
Natural Compounds as Therapeutic Agents: The Case of Human Topoisomerase IB




Abstract
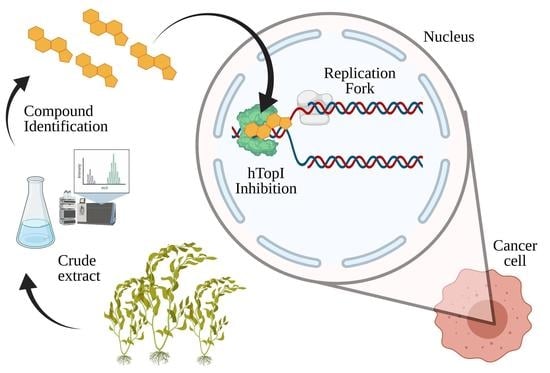
1. Introduction
2. Human DNA Topoisomerase IB as the Tumor Target
3. Natural Compounds with In Vitro and In Vivo Activity on hTopIB
3.1. Epigallocatechin-3-Gallate
3.2. Kakuol
3.3. Berberine
3.4. Pinostrobin
3.5. Sulfonoquinovosyl Diacylglyceride
3.6. Benzoxazines
3.7. Evodiamine
3.8. Cytosporolide C
4. HTopIB Inhibition by Natural Compounds Coordinated with Metals
5. Natural Compounds from Marine Organism
5.1. Bacillosporin C
5.2. Alpha-Methoxylated δ5,9 Fatty Acids
5.3. Lamellarin D
5.4. Bis(2,3-dibromo-4,5-dihydroxybenzyl) Ether
5.5. Variolin B
5.6. Discorhabdins
6. HTopIB Poisons Derived from Natural Compounds in Preclinical and Clinical Trial
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Kinghorn, A.D.; Pan, L.; Fletcher, J.N.; Chai, H. The relevance of higher plants in lead compound discovery programs. J. Nat. Prod. 2011, 74, 1539–1555. [Google Scholar] [CrossRef]
- Baker, D.D.; Chu, M.; Oza, U.; Rajgarhia, V. The value of natural products to future pharmaceutical discovery. Nat. Prod. Rep. 2007, 24, 1225–1244. [Google Scholar] [CrossRef]
- Dias, D.A.; Urban, S.; Roessner, U. A Historical Overview of Natural Products in Drug Discovery. Metabolites 2012, 2, 303–336. [Google Scholar] [CrossRef]
- Thongphasuk, P.; Stremmel, W.; Chamulitrat, W. 2,3-Dehydrosilybin Is a Better DNA Topoisomerase I Inhibitor than Its Parental Silybin. Chemotherapy 2009, 55, 42–48. [Google Scholar] [CrossRef]
- Bhandari, M.; Bhandari, A.; Bhandari, A. Recent updates on codeine. Pharm. Methods 2011, 2, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Tarver, T. The Review of Natural Products. J. Consum. Health Internet 2014, 18, 291–292. [Google Scholar] [CrossRef]
- Limmroth, V.; Katsarava, Z.; Diener, H.-C. Acetylsalicylic acid in the treatment of headache. Cephalalgia 1999, 19, 546–551. [Google Scholar] [CrossRef]
- Katz, L.; Baltz, R.H. Natural product discovery: Past, present, and future. J. Ind. Microbiol. Biotechnol. 2016, 43, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Giddings, L.A.; Newman, D.J. Microbial natural products: Molecular blueprints for antitumor drugs. J. Ind. Microbiol. Biotechnol. 2013, 40, 1181–1210. [Google Scholar] [CrossRef]
- Di Marco, A.; Cassinelli, G.; Arcamone, F. The discovery of daunorubicin. Cancer Treat. Rep. 1981, 65, 3–8. [Google Scholar] [PubMed]
- Pommier, Y.; Schwartz, R.E.; Kohn, K.W.; Zwelling, L.A. Effects of DNA Intercalating Agents on Topoisomerase II Induced DNA Strand Cleavage in Isolated Mammalian Cell Nuclei. Biochemistry 1985, 24, 6406–6410. [Google Scholar] [CrossRef]
- Tewey, K.M.; Rowe, T.C.; Yang, L.; Halligan, B.D.; Liu, L.F. Adriamycin-induced DNA damage mediated by mammalian DNA topoisomerase II. Science 1984, 226, 466–468. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Thrower, D.; Wilson, L. Effects of vinblastine, podophyllotoxin and nocodazole on mitotic spindles. Implications for the role of microtubule dynamics in mitosis. J. Cell Sci. 1992, 102, 401–416. [Google Scholar] [PubMed]
- Jordan, M.A.; Thrower, D.; Wilson, L. Mechanism of Inhibition of Cell Proliferation by Vinca Alkaloids. Cancer Res. 1991, 51, 2212–2222. [Google Scholar]
- Wilson, L. Microtubules as Targets for Drug and Toxic Chemical Action: The Mechanisms of Action of Colchicine and Vinblastine. In The Cytoskeleton; Springer: New York, NY, USA, 1986; pp. 37–52. [Google Scholar]
- Liu, L.F.; Desai, S.D.; Li, T.K.; Mao, Y.; Sun, M.; Sim, S.P. Mechanism of action of camptothecin. In Annals of the New York Academy of Sciences; New York Academy of Sciences: New York, NY, USA, 2000; Volume 922, pp. 1–10. [Google Scholar]
- Wall, M.E.; Mansukh, C. Wani Camptothecin and Taxol: Discovery to Clinic. Cancer Res. 1995, 55, 753–760. [Google Scholar]
- Wang, J.C. Interaction between DNA and an Escherichia coli protein omega. J. Mol. Biol. 1971, 55, 523–533. [Google Scholar] [CrossRef]
- Champoux, J.J.; Dulbecco, R. An activity from mammalian cells that untwists superhelical DNA-a possible swivel for DNA replication (polyoma-ethidium bromide-mouse-embryo cells-dye binding assay). Proc. Natl. Acad. Sci. USA 1972, 69, 143–146. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef]
- Wang, J.C. DNA topoisomerases: Why so many? J. Biol. Chem. 1991, 266, 6659–6662. [Google Scholar] [CrossRef]
- Soren, B.C.; Dasari, J.B.; Ottaviani, A.; Lacovelli, F.; Fiorani, P. Topoisomerase IB: A relaxing enzyme for stressed DNA. Cancer Drug Resist. 2019, 18–25. [Google Scholar] [CrossRef]
- Cretaio, E.; Pattarello, L.; Fontebasso, Y.; Banedetti, P.; Losasso, C. Human DNA topoisomerase IB: Structure and functions. Ital. J. Biochem. 2007, 56, 91–102. [Google Scholar]
- Cheng, C.; Shuman, S. A catalytic domain of eukaryotic DNA topoisomerase I. J. Biol. Chem. 1998, 273, 11589–11595. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L. A Model for the Mechanism of Human Topoisomerase I. Science 1998, 279, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Leppard, J.B.; Champoux, J.J. Human DNA topoisomerase I: Relaxation, roles, and damage control. Chromosoma 2005, 114, 75–85. [Google Scholar] [CrossRef]
- Alsner, J.; Svejstrup, J.Q.; Kjeldsen, E.; Sørensen, B.S.; Westergaard, O. Identification of an N-terminal domain of eukaryotic DNA topoisomerase I dispensable for catalytic activity but essential for in vivo function. J. Biol. Chem. 1992, 267, 12408–12411. [Google Scholar] [CrossRef]
- Lisby, M.; Olesen, J.R.; Skouboe, C.; Krogh, B.O.; Straub, T.; Boege, F.; Velmurugan, S.; Martensen, P.M.; Andersen, A.H.; Jayaram, M.; et al. Residues Within the N-terminal Domain of Human Topoisomerase I Play a Direct Role in Relaxation. J. Biol. Chem. 2001, 276, 20220–20227. [Google Scholar] [CrossRef] [PubMed]
- Redinbo, M.R.; Stewart, L.; Kuhn, P.; Champoux, J.J.; Hol, W.G. Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 1998, 279, 1504–1513. [Google Scholar] [CrossRef]
- Coletta, A.; Desideri, A. Role of the protein in the DNA sequence specificity of the cleavage site stabilized by the camptothecin topoisomerase IB inhibitor: A metadynamics study. Nucleic Acids Res. 2013, 41, 9977–9986. [Google Scholar] [CrossRef]
- Fiorani, P.; Chillemi, G.; Losasso, C.; Castelli, S.; Desideri, A. The different cleavage DNA sequence specificity explains the camptothecin resistance of the human topoisomerase I Glu418Lys mutant. Nucleic Acids Res. 2006, 34, 5093–5100. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Redinbo, M.R.; Stewart, L.; Champoux, J.J.; Hol, W.G. Structural flexibility in human topoisomerase I revealed in multiple non-isomorphous crystal structures. J. Mol. Biol. 1999, 292, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Stewart, L.; Ireton, G.C.; Champoux, J.J. A Functional Linker in Human Topoisomerase I Is Required for Maximum Sensitivity to Camptothecin in a DNA Relaxation Assay. J. Biol. Chem. 1999, 274, 32950–32960. [Google Scholar] [CrossRef]
- Fiorani, P.; Tesauro, C.; Mancini, G.; Chillemi, G.; D’Annessa, I.; Graziani, G.; Tentori, L.; Muzi, A.; Desideri, A. Evidence of the crucial role of the linker domain on the catalytic activity of human topoisomerase I by experimental and simulative characterization of the Lys681Ala mutant. Nucleic Acids Res. 2009, 37, 6849–6858. [Google Scholar] [CrossRef] [PubMed]
- Fiorani, P.; Bruselles, A.; Falconi, M.; Chillemi, G.; Desideri, A.; Benedetti, P. Single mutation in the linker domain confers protein flexibility and camptothecin resistance to human topoisomerase I. J. Biol. Chem. 2003, 278, 43268–43275. [Google Scholar] [CrossRef]
- D’Annessa, I.; Tesauro, C.; Fiorani, P.; Chillemi, G.; Castelli, S.; Vassallo, O.; Capranico, G.; Desideri, A. Role of Flexibility in Protein-DNA-Drug Recognition: The Case of Asp677Gly-Val703Ile Topoisomerase Mutant Hypersensitive to Camptothecin. J. Amino Acids 2012, 2012, 206083. [Google Scholar] [CrossRef][Green Version]
- Tesauro, C.; Morozzo della Rocca, B.; Ottaviani, A.; Coletta, A.; Zuccaro, L.; Arnò, B.; D’Annessa, I.; Fiorani, P.; Desideri, A. Molecular mechanism of the camptothecin resistance of Glu710Gly topoisomerase IB mutant analyzed in vitro and in silico. Mol. Cancer 2013, 12, 100. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Kussie, P.; Pavletich, N.; Shuman, S. Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell 1998, 92, 841–850. [Google Scholar] [CrossRef]
- Krogh, B.O.; Shuman, S. Catalytic mechanism of DNA topoisomerase IB. Mol. Cell 2000, 5, 1035–1041. [Google Scholar] [CrossRef]
- Wang, Z.; D’Annessa, I.; Tesauro, C.; Croce, S.; Ottaviani, A.; Fiorani, P.; Desideri, A. Mutation of Gly717Phe in human topoisomerase 1B has an effect on enzymatic function, reactivity to the camptothecin anticancer drug and on the linker domain orientation. Biochim. Biophys. Acta 2015, 1854, 860–868. [Google Scholar] [CrossRef]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y. Drugging Topoisomerases: Lessons and Challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef]
- Cinelli, M.A. Topoisomerase 1B poisons: Over a half-century of drug leads, clinical candidates, and serendipitous discoveries. Med. Res. Rev. 2019, 39, 1294–1337. [Google Scholar] [CrossRef]
- Strumberg, D.; Pilon, A.A.; Smith, M.; Hickey, R.; Malkas, L.; Pommier, Y. Conversion of topoisomerase I cleavage complexes on the leading strand of ribosomal DNA into 5’-phosphorylated DNA double-strand breaks by replication runoff. Mol. Cell. Biol. 2000, 20, 3977–3987. [Google Scholar] [CrossRef] [PubMed]
- Eng, W.K.; Faucette, L.; Johnson, R.K.; Sternglanz, R. Evidence that DNA topoisomerase I is necessary for the cytotoxic effects of camptothecin. Mol. Pharmacol. 1988, 6, 755–760. [Google Scholar]
- Hsiang, Y.H.; Hertzberg, R.; Hecht, S.; Liu, L.F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 27, 14873–14878. [Google Scholar] [CrossRef]
- Castelli, S.; Coletta, A.; D’Annessa, I.; Fiorani, P.; Tesauro, C.; Desideri, A. Interaction between natural compounds and human topoisomerase I. Biol. Chem. 2012, 393, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.-T.; Liu, L.; Shao, C.-L.; Yu, R.-L.; Chen, F.-L.; Yue, S.-J.; Wang, M.; Guo, Z.-L.; Fan, Y.-C.; Guan, H.-S.; et al. Discovery of DNA Topoisomerase I Inhibitors with Low-Cytotoxicity Based on Virtual Screening from Natural Products. Mar. Drugs 2017, 15, 217. [Google Scholar] [CrossRef]
- Tillhon, M.; Guamán Ortiz, L.M.; Lombardi, P.; Scovassi, A.I. Berberine: New perspectives for old remedies. Biochem. Pharmacol. 2012, 84, 1260–1267. [Google Scholar] [CrossRef]
- Jadaun, A.; Subbarao, N.; Dixit, A. Allosteric inhibition of topoisomerase I by pinostrobin: Molecular docking, spectroscopic and topoisomerase I activity studies. J. Photochem. Photobiol. B Biol. 2017, 167, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Honkanen, E.; Virtanen, A.I.; Tweit, R.C.; Dodson, R.M. The Synthesis of Precursor II of Benzoxazolinone Formed in Rye Plants, and the Enzymic Hydrolysis of Precursor I, the Glucoside. Acta Chem. Scand. 1960, 14, 504–507. [Google Scholar] [CrossRef]
- Foto, E.; Özen, Ç.; Zilifdar, F.; Tekiner-Gülbaş, B.; Yıldız, İ.; Akı-Yalçın, E.; Diril, N.; Yalçın, İ. Benzoxazines as new human topoisomerase I inhibitors and potential poisons. DARU J. Pharm. Sci. 2020, 28, 65–73. [Google Scholar] [CrossRef]
- Chan, A.; Chang, W.-S.; Chen, L.-M.; Lee, C.-M.; Chen, C.-E.; Lin, C.-M.; Hwang, J.-L. Evodiamine Stabilizes Topoisomerase I-DNA Cleavable Complex to Inhibit Topoisomerase I Activity. Molecules 2009, 14, 1342–1352. [Google Scholar] [CrossRef]
- El-Atawy, M.A.; Omar, A.Z.; Hagar, M.; Shashira, E.M. Transalkylidation reaction: Green, catalyst-free synthesis of thiosemicarbazones and solving the NMR conflict between their acyclic structure and intramolecular cycloaddition products. Green Chem. Lett. Rev. 2019, 12, 364–376. [Google Scholar] [CrossRef]
- Abu, N.; Ho, W.Y.; Yeap, S.K.; Akhtar, M.N.; Abdullah, M.P.; Omar, A.R.; Alitheen, N.B. The flavokawains: Uprising medicinal chalcones. Cancer Cell Int. 2013, 13, 102. [Google Scholar] [CrossRef] [PubMed]
- Nagle, D.G.; Ferreira, D.; Zhou, Y.D. Epigallocatechin-3-gallate (EGCG): Chemical and biomedical perspectives. Phytochemistry 2006, 67, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.J.; Gupta, S.; Belfi, C.A.; Gosky, D.M.; Mukhtar, H. Green tea constituent (-)-epigallocatechin-3-gallate inhibits topoisomerase I activity in human colon carcinoma cells. Biochem. Biophys. Res. Commun. 2001, 288, 101–105. [Google Scholar] [CrossRef]
- Singh, R.; Ahmed, S.; Islam, N.; Goldberg, V.M.; Haqqi, T.M. Epigallocatechin-3-gallate inhibits interleukin-1β-induced expression of nitric oxide synthase and production of nitric oxide in human chondrocytes: Suppression of nuclear factor κB activation by degradation of the inhibitor of nuclear factor κB. Arthritis Rheum. 2002, 46, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Kondo, T.; Ohta, T.; Igura, K.; Hara, Y.; Kaji, K. Tea catechins inhibit angiogenesis in vitro, measured by human endothelial cell growth, migration and tube formation, through inhibition of VEGF receptor binding. Cancer Lett. 2002, 180, 139–144. [Google Scholar] [CrossRef]
- Lee, J.Y.; Moon, S.S.; Hwang, B.K. Isolation and antifungal activity of kakuol, a propiophenone derivative fromAsarum sieboldii rhizome. Pest Manag. Sci. 2005, 61, 821–825. [Google Scholar] [CrossRef]
- Castelli, S.; Vieira, S.; D’Annessa, I.; Katkar, P.; Musso, L.; Dallavalle, S.; Desideri, A. A derivative of the natural compound kakuol affects DNA relaxation of topoisomerase IB inhibiting the cleavage reaction. Arch. Biochem. Biophys. 2013, 530, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Neag, M.A.; Mocan, A.; Echeverría, J.; Pop, R.M.; Bocsan, C.I.; Crisan, G.; Buzoianu, A.D. Berberine: Botanical Occurrence, traditional uses, extraction methods, and relevance in cardiovascular, metabolic, hepatic, and renal disorders. Front. Pharmacol. 2018, 9, 557. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Johnson, B.R.; Wenckus, C.S.; Fayad, M.I.; Wu, C.D. Efficacy of berberine, an antimicrobial plant alkaloid, as an endodontic irrigant against a mixed-culture biofilm in an in vitro tooth model. J. Endod. 2012, 38, 1114–1117. [Google Scholar] [CrossRef] [PubMed]
- Lou, T.; Zhang, Z.; Xi, Z.; Liu, K.; Li, L.; Liu, B.; Huang, F. Berberine inhibits inflammatory response and ameliorates insulin resistance in hepatocytes. Inflammation 2011, 34, 659–667. [Google Scholar] [CrossRef]
- Lau, C.W.; Yao, X.Q.; Chen, Z.Y.; Ko, W.H.; Huang, Y. Cardiovascular actions of berberine. Cardiovasc. Drug Rev. 2001, 19, 234–244. [Google Scholar] [CrossRef]
- Vieira, S.; Castelli, S.; Falconi, M.; Takarada, J.; Fiorillo, G.; Buzzetti, F.; Lombardi, P.; Desideri, A. Role of 13-(di)phenylalkyl berberine derivatives in the modulation of the activity of human topoisomerase IB. Int. J. Biol. Macromol. 2015, 77, 68–75. [Google Scholar] [CrossRef]
- Christena, L.R.; Subramaniam, S.; Vidhyalakshmi, M.; Mahadevan, V.; Sivasubramanian, A.; Nagarajan, S. Dual role of pinostrobin-a flavonoid nutraceutical as an efflux pump inhibitor and antibiofilm agent to mitigate food borne pathogens. RSC Adv. 2015, 5, 61881–61887. [Google Scholar] [CrossRef]
- Patel, N.K.; Bhutani, K.K. Pinostrobin and Cajanus lactone isolated from Cajanus cajan (L.) leaves inhibits TNF-α and IL-1β production: In vitro and in vivo experimentation. Phytomedicine 2014, 21, 946–953. [Google Scholar] [CrossRef]
- Lopez-Martinez, L.X.; Parkin, K.L.; Garcia, H.S. Antioxidant and quinone reductase inducing activities of ethanolic fractions from purple maize. LWT Food Sci. Technol. 2014, 59, 270–275. [Google Scholar] [CrossRef]
- Smolarz, H.D.; Bogucka-Kocka, A.; Mendyk, E.; Kocki, J. Pinostrobin—An Anti-Leukemic Flavonoid from Polygonum lapathifolium L. ssp. nodosum (Pers.) Dans. Zeitschrift fur Naturforsch. Sect. C J. Biosci. 2006, 61, 64–68. [Google Scholar] [CrossRef]
- Benson, A.A.; Daniel, H.; Wiser, R. A sulfolipid in plants. Proc. Natl. Acad. Sci. USA 1959, 45, 1582–1587. [Google Scholar] [CrossRef] [PubMed]
- Bharitkar, Y.P.; Bathini, S.; Ojha, D.; Ghosh, S.; Mukherjee, H.; Kuotsu, K.; Chattopadhyay, D.; Mondal, N.B. Antibacterial and antiviral evaluation of sulfonoquinovosyldiacylglyceride: A glycolipid isolated from Azadirachta indica leaves. Lett. Appl. Microbiol. 2014, 58, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.K.; Pradhan, B.S.; Banerjee, S.; Bikash Mondal, N.; Majumder, S.S.; Bhattacharyya, M.; Chakrabarti, S.; Roychoudhury, S.; Kumar Majumder, H. Sulfonoquinovosyl diacylglyceride selectively targets acute lymphoblastic leukemia cells and exerts potent anti-leukemic effects in vivo OPEN. Nat. Publ. Gr. 2015. [Google Scholar] [CrossRef]
- Tipton, C.L.; Ming-Chung, W.; Tsao, F.H.C.; Chang-chu, L.T.; Husted, R.R. Biosynthesis of 1,4-benzoxazin-3-ones in Zea mays. Phytochemistry 1973, 12, 347–352. [Google Scholar] [CrossRef]
- Willmott, C.J.R.; Maxwell, A. A single point mutation in the DNA gyrase A protein greatly reduces binding of fluoroquinolones to the gyrase-DNA complex. Antimicrob. Agents Chemother. 1993, 37, 126–127. [Google Scholar] [CrossRef]
- Wu, J.-H.; Chang, F.-R.; Hayashi, K.; Shiraki, H.; Liaw, C.-C.; Nakanishi, Y.; Bastow, K.F.; Yu, D.; Chen, I.-S.; Lee, K.-H. Antitumor agents. Part 218: Cappamensin A, a new In vitro anticancer principle, from Capparis sikkimensis. Bioorg. Med. Chem. Lett. 2003, 13, 2223–2225. [Google Scholar] [CrossRef]
- Liao, J.F.; Chiou, W.F.; Shen, Y.C.; Wang, G.J.; Chen, C.F. Anti-inflammatory and anti-infectious effects of Evodia rutaecarpa (Wuzhuyu) and its major bioactive components. Chin. Med. 2011, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Majumder, H.; Roychoudhury, S. Natural Compounds as Anticancer Agents Targeting DNA Topoisomerases. Curr. Genom. 2016, 18, 75–92. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Hu, C. Evodiamine: A Novel Anti-Cancer Alkaloid from Evodia rutaecarpa. Molecules 2009, 14, 1852–1859. [Google Scholar] [CrossRef]
- Dong, G.; Sheng, C.; Wang, S.; Miao, Z.; Yao, J.; Zhang, W. Selection of evodiamine as a novel topoisomerase i inhibitor by structure-based virtual screening and hit optimization of evodiamine derivatives as antitumor agents. J. Med. Chem. 2010, 53, 7521–7531. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.T.J.; George, J.H. Structural reassignment of cytosporolides A-C via Biomimetic synthetic studies and reinterpretation of NMR Data. Org. Lett. 2011, 13, 5318–5321. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Niu, S.; Sun, B.; Liu, S.; Liu, X.; Che, Y. Cytosporolides A-C, antimicrobial meroterpenoids with a unique peroxylactone skeleton from cytospora sp. Org. Lett. 2010, 12, 3144–3147. [Google Scholar] [CrossRef] [PubMed]
- Otake, K.; Yamada, K.; Miura, K.; Sasazawa, Y.; Miyazaki, S.; Niwa, Y.; Ogura, A.; Takao, K.-I.; Simizu, S. Identification of topoisomerases as molecular targets of cytosporolide C and its analog. Bioorganic Med. Chem. 2019, 27, 3334–3338. [Google Scholar] [CrossRef] [PubMed]
- Uivarosi, V.; Munteanu, A. Flavonoid Complexes as Promising Anticancer Metallodrugs. In Flavonoids—From Biosynthesis to Human Health; InTech: London, UK, 2017. [Google Scholar]
- Vutey, V.; Castelli, S.; D’Annessa, I.; Sâmia, L.B.P.; Souza-Fagundes, E.M.; Beraldo, H.; Desideri, A. Human topoisomerase IB is a target of a thiosemicarbazone copper(II) complex. Arch. Biochem. Biophys. 2016, 606, 34–40. [Google Scholar] [CrossRef]
- Li, C.W.; Xia, M.W.; Cui, C.B.; Peng, J.X.; Li, D.H. A novel oxaphenalenone, penicimutalidine: Activated production of oxaphenalenones by the diethyl sulphate mutagenesis of marine-derived fungus Penicillium purpurogenum G59. RSC Adv. 2016, 6, 82277–82281. [Google Scholar] [CrossRef]
- Andersen, R.J.; Faulkner, D.J.; Cun-heng, H.; Van Duyne, G.D.; Clardy, J. Metabolites of the Marine Prosobranch mollusc lamellaria sp. J. Am. Chem. Soc. 1985, 107, 5492–5495. [Google Scholar] [CrossRef]
- Tan, K.W.; Seng, H.L.; Lim, F.S.; Cheah, S.C.; Ng, C.H.; Koo, K.S.; Mustafa, M.R.; Ng, S.W.; Maah, M.J. Towards a selective cytotoxic agent for prostate cancer: Interaction of zinc complexes of polyhydroxybenzaldehyde thiosemicarbazones with topoisomerase i. Polyhedron 2012, 38, 275–284. [Google Scholar] [CrossRef]
- Rozmer, Z.; Perjési, P. Naturally occurring chalcones and their biological activities. Phytochem. Rev. 2016, 15, 87–120. [Google Scholar] [CrossRef]
- Isa, N.M.; Abdelwahab, S.I.; Mohan, S.; Abdul, A.B.; Sukari, M.A.; Taha, M.M.E.; Syam, S.; Narrima, P.; Cheah, S.C.; Ahmad, S.; et al. In vitro anti-inflammatory, cytotoxic and antioxidant activities of boesenbergin A, a chalcone isolated from Boesenbergia rotunda (L.) (fingerroot). Brazilian J. Med. Biol. Res. 2012, 45, 524–530. [Google Scholar] [CrossRef]
- Ajiboye, T.O.; Yakubu, M.T.; Oladiji, A.T. Cytotoxic, Antimutagenic, and Antioxidant Activities of Methanolic Extract and Chalcone Dimers (Lophirones B and C) Derived from Lophira alata (Van Tiegh. Ex Keay) Stem Bark. J. Evid. Based Complement. Altern. Med. 2014, 19, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.W.; Liu, Y. Molecular structure and stereochemistry of silybin A, silybin B, isosilybin A, and isosilybin B, isolated from Silybum marianum (milk thistle). J. Nat. Prod. 2003, 66, 1171–1174. [Google Scholar] [CrossRef] [PubMed]
- Cufí, S.; Bonavia, R.; Vazquez-Martin, A.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyàs, E.; Martin-Castillo, B.; Barrajón-Catalán, E.; Visa, J.; Segura-Carretero, A.; et al. Silibinin meglumine, a water-soluble form of milk thistle silymarin, is an orally active anti-cancer agent that impedes the epithelial-to-mesenchymal transition (EMT) in EGFR-mutant non-small-cell lung carcinoma cells. Food Chem. Toxicol. 2013, 60, 360–368. [Google Scholar] [CrossRef]
- Kasala, E.R.; Bodduluru, L.N.; Madana, R.M.; Athira, K.V.; Gogoi, R.; Barua, C.C. Chemopreventive and therapeutic potential of chrysin in cancer: Mechanistic perspectives. Toxicol. Lett. 2015, 233, 214–225. [Google Scholar] [CrossRef]
- León, I.E.; Cadavid-Vargas, J.F.; Tiscornia, I.; Porro, V.; Castelli, S.; Katkar, P.; Desideri, A.; Bollati-Fogolin, M.; Etcheverry, S.B. Oxidovanadium(IV) complexes with chrysin and silibinin: Anticancer activity and mechanisms of action in a human colon adenocarcinoma model. J. Biol. Inorg. Chem. 2015, 20, 1175–1191. [Google Scholar] [CrossRef]
- Marco, E.; Laine, W.; Tardy, C.; Lansiaux, A.; Iwao, M.; Ishibashi, F.; Bailly, C.; Gago, F. Molecular determinants of topoisomerase I poisoning by lamellarins: Comparison with camptothecin and structure-activity relationships. J. Med. Chem. 2005, 48, 3796–3807. [Google Scholar] [CrossRef]
- Perry, N.B.; Ettouati, L.; Litaudon, M.; Blunt, J.W.; Munro, M.H.G.; Parkin, S.; Hope, H. Alkaloids from the antarctic sponge Kirkpatrickia varialosa. Part 1: Variolin b, a new antitumour and antiviral compound. Tetrahedron 1994, 50, 3987–3992. [Google Scholar] [CrossRef]
- Furrow, F.B.; Amsler, C.D.; McClintock, J.B.; Baker, B.J. Surface sequestration of chemical feeding deterrents in the Antarctic sponge Latrunculia apicalis as an optimal defense against sea star spongivory. Mar. Biol. 2003, 443–449. [Google Scholar] [CrossRef]
- Beretta, G.L.; Gatti, L.; Perego, P.; Zaffaroni, N. Camptothecin resistance in cancer: Insights into the molecular mechanisms of a DNA-damaging drug. Curr. Med. Chem. 2013, 20, 1541–1565. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.Y.; Song, I.C.; Ko, Y.B.; Lee, H.J. The combination of cisplatin and topotecan as a second-line treatment for patients with advanced/recurrent uterine cervix cancer. Medicine 2018, 97. [Google Scholar] [CrossRef]
- Fang, S.-M.; Wu, C.-J.; Li, C.-W.; Cui, C.-B. A Practical Strategy to Discover New Antitumor Compounds by Activating Silent Metabolite Production in Fungi by Diethyl Sulphate Mutagenesis. Mar. Drugs 2014, 12, 1788–1814. [Google Scholar] [CrossRef] [PubMed]
- Djerassi, C.; Lam, W.K. Sponge Phospholipids. Acc. Chem. Res. 1991, 24, 69–75. [Google Scholar] [CrossRef]
- Carballeira, N.M.; Montano, N.; Amador, L.A.; Rodríguez, A.D.; Golovko, M.Y.; Golovko, S.A.; Reguera, R.M.; Álvarez-Velilla, R.; Balaña-Fouce, R. Novel Very Long-Chain α-Methoxylated Δ5,9 Fatty Acids from the Sponge Asteropus Niger Are Effective Inhibitors of Topoisomerases IB. Lipids 2016, 51, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Quesada, A.R.; García Grávalos, M.D.; Fernández Puentes, J.L. Polyaromatic alkaloids from marine invertebrates as cytotoxic compounds and inhibitors of multidrug resistance caused by P-glycoprotein. Br. J. Cancer 1996, 74, 677–682. [Google Scholar] [CrossRef]
- Imperatore, C.; Aiello, A.; D’Aniello, F.; Senese, M.; Menna, M. Alkaloids from marine invertebrates as important leads for anticancer drugs discovery and development. Molecules 2014, 19, 20391–20423. [Google Scholar] [CrossRef] [PubMed]
- Facompré, M.; Tardy, C.; Bal-Mahieu, C.; Colson, P.; Perez, C.; Manzanares, I.; Cuevas, C.; Bailly, C. Lamellarin D: A Novel Potent Inhibitor of Topoisomerase I. Cancer Res. 2003, 63, 7392–7399. [Google Scholar] [PubMed]
- Liu, M.; Wang, G.; Xiao, L.; Xu, A.; Liu, X.; Xu, P.; Lin, X. Bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, a marine algae derived bromophenol, inhibits the growth of Botrytis cinerea and interacts with DNA molecules. Mar. Drugs 2014, 12, 3838–3851. [Google Scholar] [CrossRef]
- Xu, N.; Fan, X.; Yan, X.; Li, X.; Niu, R.; Tseng, C.K. Antibacterial bromophenols from the marine red alga Rhodomela confervoides. Phytochemistry 2003, 62, 1221–1224. [Google Scholar] [CrossRef]
- Shi, D.; Xu, F.; He, J.; Li, J.; Fan, X.; Han, L. Inhibition of bromophenols against PTP1B and anti-hyperglycemic effect of Rhodomela confervoides extract in diabetic rats. Chin. Sci. Bull. 2008, 53, 2476–2479. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, W.; Wei, J.; Qiu, L.; Lin, X. Marine bromophenol bis(2,3-dibromo-4,5-dihydroxybenzyl) ether, induces mitochondrial apoptosis in K562 cells and inhibits topoisomerase I in vitro. Toxicol. Lett. 2012, 211, 126–134. [Google Scholar] [CrossRef]
- Canals, A.; Arribas-Bosacoma, R.; Albericio, F.; Álvarez, M.; Aymamí, J.; Coll, M. Intercalative DNA binding of the marine anticancer drug variolin B. Sci. Rep. 2017, 7, 1–5. [Google Scholar] [CrossRef]
- Simone, M.; Erba, E.; Damia, G.; Vikhanskaya, F.; Di Francesco, A.M.; Riccardi, R.; Bailly, C.; Cuevas, C.; Fernandez Sousa-Faro, J.M.; D’Incalci, M. Variolin B and its derivate deoxy-variolin B: New marine natural compounds with cyclin-dependent kinase inhibitor activity. Eur. J. Cancer 2005, 41, 2366–2377. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, B.; Bergquist, P.R.; Battershill, C.N. Taxonomic revision of the genus Latrunculia Du Bocage (Porifera: Demospongiae: Latrunculiidae) in New Zealand. N. Z. J. Mar. Freshw. Res. 2002, 36, 151–184. [Google Scholar] [CrossRef]
- Reyes, F.; Martín, R.; Rueda, A.; Fernández, R.; Montalvo, D.; Gómez, C.; Sánchez-Puelles, J.M. Discorhabdins I and L, Cytotoxic Alkaloids from the Sponge Latrunculia brevis. J. Nat. Prod. 2004, 67, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Peifer, C.; Janussen, D.; Tasdemir, D. New discorhabdin alkaloids from the antarctic deep-sea sponge latrunculia biformis. Mar. Drugs 2019, 17, 439. [Google Scholar] [CrossRef] [PubMed]
- Kawato, Y.; Aonuma, M.; Hirota, Y.; Kuga, H.; Sato, K. Intracellular Roles of SN-38, a Metabolite of the Camptothecin Derivative CPT-11, in the Antitumor Effect of CPT-11. Cancer Res. 1991, 51, 4187–4191. [Google Scholar]
- Crea, F.; Giovannetti, E.; Cortesi, F.; Mey, V.; Nannizzi, S.; Gallegos Ruiz, M.I.; Ricciardi, S.; Del Tacca, M.; Peters, G.J.; Danesi, R. Epigenetic mechanisms of irinotecan sensitivity in colorectal cancer cell lines. Mol. Cancer Ther. 2009, 8, 1964–1973. [Google Scholar] [CrossRef] [PubMed]
- Kingsbury, W.D.; Boehm, J.C.; Jakas, D.R.; Holden, K.G.; Gallagher, G.; Caranfa, M.J.; McCabe, F.L.; Faucette, L.F.; Johnson, R.K.; Hertzberg, R.P.; et al. Synthesis of Water-Soluble (Aminoalkyl)camptothecin Analogues: Inhibition of Topoisomerase I and Antitumor Activity. J. Med. Chem. 1991, 34, 98–107. [Google Scholar] [CrossRef]
- Paton, F.; Paulden, M.; Saramago, P.; Manca, A.; Misso, K.; Palmer, S.; Eastwood, A. Topotecan for the treatment of recurrent and stage IVB carcinoma of the cervix. Health Technol. Assess. 2010, 14 (Suppl. S1), 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Kim, S.W.; Suh, C.; Lee, J.S.; Lee, J.H.; Lee, S.J.; Ryoo, B.Y.; Park, K.; Kim, J.S.; Heo, D.S.; et al. Belotecan, new camptothecin analogue, is active in patients with small-cell lung cancer: Results of a multicenter early phase II study. Ann. Oncol. 2008, 19, 123–127. [Google Scholar] [CrossRef]
- Crul, M. CKD-602 Chong Kun Dang. Curr. Opin. Investig. Drugs 2003, 4, 1455–1459. [Google Scholar] [PubMed]
- Kim, G.M.; Kim, Y.S.; Ae Kang, Y.; Jeong, J.H.; Kim, S.M.; Hong, Y.K.; Sung, J.H.; Lim, S.T.; Kim, J.H.; Kim, S.K.; et al. Efficacy and toxicity of belotecan for relapsed or refractory small cell lung cancer patients. J. Thorac. Oncol. 2012, 7, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Hun Choi, C.; Lee, Y.-Y.; Song, T.-J.; Park, H.-S.; Kyu Kim, M.; Kim, T.-J.; Lee, J.-W.; Lee, J.-H.; Bae, D.-S.; Kim, B.-G. Phase II Study of Belotecan, a Camptothecin Analogue, in Combination with Carboplatin for the Treatment of Recurrent Ovarian Cancer. Cancer 2011, 117, 2104–2111. [Google Scholar] [CrossRef]
- Rasheed, Z.A.; Rubin, E.H. Mechanisms of resistance to topoisomerase I-targeting drugs. Oncogene 2003, 7296–7304. [Google Scholar] [CrossRef]
- Li, F.; Jiang, T.; Li, Q.; Ling, X. Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: Did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am. J. Cancer Res. 2017, 7, 2350–2394. [Google Scholar] [PubMed]
- Topoisomerase I Inhibitor—List Results—ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=topoisomerase+I+inhibitor&cntry=&state=&city=&dist=&Search=Search (accessed on 19 January 2021).
- Cao, Z.; Kozielski, A.; Liu, X.; Wang, Y.; Vardeman, D.; Giovanella, B. Crystalline camptothecin-20(S)-O-propionate hydrate: A novel anticancer agent with strong activity against 19 human tumor xenografts. Cancer Res. 2009, 69, 4742–4749. [Google Scholar] [CrossRef]
- Beck, D.E.; Abdelmalak, M.; Lv, W.; Reddy, P.V.N.; Tender, G.S.; O’Neill, E.; Agama, K.; Marchand, C.; Pommier, Y.; Cushman, M. Discovery of potent indenoisoquinoline topoisomerase i poisons lacking the 3-nitro toxicophore. J. Med. Chem. 2015, 58, 3997–4015. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Chen, Y.; Yang, H.; Zhang, H.L.; Agama, K.; Pommier, Y.; An, L.K. The antitumor activity of CYB-L10, a human topoisomerase IB catalytic inhibitor. J. Enzyme Inhib. Med. Chem. 2019, 34, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, Y. DNA topoisomerase I-targeting drugs. Nihon Rinsho. 2015, 73, 174–177. [Google Scholar] [CrossRef]
- Ireton, G.C. The Domain Organization of Human Topoisomerase I. J. Biol. Chem. 1996, 271, 7602–7608. [Google Scholar] [CrossRef]
- Beutler, J.A. Natural products as a foundation for drug discovery. Curr. Protoc. Pharmacol. 2009, 1–21. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Source | Type of Inhibitor | Reference |
|---|---|---|---|
| EGCG | Camellia sinensis | Not studied | [51] |
| Kakuol | Asarum sieboldii | Catalytic Inhibitor | [52] |
| Berberine | Coptis chinensis and Berberis vulgaris | Catalytic Inhibitor | [53] |
| Pinostrobin | Honey and dietary vegetables | Poison | [54] |
| SQDG | Azadirachta indica | Catalytic Inhibitor | [55] |
| Benzoxazines | Capparis sikimensis | Catalytic Inhibitor | [56] |
| Evodiamine | Evodia rutaecarpa | Poison | [57] |
| Cytosporolide C | Cytospora sp | Not studied | [58] |
| Compound Name | Source | Type of Inhibitor | Reference |
|---|---|---|---|
| Zinc complexes of polyhydroxybenzaldehyde thiosemicarbazones | Plants natural product | Catalytic Inhibitor | [88] |
| Chalcones-Thiosemicarbazone copper(II) complex | Piper methysticum, Boesen-bergia rotunda, Lophira alata | Catalytic Inhibitor | [89] |
| Silibinin oxidovanadium (IV) | Silybum marianum | Catalytic Inhibitor | [90] |
| Compound Name | Source | Type of Inhibitor | Reference |
|---|---|---|---|
| Bacillosporin C | Penicillium purpurogenum species | Not studied | [67] |
| α-Methoxylated Δ5,9 fatty acids | Asteropus niger | Catalytic Inhibitor | [99] |
| Lamellarin D | Lamellaria spp. | Poison | [100] |
| BDDE | Leathesia nana, Rhodomela confervoides | Catalytic Inhibitor | [101] |
| Deoxyvariolin B | Kirckpatrickia variolosa | Not studied | [102] |
| Discorhabdins | Latrunculia biformis | Not studied | [103] |
| Study | Study Purpose | Time Frame | Sample Size | NCT |
|---|---|---|---|---|
| Phase I Study Clinical Trial of Camptothecin-20-O-Propionate Hydrate (CZ48) Malignant Lymphoma of Extranodal and/or Solid Organ Site and Solid Tumor | Describe the dose limiting toxicities and adverse event profile of Camptothecin-20-O-Propionate hydrate (CZ48) administered orally every day for 4 weeks | July 2008–February 2020 | 65 participants | NCT02575638 |
| A Phase I Study Indenoisoquinoline LMP744 in Adults With Relapsed Solid Tumors and Lymphomas | Establish the safety, tolerability and the maximum tolerated dose (MTD) of LMP744 administered intravenously (IV) in patients with refractory solid tumors and lymphomas | February 2017–ongoing (estimated completion October 2022) | 53 participants | NCT03030417 |
| Phase II Study Evaluate the Efficacy and Safety of TLC388 (Lipotecan®) as Second-line Treatment in Subjects With Poorly Differentiated Neuroendocrine Carcinomas | Evaluate the efficacy and safety of Lipotecan® monotherapy in subjects with poorly differentiated neuroendocrine carcinomas. Only those subjects who have failed to first line chemotherapy | July 2015–ongoing (last update 3 April 2019 | 23 participants | NCT02457273 |
| Phase II Study Study of Etirinotecan Pegol (NKTR-102) in the treatment of patients with metastatic and Recurrent Non-Small Cell Lung Cancer (NSCLC) after failure of 2nd line treatment. | Estimate the objective response rate (Complete Response or Partial Response, as measured by RECIST version 1.1) for patients with metastatic or recurrent NSCLC being treated with etirinotecan pegol after failure of second-line therapy. | January 2013–ongoing (last update April 2020) | 40 participants | NCT01773109 |
| A Phase II Study LY01610 (Irinotecan Hydrochloride Liposome Injection) in Patients with Small Cell Lung Cancer | Evaluate the efficacy and safety of LY01610 in subjects with extensive small cell lung cancer that progressed after first-line anti-tumor therapy | November 2019–Ongoing (estimated completion September 2022) | 90 participants | NCT04381910 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ottaviani, A.; Iacovelli, F.; Fiorani, P.; Desideri, A. Natural Compounds as Therapeutic Agents: The Case of Human Topoisomerase IB. Int. J. Mol. Sci. 2021, 22, 4138. https://doi.org/10.3390/ijms22084138
Ottaviani A, Iacovelli F, Fiorani P, Desideri A. Natural Compounds as Therapeutic Agents: The Case of Human Topoisomerase IB. International Journal of Molecular Sciences. 2021; 22(8):4138. https://doi.org/10.3390/ijms22084138
Chicago/Turabian StyleOttaviani, Alessio, Federico Iacovelli, Paola Fiorani, and Alessandro Desideri. 2021. "Natural Compounds as Therapeutic Agents: The Case of Human Topoisomerase IB" International Journal of Molecular Sciences 22, no. 8: 4138. https://doi.org/10.3390/ijms22084138
APA StyleOttaviani, A., Iacovelli, F., Fiorani, P., & Desideri, A. (2021). Natural Compounds as Therapeutic Agents: The Case of Human Topoisomerase IB. International Journal of Molecular Sciences, 22(8), 4138. https://doi.org/10.3390/ijms22084138