
Dynamic Distribution of HIG2A between the Mitochondria and the Nucleus in Response to Hypoxia and Oxidative Stress


Abstract
:1. Introduction
2. Results
2.1. Effect of Oxidative Stress on HIG2A Protein Levels in the HEK293 Cell Line
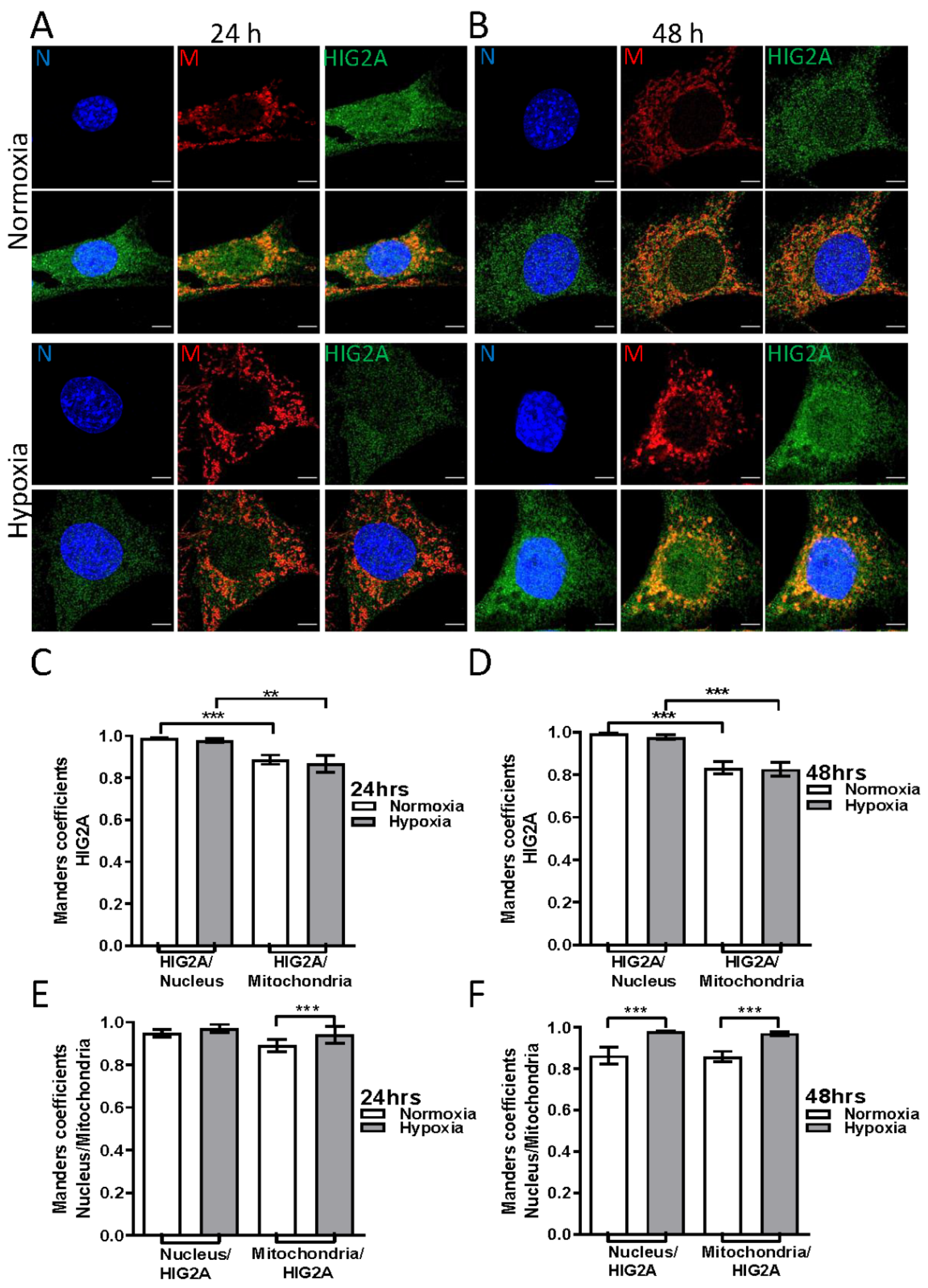
2.2. Effect of Hypoxic Stress on the Subcellular Localization of HIG2A in HEK293 and C2C12 Cell Lines
2.3. Effect of Mitochondrial Stress on the Subcellular Localization of HIG2A in the Nucleus of HEK293 Cells: Live Cell Image Analysis
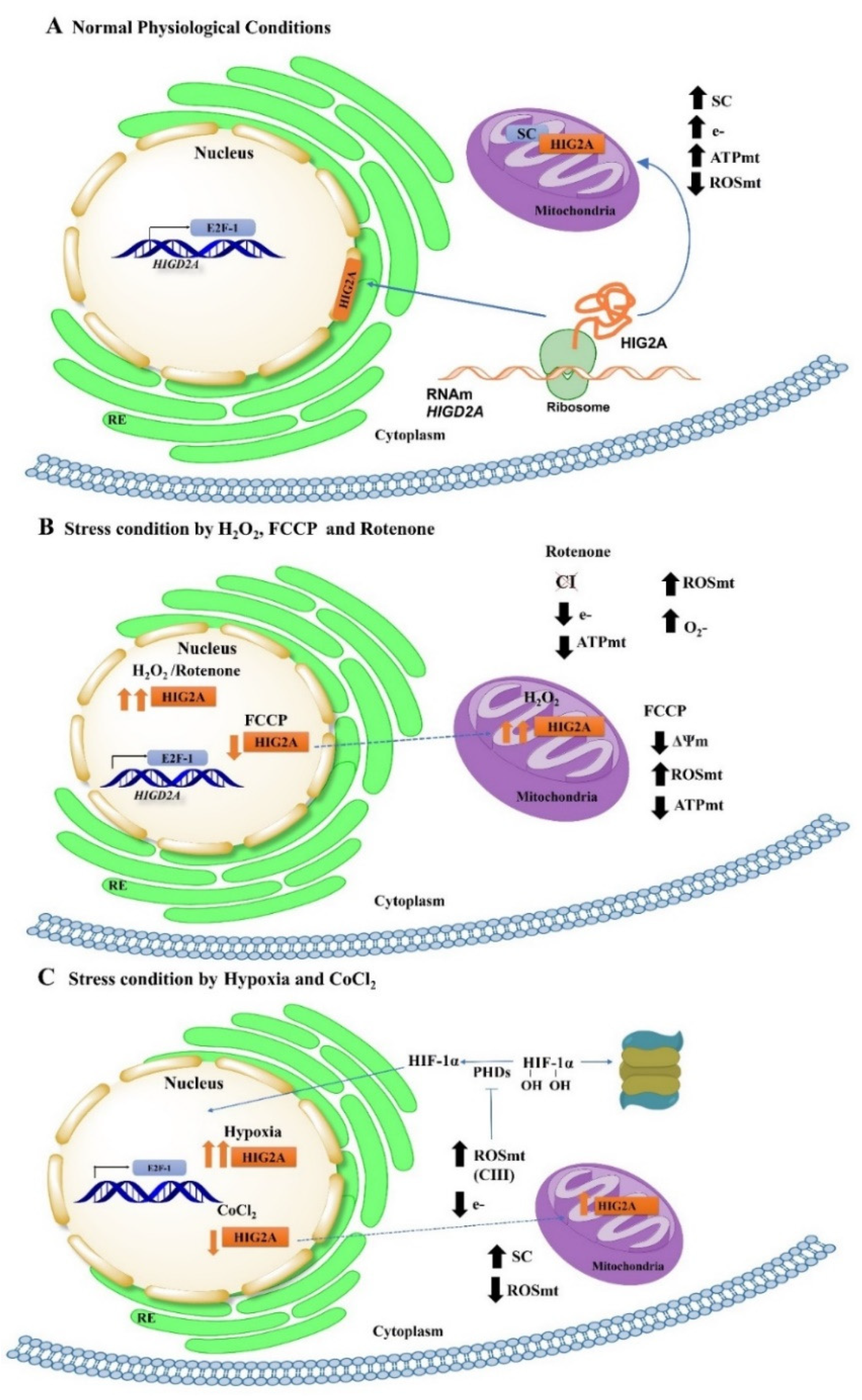
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Hypoxia Induction
4.3. Induction of Cellular and Mitochondrial Stress by Pharmacological Treatments
4.4. Indirect Immunofluorescence (IFA)
4.5. Image Acquisition and Processing
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef] [Green Version]
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial Respiratory Supercomplex Association Limits Production of Reactive Oxygen Species from Complex, I. Antioxid. Redox Signal. 2013, 19, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Diaz, F.; Antonio Enriquez, J.; Moraes, C.T. Cells Lacking Rieske Iron-Sulfur Protein Have a Reactive Oxygen Species-Associated Decrease in Respiratory Complexes I and IV. Mol. Cell. Biol. 2012, 32, 415–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acín-Pérez, R.; Fernández-Silva, P.; Peleato, M.L.; Pérez-Martos, A.; Enriquez, J.A. Respiratory Active Mitochondrial Supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Genova, M.L.; Lenaz, G. A critical appraisal of the role of respiratory supercomplexes in mitochondria. Biol. Chem. 2013, 394, 631–639. [Google Scholar] [CrossRef]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, M.A.; et al. Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef] [PubMed]
- Schagger, H.; de Coo, R.; Bauer, M.F.; Hofmann, S.; Godinot, C.; Brandt, U. Significance of respirasomes for the assembly/stability of human respiratory chain complex I. J. Biol. Chem. 2004, 279, 36349–36353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-C.; Taylor, E.B.; Dephoure, N.; Heo, J.-M.; Tonhato, A.; Papandreou, I.; Nath, N.; Denko, N.C.; Gygi, S.P.; Rutter, J. Identification of a protein mediating respiratory supercomplex stability. Cell Metab. 2012, 15, 348–360. [Google Scholar] [CrossRef] [Green Version]
- Strogolova, V.; Furness, A.; Robb-McGrath, M.; Garlich, J.; Stuart, R.A. Rcf1 and Rcf2, Members of the Hypoxia-Induced Gene 1 Protein Family, Are Critical Components of the Mitochondrial Cytochrome bc(1)-Cytochrome c Oxidase Supercomplex. Mol. Cell. Biol. 2012, 32, 1363–1373. [Google Scholar] [CrossRef] [Green Version]
- Vukotic, M.; Oeljeklaus, S.; Wiese, S.; Vögtle, F.N.; Meisinger, C.; Meyer, H.E.; Zieseniss, A.; Katschinski, D.M.; Jans, D.C.; Jakobs, S.; et al. Rcf1 Mediates Cytochrome Oxidase Assembly and Respirasome Formation, Revealing Heterogeneity of the Enzyme Complex. Cell Metab. 2012, 15, 336–347. [Google Scholar] [CrossRef] [Green Version]
- Timón-Gómez, A.; Garlich, J.; Stuart, R.A.; Ugalde, C.; Barrientos, A. Distinct Roles of Mitochondrial HIGD1A and HIGD2A in Respiratory Complex and Supercomplex Biogenesis. Cell Rep. 2020, 31, 107607. [Google Scholar] [CrossRef]
- Salazar, C.; Elorza, A.A.; Cofre, G.; Ruiz-Hincapie, P.; Shirihai, O.; Ruiz, L.M. The OXPHOS supercomplex assembly factor HIG2A responds to changes in energetic metabolism and cell cycle. J. Cell. Physiol. 2019, 234, 17405–17419. [Google Scholar] [CrossRef]
- An, H.-J.; Shin, H.; Jo, S.-G.; Kim, Y.J.; Lee, J.-O.; Paik, S.-G.; Lee, H. The survival effect of mitochondrial Higd-1a is associated with suppression of cytochrome C release and prevention of caspase activation. Biochim. Biophys. Acta 2011, 1813, 2088–2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gewurz, B.E.; Towfic, F.; Mar, J.C.; Shinners, N.P.; Takasaki, K.; Zhao, B.; Cahir-McFarland, E.D.; Quackenbush, J.; Xavier, R.J.; Kieff, E. Genome-wide siRNA screen for mediators of NF-kappa B activation. Proc. Natl. Acad. Sci. USA 2012, 109, 2467–2472. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; White, E.A.; Sowa, M.E.; Powell, M.L.C.; Ottinger, M.; Harper, J.W.; Howley, P.M. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 3752–3757. [Google Scholar] [CrossRef] [Green Version]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.-K.; Elledge, S.J. A Genome-wide RNAi Screen Identifies Multiple Synthetic Lethal Interactions with the Ras Oncogene. Cell 2009, 137, 835–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salazar, C.; Yañez, O.; Elorza, A.A.; Cortes, N.; García-Beltrán, O.; Tiznado, W.; Ruiz, L.M. Biosystem Analysis of the Hypoxia Inducible Domain Family Member 2A: Implications in Cancer Biology. Genes 2020, 11, 206. [Google Scholar] [CrossRef] [Green Version]
- Tanner, S.; Shen, Z.; Ng, J.; Florea, L.; Guigo, R.; Briggs, S.P.; Bafna, V. Improving gene annotation using peptide mass spectrometry. Genome Res. 2007, 17, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K.; et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [Green Version]
- Quirós, P.M.; Mottis, A.; Auwerx, J. Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 2016, 17, 213. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Butow, R.A. RTG1 and RTG2: Two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell 1993, 72, 61–71. [Google Scholar] [CrossRef]
- Torelli, N.Q.; Ferreira-Júnior, J.R.; Kowaltowski, A.J.; da Cunha, F.M. RTG1- and RTG2-dependent retrograde signaling controls mitochondrial activity and stress resistance in Saccharomyces cerevisiae. Free Radic. Biol. Med. 2015, 81, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Arnould, T.; Michel, S.; Renard, P. Mitochondria Retrograde Signaling and the UPR mt: Where Are We in Mammals? Int. J. Mol. Sci. 2015, 16, 18224–18251. [Google Scholar] [CrossRef] [Green Version]
- Guha, M.; Tang, W.; Sondheimer, N.; Avadhani, N.G. Role of calcineurin, hnRNPA2 and Akt in mitochondrial respiratory stress-mediated transcription activation of nuclear gene targets. Biochim. Biophys. Acta 2010, 1797, 1055–1065. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [Green Version]
- Formentini, L.; Sánchez-Aragó, M.; Sánchez-Cenizo, L.; Cuezva, J.M. The Mitochondrial ATPase Inhibitory Factor 1 Triggers a ROS-Mediated Retrograde Prosurvival and Proliferative Response. Mol. Cell 2012, 45, 731–742. [Google Scholar] [CrossRef] [Green Version]
- Chae, S.; Ahn, B.Y.; Byun, K.; Cho, Y.M.; Yu, M.-H.; Lee, B.; Hwang, D.; Park, K.S. A Systems Approach for Decoding Mitochondrial Retrograde Signaling Pathways. Sci. Signal. 2013, 6, rs4. [Google Scholar] [CrossRef]
- Cardamone, M.D.; Tanasa, B.; Cederquist, C.T.; Huang, J.; Mahdaviani, K.; Li, W.; Rosenfeld, M.G.; Liesa, M.; Perissi, V. Mitochondrial Retrograde Signaling in Mammals Is Mediated by the Transcriptional Cofactor GPS2 via Direct Mitochondria-to-Nucleus Translocation. Mol. Cell 2018, 69, 757–772.e757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazwinski, S.M. The Retrograde Response: When Mitochondrial Quality Control Is Not Enough. Biochim. Biophys. Acta 2013, 1833, 400–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazwinski, S.M. Chapter Five—The Retrograde Response: A Conserved Compensatory Reaction to Damage from Within and from Without. In Progress in Molecular Biology and Translational Science; Osiewacz, H.D., Ed.; Academic Press: Cambridge, MA, USA, 2014; Volume 127, pp. 133–154. [Google Scholar]
- Ameri, K.; Rajah, A.M.; Nguyen, V.; Sanders, T.A.; Jahangiri, A.; DeLay, M.; Donne, M.; Choi, H.J.; Tormos, K.V.; Yeghiazarians, Y.; et al. Nuclear Localization of the Mitochondrial Factor HIGD1A during Metabolic Stress. PLoS ONE 2013, 8, e62758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piret, J.-P.; Mottet, D.; Raes, M.; Michiels, C. CoCl2, a Chemical Inducer of Hypoxia-Inducible Factor-1, and Hypoxia Reduce Apoptotic Cell Death in Hepatoma Cell Line HepG2. Ann. N. Y. Acad. Sci. 2002, 973, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Kovářová, N.; Pecina, P.; Nůsková, H.; Vrbacký, M.; Zeviani, M.; Mráček, T.; Viscomi, C.; Houštěk, J. Tissue- and species-specific differences in cytochrome c oxidase assembly induced by SURF1 defects. Biochim. Biophys. Acta 2016, 1862, 705–715. [Google Scholar] [CrossRef] [Green Version]
- Kovarova, N.; Vrbacka, A.C.; Pecina, P.; Stranecky, V.; Pronicka, E.; Kmoch, S.; Houstek, J. Adaptation of respiratory chain biogenesis to cytochrome c oxidase deficiency caused by SURF1 gene mutations. Biochim. Biophys. Acta 2012, 1822, 1114–1124. [Google Scholar] [CrossRef] [Green Version]
- Ameri, K.; Jahangiri, A.; Rajah, A.M.; Tormos, K.V.; Nagarajan, R.; Pekmezci, M.; Nguyen, V.; Wheeler, M.L.; Murphy, M.P.; Sanders, T.A.; et al. HIGD1A regulates oxygen consumption, ROS production and AMPK activity during glucose deprivation to modulate cell survival and tumor growth. Cell Rep. 2015, 10, 891–899. [Google Scholar] [CrossRef] [Green Version]
- Dewilde, S.; Vercelli, A.; Chiarle, R.; Poli, V. Of alphas and betas: Distinct and overlapping functions of STAT3 isoforms. Front. Biosci.-Landmark 2008, 13, 6501–6514. [Google Scholar] [CrossRef] [Green Version]
- Avalle, L.; Poli, V. Nucleus, Mitochondrion, or Reticulum? STAT3 à La Carte. Int. J. Mol. Sci. 2018, 19, 2820. [Google Scholar] [CrossRef] [Green Version]
- Maritano, D.; Sugrue, M.L.; Tininini, S.; Dewilde, S.; Strobl, B.; Fu, X.; Murray-Tait, V.; Chiarle, R.; Poli, V. The STAT3 isoforms α and β have unique and specific functions. Nat. Immunol. 2004, 5, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Gough, D.J.; Corlett, A.; Schlessinger, K.; Wegrzyn, J.; Larner, A.C.; Levy, D.E. Mitochondrial STAT3 Supports Ras-Dependent Oncogenic Transformation. Science 2009, 324, 1713. [Google Scholar] [CrossRef] [Green Version]
- Wegrzyn, J.; Potla, R.; Chwae, Y.-J.; Sepuri, N.B.V.; Zhang, Q.; Koeck, T.; Derecka, M.; Szczepanek, K.; Szelag, M.; Gornicka, A.; et al. Function of Mitochondrial Stat3 in Cellular Respiration. Science 2009, 323, 793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, Ó.; Ondo-Méndez, A.; Garzón, R. Activación de STAT3 por hipoxia en modelos in vitro de cáncer de cuello uterino y en células endoteliales. Biomédica 2017, 37, 119–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.H.; Son, J.M.; Benayoun, B.A.; Lee, C. The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell Metab. 2018, 28, 516–524.e517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yong, C.Q.; Tang, B.L. A Mitochondrial Encoded Messenger at the Nucleus. Cells 2018, 7, 105. [Google Scholar] [CrossRef] [Green Version]
- Melber, A.; Haynes, C.M. UPR(mt) regulation and output: A stress response mediated by mitochondrial-nuclear communication. Cell Res. 2018, 28, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Kornmann, B. Quality control in mitochondria: Use it, break it, fix it, trash it. F1000Prime Rep. 2014, 6, 15. [Google Scholar] [CrossRef]
- Pellegrino, M.W.; Nargund, A.M.; Haynes, C.M. Signaling the Mitochondrial Unfolded Protein Response. Biochim. Biophys. Acta 2013, 1833, 410–416. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.M.; Haynes, C.M. UPR(mt)-mediated cytoprotection and organismal aging. Biochim. Biophys. Acta 2015, 1847, 1448–1456. [Google Scholar] [CrossRef] [Green Version]
- Nargund, A.M.; Pellegrino, M.W.; Fiorese, C.J.; Baker, B.M.; Haynes, C.M. Mitochondrial Import Efficiency of ATFS-1 Regulates Mitochondrial UPR Activation. Science 2012, 337, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Fiorese, C.J.; Schulz, A.M.; Lin, Y.-F.; Rosin, N.; Pellegrino, M.W.; Haynes, C.M. The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr. Biol. 2016, 26, 2037–2043. [Google Scholar] [CrossRef] [Green Version]
- English, J.; Son, J.M.; Cardamone, M.D.; Lee, C.; Perissi, V. Decoding the rosetta stone of mitonuclear communication. Pharmacol. Res. 2020, 161, 105161. [Google Scholar] [CrossRef]
- Iwata, T.; Mizusawa, N.; Taketani, Y.; Itakura, M.; Yoshimoto, K. Parafibromin tumor suppressor enhances cell growth in the cells expressing SV40 large T antigen. Oncogene 2007, 26, 6176–6183. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.H.; DeCaprio, J.A. Cellular transformation by SV40 large T antigen: Interaction with host proteins. Semin. Cancer Biol. 2001, 11, 15–22. [Google Scholar] [CrossRef]
- Fanning, E.; Knippers, R. Structure and Function of Simian Virus 40 Large Tumor Antigen. Annu. Rev. Biochem. 1992, 61, 55–85. [Google Scholar] [CrossRef]
- Chen, Y.; Fu, L.L.; Wen, X.; Wang, X.Y.; Liu, J.; Cheng, Y.; Huang, J. Sirtuin-3 (SIRT3), a therapeutic target with oncogenic and tumor-suppressive function in cancer. Cell Death Dis. 2014, 5, e1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Porporato, P.E.; Filigheddu, N.; Pedro, J.M.B.-S.; Kroemer, G.; Galluzzi, L. Mitochondrial metabolism and cancer. Cell Res. 2018, 28, 265–280. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashton, T.M.; McKenna, W.G.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez, F.; Lim, J.-H.; Chim, H.; Bhalla, K.; Girnun, G.; Pierce, K.; Clish, C.B.; Granter, S.R.; Widlund, H.R.; Spiegelman, B.M.; et al. PGC1&; Expression Defines a Subset of Human Melanoma Tumors with Increased Mitochondrial Capacity and Resistance to Oxidative Stress. Cancer Cell 2013, 23, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Lai, R.K.-H.; Xu, I.M.-J.; Chiu, D.K.-C.; Tse, A.P.-W.; Wei, L.L.; Law, C.-T.; Lee, D.; Wong, C.-M.; Wong, M.P.; Ng, I.O.-L.; et al. NDUFA4L2 Fine-tunes Oxidative Stress in Hepatocellular Carcinoma. Clin. Cancer Res. 2016, 22, 3105. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Nie, S.-L.; Zhou, J.-M.; Liu, F.; Hu, Y.-B.; Jiang, J.-R.; Li, N.; Liu, J.-S. Overexpression of NDUFA4L2 is associated with poor prognosis in patients with colorectal cancer. ANZ J. Surg. 2017, 87, E251–E255. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.A.; Robinson, T.J.; Liu, J.C.; Shrestha, M.; Voisin, V.; Ju, Y.; Chung, P.E.D.; Pellecchia, G.; Fell, V.L.; Bae, S.; et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J. Clin. Investig. 2016, 126, 3739–3757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitaker-Menezes, D.; Martinez-Outschoorn, U.E.; Flomenberg, N.; Birbe, R.; Witkiewicz, A.K.; Howell, A.; Pavlides, S.; Tsirigos, A.; Ertel, A.; Pestell, R.G.; et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ. Cell Cycle 2011, 10, 4047–4064. [Google Scholar] [CrossRef] [Green Version]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Birkenmeier, K.; Dröse, S.; Wittig, I.; Winkelmann, R.; Käfer, V.; Döring, C.; Hartmann, S.; Wenz, T.; Reichert, A.S.; Brandt, U.; et al. Hodgkin and Reed–Sternberg cells of classical Hodgkin lymphoma are highly dependent on oxidative phosphorylation. Int. J. Cancer 2016, 138, 2231–2246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadava, N.; Nicholls, D.G. Spare respiratory capacity rather than oxidative stress regulates glutamate excitotoxicity after partial respiratory inhibition of mitochondrial complex I with rotenone. J. Neurosci. 2007, 27, 7310–7317. [Google Scholar] [CrossRef] [Green Version]
- Greenamyre, J.T.; Betarbet, R.; Sherer, T.B. The rotenone model of Parkinson’s disease: Genes, environment and mitochondria. Parkinsonism Relat. Disord. 2003, 9, 59–64. [Google Scholar] [CrossRef]
- Kurosu, T.; Tsuji, K.; Kida, A.; Koyama, T.; Yamamoto, M.; Miura, O. Rottlerin synergistically enhances imatinib-induced apoptosis of BCR/ABL-expressing cells through its mitochondrial uncoupling effect independent of protein kinase C-δ. Oncogene 2007, 26, 2975–2987. [Google Scholar] [CrossRef] [Green Version]
- Tello, D.; Balsa, E.; Acosta-Iborra, B.; Fuertes-Yebra, E.; Elorza, A.; Ordóñez, Á.; Corral-Escariz, M.; Soro, I.; López-Bernardo, E.; Perales-Clemente, E.; et al. Induction of the Mitochondrial NDUFA4L2 Protein by HIF-1α Decreases Oxygen Consumption by Inhibiting Complex I Activity. Cell Metab. 2011, 14, 768–779. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, R.; Zhang, H.; Kim, J.-W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 Regulates Cytochrome Oxidase Subunits to Optimize Efficiency of Respiration in Hypoxic Cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Cogliati, S.; Calvo, E.; Loureiro, M.; Guaras, A.M.; Nieto-Arellano, R.; Garcia-Poyatos, C.; Ezkurdia, I.; Mercader, N.; Vázquez, J.; Enriquez, J.A. Mechanism of super-assembly of respiratory complexes III and IV. Nature 2016, 539, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Horie-Inoue, K.; Suzuki, T.; Hobo, R.; Nakasato, N.; Takeda, S.; Inoue, S. Mitochondrial supercomplex assembly promotes breast and endometrial tumorigenesis by metabolic alterations and enhanced hypoxia tolerance. Nat. Commun. 2019, 10, 4108. [Google Scholar] [CrossRef]
- Wu, D.; Yotnda, P. Induction and testing of hypoxia in cell culture. J. Vis. Exp. 2011, 54, e2899. [Google Scholar] [CrossRef] [Green Version]
- Kamogashira, T.; Hayashi, K.; Fujimoto, C.; Iwasaki, S.; Yamasoba, T. Functionally and morphologically damaged mitochondria observed in auditory cells under senescence-inducing stress. NPJ Aging Mech. Dis. 2017, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Kumar, N.; Roginskaya, V.; Fouquerel, E.; Opresko, P.L.; Shiva, S.; Watkins, S.C.; Kolodieznyi, D.; Bruchez, M.P.; Van Houten, B. Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc. Natl. Acad. Sci. USA 2019, 116, 18435–18444. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Wang, Z.; Zhang, M.; Huang, C.; Song, Y.; Xu, F.; Zhang, J.; Li, J.; He, M.; Li, Y.; et al. SQR mediates therapeutic effects of H(2)S by targeting mitochondrial electron transport to induce mitochondrial uncoupling. Sci. Adv. 2020, 6, eaaz5752. [Google Scholar] [CrossRef]
- Belin, B.J.; Lee, T.; Mullins, R.D. DNA damage induces nuclear actin filament assembly by Formin-2 and Spire-½ that promotes efficient DNA repair. eLife 2015, 4, e07735. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Winlove, C.P.; Michel, C.C. Oxygen Partial Pressure in Outer Layers of Skin of Human Finger Nail Folds. J. Physiol. 2003, 549, 855–863. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Kittler, J.; Illingworth, J.; Föglein, J. Threshold selection based on a simple image statistic. Comput. Vis. Graph. Image Process. 1985, 30, 125–147. [Google Scholar] [CrossRef]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and Quantitative Measurement of Protein-Protein Colocalization in Live Cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinchuk, V.; Wu, Y.; Grossenbacher-Zinchuk, O. Bridging the gap between qualitative and quantitative colocalization results in fluorescence microscopy studies. Sci. Rep. 2013, 3, 1365. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | HIG2A/Nucleus [M2] | HIG2A/Mitochondria [M1] | p-Value | Nucleus/HIG2A [M1] | Mitochondria/HIG2A [M2] |
|---|---|---|---|---|---|
| Normoxia 24 h | 0.95 | 0.85 | ** p ≤ 0.0011 | 0.84 | 0.90 |
| Hypoxia 24 h | 0.94 | 0.88 | ** p ≤ 0.0011 | 0.79 | 0.95 |
| Normoxia 48 h | 0.96 | 0.78 | *** p ≤ 0.0001 | 0.89 | 0.95 |
| Hypoxia 48 h | 0.94 | 0.82 | ** p ≤ 0.0029 | 0.85 | 0.93 |
| Treatment | HIG2A/Nucleus [M2] | HIG2A/Mitochondria [M1] | p-Value | Nucleus/HIG2A [M1] | Mitochondria/HIG2A [M2] |
|---|---|---|---|---|---|
| Normoxia 24 h | 0.98 | 0.88 | *** p ≤ 0.0001 | 0.94 | 0.89 2 |
| Hypoxia 24 h | 0.97 | 0.86 | ** p ≤ 0.0073 | 0.97 | 0.94 2 |
| Normoxia 48 h | 0.99 | 0.83 | *** p ≤ 0.0001 | 0.86 1 | 0.85 3 |
| Hypoxia 48 h | 0.97 | 0.82 | *** p ≤ 0.0002 | 0.97 1 | 0.97 3 |
| Treatment | HIG2A/Nucleus [M2] | p-Value |
|---|---|---|
| Control | 0.95 | |
| FCCP 1 h | 0.88 | * p-value ≤ 0.0106 |
| FCCP 3 h | 0.90 | ** p-value ≤ 0.0061 |
| H2O2 1 h | 0.95 | |
| TG | 0.94 | |
| CoCl2 | 0.87 | * p-value ≤ 0.0142 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salazar, C.; Barros, M.; Elorza, A.A.; Ruiz, L.M. Dynamic Distribution of HIG2A between the Mitochondria and the Nucleus in Response to Hypoxia and Oxidative Stress. Int. J. Mol. Sci. 2022, 23, 389. https://doi.org/10.3390/ijms23010389
Salazar C, Barros M, Elorza AA, Ruiz LM. Dynamic Distribution of HIG2A between the Mitochondria and the Nucleus in Response to Hypoxia and Oxidative Stress. International Journal of Molecular Sciences. 2022; 23(1):389. https://doi.org/10.3390/ijms23010389
Chicago/Turabian StyleSalazar, Celia, Miriam Barros, Alvaro A. Elorza, and Lina María Ruiz. 2022. "Dynamic Distribution of HIG2A between the Mitochondria and the Nucleus in Response to Hypoxia and Oxidative Stress" International Journal of Molecular Sciences 23, no. 1: 389. https://doi.org/10.3390/ijms23010389