, H.J.; Calero, M.; Andres, M.V.; Gomez-Carrillo, B.; Leon, T.; et al. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimers Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Kost, J.; Tariot, P.N.; Aisen, P.S.; Cummings, J.L.; Vellas, B.; Sur, C.; Mukai, Y.; Voss, T.; Furtek, C.; et al. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N. Engl. J. Med. 2018, 378, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Ostrowitzki, S.; Lasser, R.A.; Dorflinger, E.; Scheltens, P.; Barkhof, F.; Nikolcheva, T.; Ashford, E.; Retout, S.; Hofmann, C.; Delmar, P.; et al. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res. Ther. 2017, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Doody, R.S.; Gavrilova, S.I.; Sano, M.; Thomas, R.G.; Aisen, P.S.; Bachurin, S.O.; Seely, L.; Hung, D.; Dimebon, I. Effect of dimebon on cognition, activities of daily living, behaviour, and global function in patients with mild-to-moderate Alzheimer’s disease: A randomised, double-blind, placebo-controlled study. Lancet 2008, 372, 207–215. [Google Scholar] [CrossRef]
- Kang, R.; Tang, D. PKR-dependent inflammatory signals. Sci. Signal. 2012, 5, pe47. [Google Scholar] [CrossRef] [Green Version]
- Moradi Majd, R.; Mayeli, M.; Rahmani, F. Pathogenesis and promising therapeutics of Alzheimer disease through eIF2alpha pathway and correspondent kinases. Metab. Brain Dis. 2020, 35, 1241–1250. [Google Scholar] [CrossRef]
- Ahmed, T.; Zulfiqar, A.; Arguelles, S.; Rasekhian, M.; Nabavi, S.F.; Silva, A.S.; Nabavi, S.M. Map kinase signaling as therapeutic target for neurodegeneration. Pharmacol. Res. 2020, 160, 105090. [Google Scholar] [CrossRef]
- Sindou, P.; Lesort, M.; Couratier, P.; Yardin, C.; Esclaire, F.; Hugon, J. Glutamate increases tau phosphorylation in primary neuronal cultures from fetal rat cerebral cortex. Brain Res. 1994, 646, 124–128. [Google Scholar] [CrossRef]
- Marchal, J.A.; Lopez, G.J.; Peran, M.; Comino, A.; Delgado, J.R.; Garcia-Garcia, J.A.; Conde, V.; Aranda, F.M.; Rivas, C.; Esteban, M.; et al. The impact of PKR activation: From neurodegeneration to cancer. FASEB J. 2014, 28, 1965–1974. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Walter, P. The integrated stress response: From mechanism to disease. Science 2020, 368, eaat5314. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Newton, K. RIPK1 and RIPK3: Critical regulators of inflammation and cell death. Trends Cell Biol. 2015, 25, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Chang, R.C.; Suen, K.C.; Ma, C.H.; Elyaman, W.; Ng, H.K.; Hugon, J. Involvement of double-stranded RNA-dependent protein kinase and phosphorylation of eukaryotic initiation factor-2alpha in neuronal degeneration. J. Neurochem. 2002, 83, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Suen, K.C.; Yu, M.S.; So, K.F.; Chang, R.C.; Hugon, J. Upstream signaling pathways leading to the activation of double-stranded RNA-dependent serine/threonine protein kinase in beta-amyloid peptide neurotoxicity. J. Biol. Chem. 2003, 278, 49819–49827. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.M.; Whitmore, M.; Xu, Z.; Jiang, Z.; Li, X.; Williams, B.R. Protein kinase R (PKR) interacts with and activates mitogen-activated protein kinase kinase 6 (MKK6) in response to double-stranded RNA stimulation. J. Biol. Chem. 2004, 279, 37670–37676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raingeaud, J.; Whitmarsh, A.J.; Barrett, T.; Derijard, B.; Davis, R.J. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell Biol. 1996, 16, 1247–1255. [Google Scholar] [CrossRef] [Green Version]
- Menon, M.B.; Gropengiesser, J.; Fischer, J.; Novikova, L.; Deuretzbacher, A.; Lafera, J.; Schimmeck, H.; Czymmeck, N.; Ronkina, N.; Kotlyarov, A.; et al. p38(MAPK)/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nat. Cell Biol. 2017, 19, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Menon, M.B.; Gaestel, M. MK2-TNF-Signaling Comes Full Circle. Trends Biochem. Sci. 2018, 43, 170–179. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Delanghe, T.; Rojas-Rivera, D.; Priem, D.; Delvaeye, T.; Bruggeman, I.; Van Herreweghe, F.; Vandenabeele, P.; Bertrand, M.J.M. MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 2017, 19, 1237–1247. [Google Scholar] [CrossRef]
- Bergeron, J.; Benlimame, N.; Zeng-Rong, N.; Xiao, D.; Scrivens, P.J.; Koromilas, A.E.; Alaoui-Jamali, M.A. Identification of the interferon-inducible double-stranded RNA-dependent protein kinase as a regulator of cellular response to bulky adducts. Cancer Res. 2000, 60, 6800–6804. [Google Scholar]
- Page, G.; Rioux Bilan, A.; Ingrand, S.; Lafay-Chebassier, C.; Pain, S.; Perault Pochat, M.C.; Bouras, C.; Bayer, T.; Hugon, J. Activated double-stranded RNA-dependent protein kinase and neuronal death in models of Alzheimer’s disease. Neuroscience 2006, 139, 1343–1354. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Paquet, C.; Dumurgier, J.; Lapalus, P.; Gray, F.; Laplanche, J.L.; Hugon, J.; Groupe d’Investigation du Liquide Cephalorachidien Study Network. Increased cerebrospinal fluid levels of double-stranded RNA-dependant protein kinase in Alzheimer’s disease. Biol. Psychiatry 2012, 71, 829–835. [Google Scholar] [CrossRef]
- Dumurgier, J.; Mouton-Liger, F.; Lapalus, P.; Prevot, M.; Laplanche, J.L.; Hugon, J.; Paquet, C.; Groupe d’Investigation du Liquide Cephalorachidien Study Network. Cerebrospinal fluid PKR level predicts cognitive decline in Alzheimer’s disease. PLoS ONE 2013, 8, e53587. [Google Scholar] [CrossRef] [Green Version]
- Mouton-Liger, F.; Paquet, C.; Dumurgier, J.; Bouras, C.; Pradier, L.; Gray, F.; Hugon, J. Oxidative stress increases BACE1 protein levels through activation of the PKR-eIF2alpha pathway. Biochim. Biophys. Acta 2012, 1822, 885–896. [Google Scholar] [CrossRef] [Green Version]
- Tible, M.; Mouton Liger, F.; Schmitt, J.; Giralt, A.; Farid, K.; Thomasseau, S.; Gourmaud, S.; Paquet, C.; Rondi Reig, L.; Meurs, E.; et al. PKR knockout in the 5xFAD model of Alzheimer’s disease reveals beneficial effects on spatial memory and brain lesions. Aging Cell 2019, 18, e12887. [Google Scholar] [CrossRef] [Green Version]
- Bose, A.; Mouton-Liger, F.; Paquet, C.; Mazot, P.; Vigny, M.; Gray, F.; Hugon, J. Modulation of tau phosphorylation by the kinase PKR: Implications in Alzheimer’s disease. Brain Pathol. 2011, 21, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Reimer, L.; Betzer, C.; Kofoed, R.H.; Volbracht, C.; Fog, K.; Kurhade, C.; Nilsson, E.; Overby, A.K.; Jensen, P.H. PKR kinase directly regulates tau expression and Alzheimer’s disease-related tau phosphorylation. Brain Pathol. 2020, 31, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Ingrand, S.; Barrier, L.; Lafay-Chebassier, C.; Fauconneau, B.; Page, G.; Hugon, J. The oxindole/imidazole derivative C16 reduces in vivo brain PKR activation. FEBS Lett. 2007, 581, 4473–4478. [Google Scholar] [CrossRef] [Green Version]
- Mouton-Liger, F.; Rebillat, A.S.; Gourmaud, S.; Paquet, C.; Leguen, A.; Dumurgier, J.; Bernadelli, P.; Taupin, V.; Pradier, L.; Rooney, T.; et al. PKR downregulation prevents neurodegeneration and beta-amyloid production in a thiamine-deficient model. Cell Death Dis. 2015, 6, e1594. [Google Scholar] [CrossRef]
- Peel, A.L.; Rao, R.V.; Cottrell, B.A.; Hayden, M.R.; Ellerby, L.M.; Bredesen, D.E. Double-stranded RNA-dependent protein kinase, PKR, binds preferentially to Huntington’s disease (HD) transcripts and is activated in HD tissue. Hum. Mol. Genet. 2001, 10, 1531–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peel, A.L.; Sorscher, N.; Kim, J.Y.; Galvan, V.; Chen, S.; Bredesen, D.E. Tau phosphorylation in Alzheimer’s disease: Potential involvement of an APP-MAP kinase complex. Neuromol. Med. 2004, 5, 205–218. [Google Scholar] [CrossRef]
- Zhu, X.; Rottkamp, C.A.; Hartzler, A.; Sun, Z.; Takeda, A.; Boux, H.; Shimohama, S.; Perry, G.; Smith, M.A. Activation of MKK6, an upstream activator of p38, in Alzheimer’s disease. J. Neurochem. 2001, 79, 311–318. [Google Scholar] [CrossRef]
- Chang, R.C.; Wong, A.K.; Ng, H.K.; Hugon, J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 2002, 13, 2429–2432. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [Green Version]
- Leestemaker, Y.; de Jong, A.; Witting, K.F.; Penning, R.; Schuurman, K.; Rodenko, B.; Zaal, E.A.; van de Kooij, B.; Laufer, S.; Heck, A.J.R.; et al. Proteasome Activation by Small Molecules. Cell Chem. Biol. 2017, 24, 725–736.e7. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Liu, K.; Liu, F.; Chen, H.; Wang, X.; Zu, X.; Ma, X.; Wang, T.; Wu, Q.; Zheng, Y.; et al. Gossypetin is a novel MKK3 and MKK6 inhibitor that suppresses esophageal cancer growth in vitro and in vivo. Cancer Lett. 2019, 442, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, K.; Floyd, R.A.; Zheng, N.Y.; Nael, R.; Robinson, K.A.; Nguyen, X.; Pye, Q.N.; Stewart, C.A.; Geddes, J.; Markesbery, W.R.; et al. p38 kinase is activated in the Alzheimer’s disease brain. J. Neurochem. 1999, 72, 2053–2058. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Kim, N.J. Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef] [Green Version]
- Bachstetter, A.D.; Xing, B.; de Almeida, L.; Dimayuga, E.R.; Watterson, D.M.; Van Eldik, L.J. Microglial p38alpha MAPK is a key regulator of proinflammatory cytokine up-regulation induced by toll-like receptor (TLR) ligands or beta-amyloid (Abeta). J. Neuroinflammation 2011, 8, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Rottkamp, C.A.; Boux, H.; Takeda, A.; Perry, G.; Smith, M.A. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2000, 59, 880–888. [Google Scholar] [CrossRef] [Green Version]
- Munoz, L.; Ralay Ranaivo, H.; Roy, S.M.; Hu, W.; Craft, J.M.; McNamara, L.K.; Chico, L.W.; Van Eldik, L.J.; Watterson, D.M. A novel p38 alpha MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. J. Neuroinflamm. 2007, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Sun, A.; Liu, M.; Nguyen, X.V.; Bing, G. P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp. Neurol. 2003, 183, 394–405. [Google Scholar] [CrossRef]
- Goh, K.C.; deVeer, M.J.; Williams, B.R. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J. 2000, 19, 4292–4297. [Google Scholar] [CrossRef] [Green Version]
- Melone, M.A.B.; Dato, C.; Paladino, S.; Coppola, C.; Trebini, C.; Giordana, M.T.; Perrone, L. Verapamil Inhibits Ser202/Thr205 Phosphorylation of Tau by Blocking TXNIP/ROS/p38 MAPK Pathway. Pharm. Res. 2018, 35, 44. [Google Scholar] [CrossRef]
- Cui, Y.Q.; Wang, Q.; Zhang, D.M.; Wang, J.Y.; Xiao, B.; Zheng, Y.; Wang, X.M. Triptolide Rescues Spatial Memory Deficits and Amyloid-beta Aggregation Accompanied by Inhibition of Inflammatory Responses and MAPKs Activity in APP/PS1 Transgenic Mice. Curr. Alzheimer Res. 2016, 13, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Falcicchia, C.; Tozzi, F.; Arancio, O.; Watterson, D.M.; Origlia, N. Involvement of p38 MAPK in Synaptic Function and Dysfunction. Int. J. Mol. Sci. 2020, 21, 5624. [Google Scholar] [CrossRef] [PubMed]
- Gee, M.S.; Son, S.H.; Jeon, S.H.; Do, J.; Kim, N.; Ju, Y.J.; Lee, S.J.; Chung, E.K.; Inn, K.S.; Kim, N.J.; et al. A selective p38alpha/beta MAPK inhibitor alleviates neuropathology and cognitive impairment, and modulates microglia function in 5XFAD mouse. Alzheimers Res. Ther. 2020, 12, 45. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Jiang, H.; Wang, L.; Yi, H.; Li, Z.; Liu, R. Vitegnoside Mitigates Neuronal Injury, Mitochondrial Apoptosis, and Inflammation in an Alzheimer’s Disease Cell Model via the p38 MAPK/JNK Pathway. J. Alzheimers Dis. 2019, 72, 199–214. [Google Scholar] [CrossRef] [PubMed]
- Scheltens, P.; Prins, N.; Lammertsma, A.; Yaqub, M.; Gouw, A.; Wink, A.M.; Chu, H.M.; van Berckel, B.N.M.; Alam, J. An exploratory clinical study of p38alpha kinase inhibition in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2018, 5, 464–473. [Google Scholar] [CrossRef] [Green Version]
- At CTAD, Early Failures and Hints of Success, from Small Trials. Available online: https://www.alzforum.org/news/conference-coverage/ctad-early-failures-and-hints-success-small-trials (accessed on 1 January 2021).
- In Phase 2 Trial, Neflamapimod Aids Cognition in Lewy Body Dementia. Available online: https://www.alzforum.org/news/conference-coverage/phase-2-trial-neflamapimod-aids-cognition-lewy-body-dementia-0 (accessed on 1 January 2021).
- Culbert, A.A.; Skaper, S.D.; Howlett, D.R.; Evans, N.A.; Facci, L.; Soden, P.E.; Seymour, Z.M.; Guillot, F.; Gaestel, M.; Richardson, J.C. MAPK-activated protein kinase 2 deficiency in microglia inhibits pro-inflammatory mediator release and resultant neurotoxicity. Relevance to neuroinflammation in a transgenic mouse model of Alzheimer disease. J. Biol. Chem. 2006, 281, 23658–23667. [Google Scholar] [CrossRef] [Green Version]
- Goldsmith, C.S.; Kim, S.M.; Karunarathna, N.; Neuendorff, N.; Toussaint, L.G.; Earnest, D.J.; Bell-Pedersen, D. Inhibition of p38 MAPK activity leads to cell type-specific effects on the molecular circadian clock and time-dependent reduction of glioma cell invasiveness. BMC Cancer 2018, 18, 43. [Google Scholar] [CrossRef]
- Hammaker, D.; Firestein, G.S. “Go upstream, young man”: Lessons learned from the p38 saga. Ann. Rheum. Dis. 2010, 69 (Suppl. 1), i77–i82. [Google Scholar] [CrossRef]
- Dominguez, C.; Powers, D.A.; Tamayo, N. p38 MAP kinase inhibitors: Many are made, but few are chosen. Curr. Opin. Drug Discov. Devel. 2005, 8, 421–430. [Google Scholar]
- Hedstrom, U.; Norberg, M.; Evertsson, E.; Lever, S.R.; Munck Af Rosenschold, M.; Lonn, H.; Bold, P.; Kack, H.; Berntsson, P.; Vinblad, J.; et al. An Angle on MK2 Inhibition-Optimization and Evaluation of Prevention of Activation Inhibitors. ChemMedChem 2019, 14, 1701–1709. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Diwan, M.; Dastidar, S.G.; Najmi, A.K. Differential effect of p38 and MK2 kinase inhibitors on the inflammatory and toxicity biomarkers in vitro. Hum. Exp. Toxicol. 2018, 37, 521–531. [Google Scholar] [CrossRef]
- Wang, C.; Hockerman, S.; Jacobsen, E.J.; Alippe, Y.; Selness, S.R.; Hope, H.R.; Hirsch, J.L.; Mnich, S.J.; Saabye, M.J.; Hood, W.F.; et al. Selective inhibition of the p38alpha MAPK-MK2 axis inhibits inflammatory cues including inflammasome priming signals. J. Exp. Med. 2018, 215, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Wang, Z.; Liang, X.; Nie, Y.; Chang, X.; Xue, H.; Li, S.; Min, C. Intranasal MMI-0100 Attenuates Abeta1-42- and LPS-Induced Neuroinflammation and Memory Impairments via the MK2 Signaling Pathway. Front. Immunol. 2019, 10, 2707. [Google Scholar] [CrossRef] [PubMed]
- Dondelinger, Y.; Delanghe, T.; Priem, D.; Wynosky-Dolfi, M.A.; Sorobetea, D.; Rojas-Rivera, D.; Giansanti, P.; Roelandt, R.; Gropengiesser, J.; Ruckdeschel, K.; et al. Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nat. Commun. 2019, 10, 1729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, J.; Amin, P.; Ofengeim, D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. [Google Scholar] [CrossRef]
- Geng, J.; Ito, Y.; Shi, L.; Amin, P.; Chu, J.; Ouchida, A.T.; Mookhtiar, A.K.; Zhao, H.; Xu, D.; Shan, B.; et al. Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nat. Commun. 2017, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Nogusa, S.; Chen, P.; Maki, J.L.; Lerro, A.; Andrake, M.; Rall, G.F.; Degterev, A.; Balachandran, S. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc. Natl. Acad. Sci. USA 2013, 110, E3109-18. [Google Scholar] [CrossRef] [Green Version]
- Jaco, I.; Annibaldi, A.; Lalaoui, N.; Wilson, R.; Tenev, T.; Laurien, L.; Kim, C.; Jamal, K.; Wicky John, S.; Liccardi, G.; et al. MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Mol. Cell 2017, 66, 698–710.e5. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Piras, I.S.; Ferreira, E.; Huentelman, M.J.; Liang, W.S.; Readhead, B.; Dudley, J.T.; Spangenberg, E.E.; Green, K.N.; et al. Necroptosis activation in Alzheimer’s disease. Nat. Neurosci. 2017, 20, 1236–1246. [Google Scholar] [CrossRef] [PubMed]
- Koper, M.J.; Van Schoor, E.; Ospitalieri, S.; Vandenberghe, R.; Vandenbulcke, M.; von Arnim, C.A.F.; Tousseyn, T.; Balusu, S.; De Strooper, B.; Thal, D.R. Necrosome complex detected in granulovacuolar degeneration is associated with neuronal loss in Alzheimer’s disease. Acta Neuropathol. 2020, 139, 463–484. [Google Scholar] [CrossRef] [PubMed]
- Najjar, M.; Saleh, D.; Zelic, M.; Nogusa, S.; Shah, S.; Tai, A.; Finger, J.N.; Polykratis, A.; Gough, P.J.; Bertin, J.; et al. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity 2016, 45, 46–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ofengeim, D.; Mazzitelli, S.; Ito, Y.; DeWitt, J.P.; Mifflin, L.; Zou, C.; Das, S.; Adiconis, X.; Chen, H.; Zhu, H.; et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E8788–E8797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grievink, H.W.; Heuberger, J.; Huang, F.; Chaudhary, R.; Birkhoff, W.A.J.; Tonn, G.R.; Mosesova, S.; Erickson, R.; Moerland, M.; Haddick, P.C.G.; et al. DNL104, a Centrally Penetrant RIPK1 Inhibitor, Inhibits RIP1 Kinase Phosphorylation in a Randomized Phase I Ascending Dose Study in Healthy Volunteers. Clin. Pharmacol. Ther. 2020, 107, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Heppt, M.V.; Tietze, J.K.; Graf, S.A.; Berking, C. Combination therapy of melanoma using kinase inhibitors. Curr. Opin. Oncol. 2015, 27, 134–140. [Google Scholar] [CrossRef]
- Gourmaud, S.; Mouton-Liger, F.; Abadie, C.; Meurs, E.F.; Paquet, C.; Hugon, J. Dual Kinase Inhibition Affords Extended in vitro Neuroprotection in Amyloid-beta Toxicity. J. Alzheimers Dis. 2016, 54, 1659–1670. [Google Scholar] [CrossRef]
- Alam, J.; Blackburn, K.; Patrick, D. Neflamapimod: Clinical Phase 2b-Ready Oral Small Molecule Inhibitor of p38alpha to Reverse Synaptic Dysfunction in Early Alzheimer’s Disease. J. Prev. Alzheimers Dis. 2017, 4, 273–278. [Google Scholar]
- Hampel, H.; Ewers, M.; Burger, K.; Annas, P.; Mortberg, A.; Bogstedt, A.; Frolich, L.; Schroder, J.; Schonknecht, P.; Riepe, M.W.; et al. Lithium trial in Alzheimer’s disease: A randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 2009, 70, 922–931. [Google Scholar] [CrossRef]
- Karikari, T.K.; Pascoal, T.A.; Ashton, N.J.; Janelidze, S.; Benedet, A.L.; Rodriguez, J.L.; Chamoun, M.; Savard, M.; Kang, M.S.; Therriault, J.; et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: A diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020, 19, 422–433. [Google Scholar] [CrossRef]
- Hampel, H.; Vergallo, A.; Caraci, F.; Cuello, A.C.; Lemercier, P.; Vellas, B.; Giudici, K.V.; Baldacci, F.; Hanisch, B.; Haberkamp, M.; et al. Future avenues for Alzheimer’s disease detection and therapy: Liquid biopsy, intracellular signaling modulation, systems pharmacology drug discovery. Neuropharmacology 2020, 185, 108081. [Google Scholar] [CrossRef]
- Khan, A.; Corbett, A.; Ballard, C. Emerging treatments for Alzheimer’s disease for non-amyloid and non-tau targets. Expert Rev. Neurother. 2017, 17, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Haller, V.; Nahidino, P.; Forster, M.; Laufer, S.A. An updated patent review of p38 MAP kinase inhibitors (2014–2019). Expert Opin. Ther. Pat. 2020, 30, 453–466. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| PKR | Accumulation of Neuronal PKR in AD Brains | Chang et al., 2002 |
|---|---|---|
| PKR | Increased levels of CSF PKR in AD patients | Mouton Liger et al., 2012 |
| PKR | CSF PKR levels correlate with cognitive decline | Dumurgier et al., 2013 |
| MKK6 | Increased stainings of MKK6 in AD brains | Zhu et al., 2001 |
| P38 | Increased stainings of P38 in AD brains | Hensley et al., 1998 |
| P38 | Increased levels of p38 in AD brains | Zhu et al., 2000 |
| P38 | Early activation of p38 in AD brains | Sun et al., 2003 |
| RIPK1 | RIPK1 mediates microglial activation in AD | Offengeim et al., 2017 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hugon, J.; Paquet, C. The PKR/P38/RIPK1 Signaling Pathway as a Therapeutic Target in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 3136. https://doi.org/10.3390/ijms22063136
Hugon J, Paquet C. The PKR/P38/RIPK1 Signaling Pathway as a Therapeutic Target in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(6):3136. https://doi.org/10.3390/ijms22063136
Chicago/Turabian StyleHugon, Jacques, and Claire Paquet. 2021. "The PKR/P38/RIPK1 Signaling Pathway as a Therapeutic Target in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 6: 3136. https://doi.org/10.3390/ijms22063136