
Commercial SARS-CoV-2 Targeted, Protease Inhibitor Focused and Protein–Protein Interaction Inhibitor Focused Molecular Libraries for Virtual Screening and Drug Design



Abstract
:1. Introduction
2. In Silico Library Design for Medicinal Chemistry
3. Methods
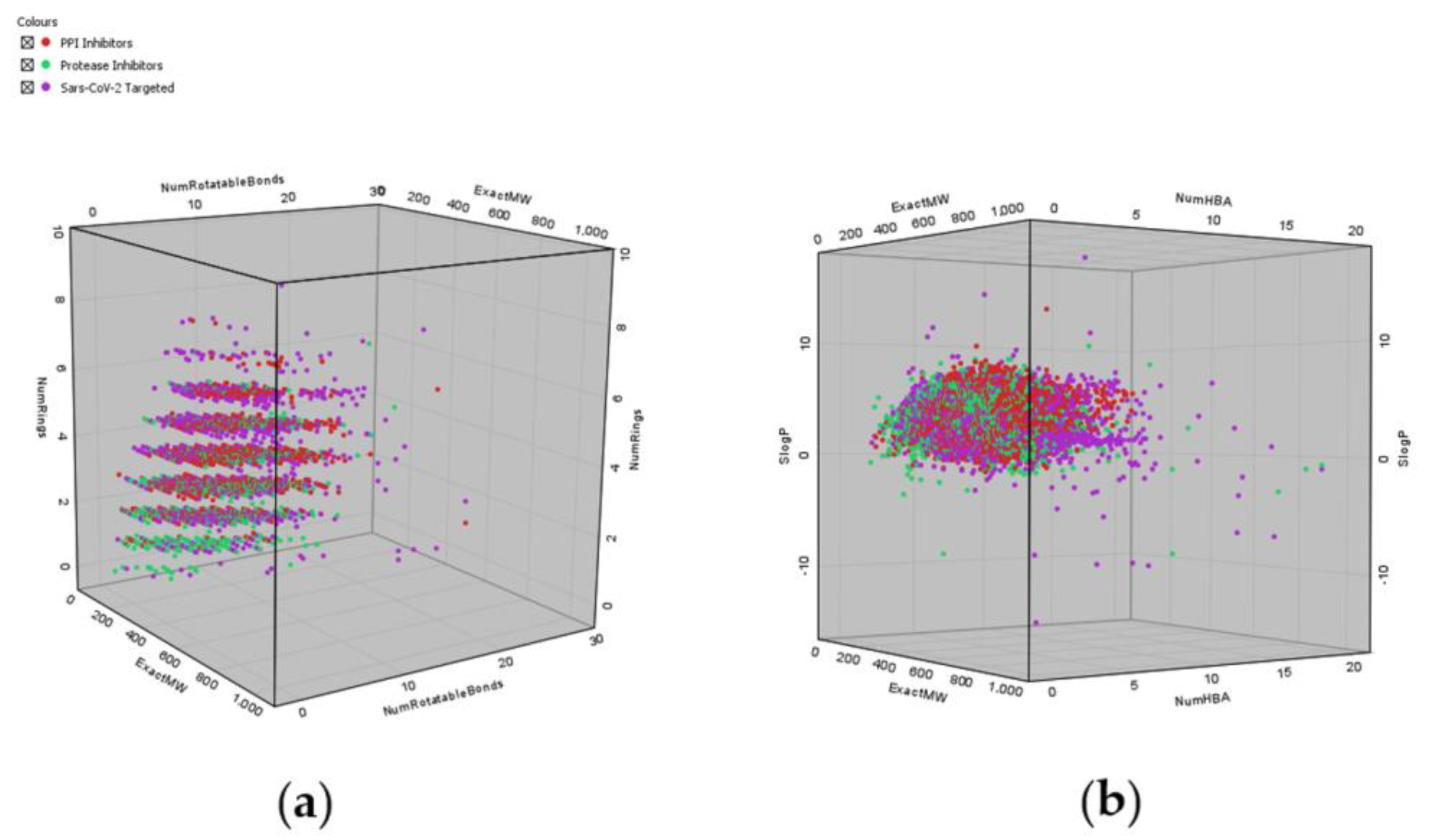
4. Examples of Commercial Targeted Libraries
4.1. SARS-CoV-2- or COVID-19-Targeted Libraries
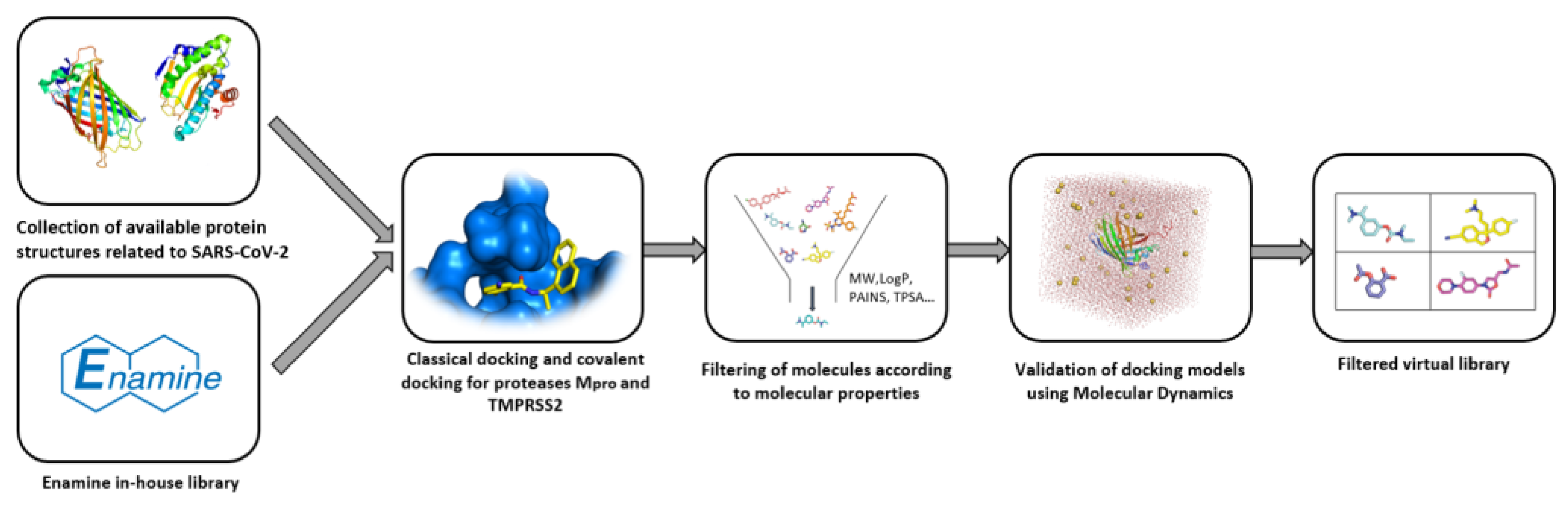
4.1.1. Enamine
4.1.2. Otava
4.1.3. Chembridge
4.1.4. Life Chemicals
4.1.5. TargetMol
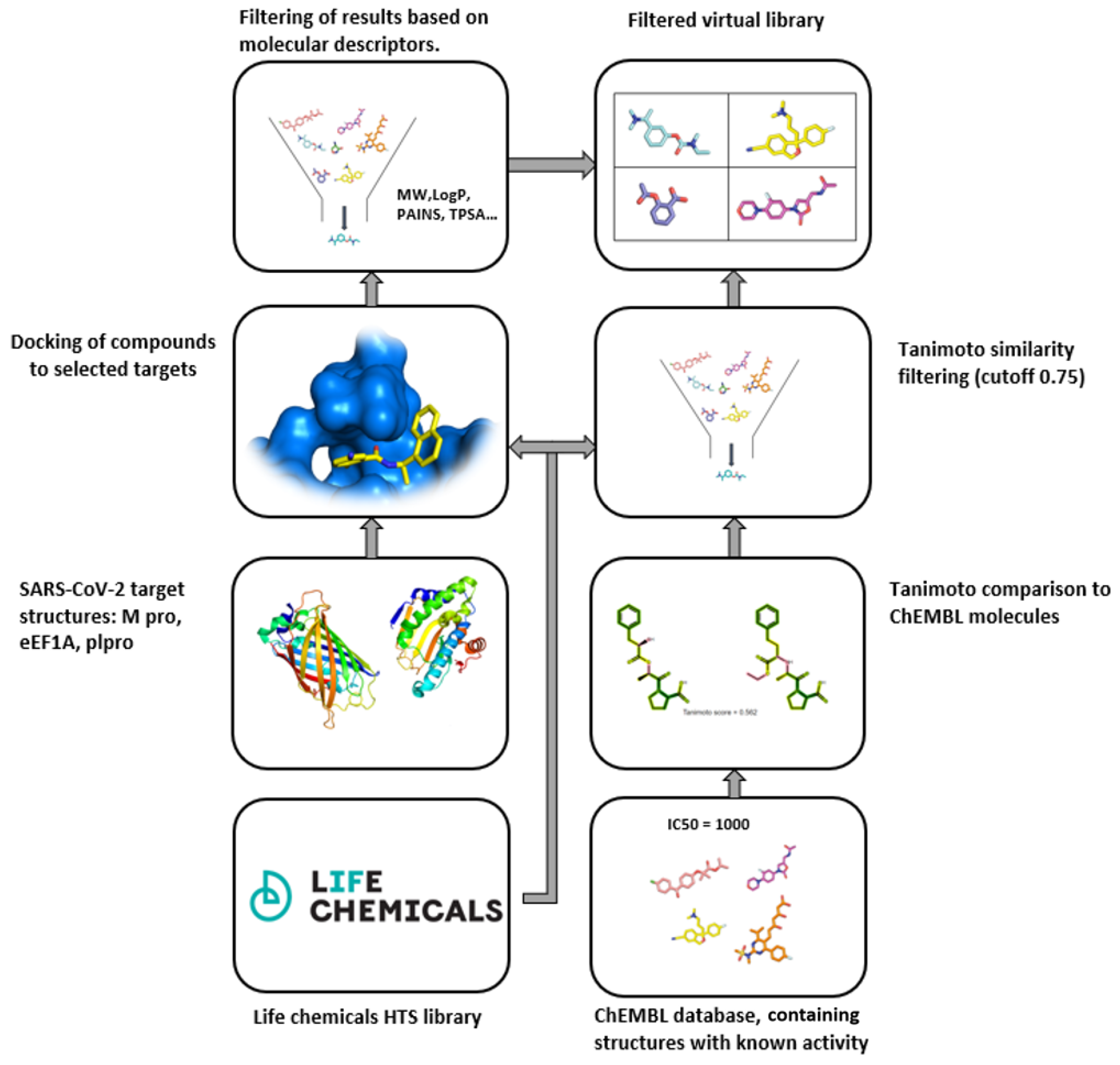
4.2. Protease-Inhibitor-Focused Libraries
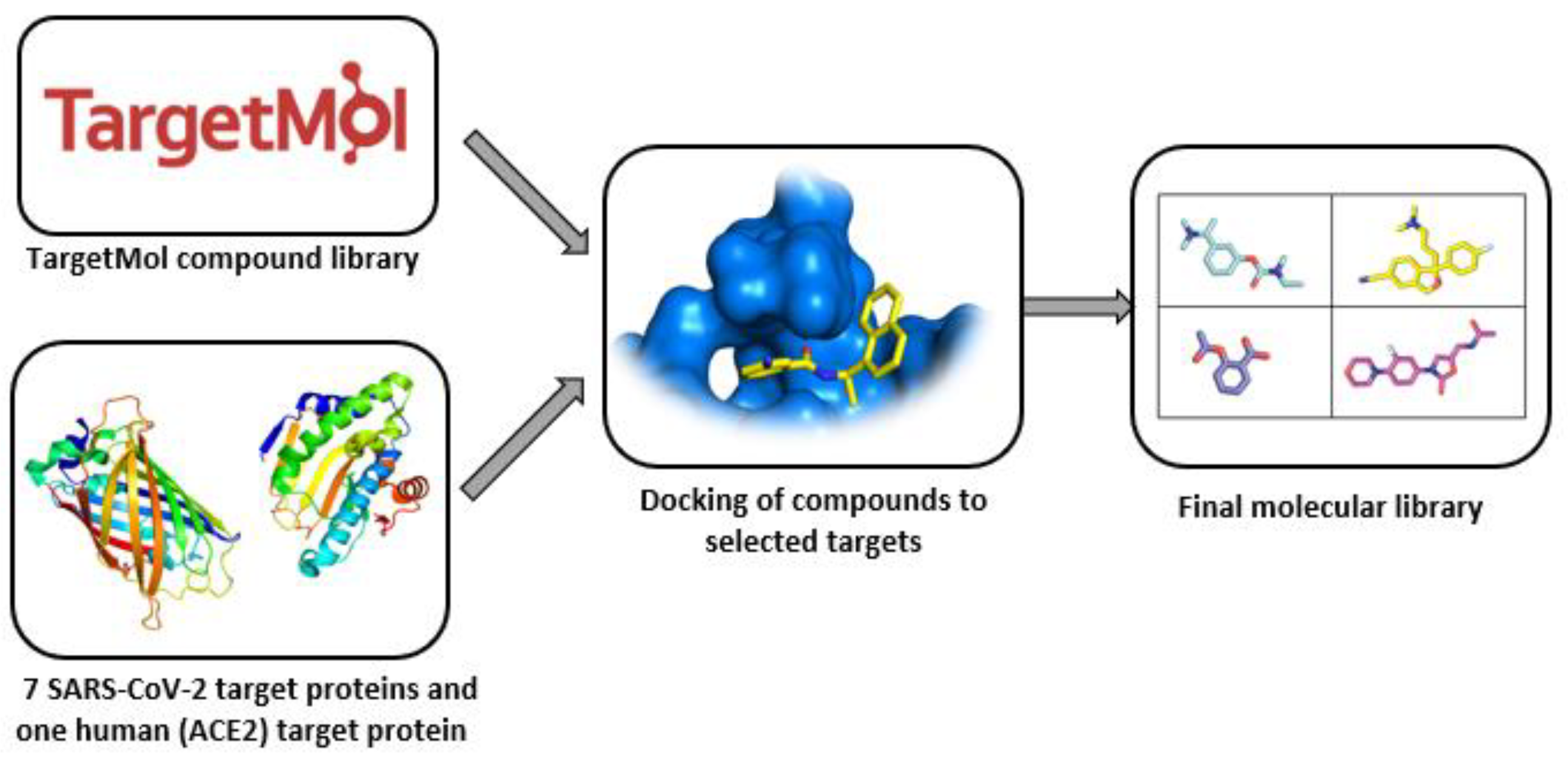
4.3. Protein–Protein-Interaction-Inhibitor-Focused Libraries
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HTVS | high-throughput virtual screening |
| CADD | computer-aided drug design |
| QSAR | quantitative structure-activity relation |
| PBVS | pharmacophore-based virtual screening |
| DVBS | docking-based virtual screening |
| SDF | Structure Data Format |
| MOL | MDL Molfile |
| PPI | protein–protein interaction |
| MW | Molecular Weight |
| TPSA | Total Polar Surface Area |
| HBA | number of hydrogen bond acceptors |
| HBD | number of hydrogen bond donors |
References
- Mohs, R.C.; Greig, N.H. Drug Discovery and Development: Role of Basic Biological Research. Alzheimers Dement. Transl. Res. Clin. Interv. 2017, 3, 651–657. [Google Scholar] [CrossRef]
- Lionta, E.; Spyrou, G.; Vassilatis, D.; Cournia, Z. Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem. 2014, 14, 1923–1938. [Google Scholar] [CrossRef]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to Improve R&D Productivity: The Pharmaceutical Industry’s Grand Challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-F.; Zhong, W.-Z. Drug Design and Discovery: Principles and Applications. Molecules 2017, 22, 279. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganesan, A.; Coote, M.L.; Barakat, K. Molecular Dynamics-Driven Drug Discovery: Leaping Forward with Confidence. Drug Discov. Today 2017, 22, 249–269. [Google Scholar] [CrossRef]
- Damale, M.G.; Harke, S.N.; Kalam Khan, F.A.; Shinde, D.B.; Sangshetti, J.N. Recent Advances in Multidimensional QSAR (4D-6D): A Critical Review. Mini Rev. Med. Chem. 2014, 14, 35–55. [Google Scholar] [CrossRef]
- Ballester, P.J. Machine Learning for Molecular Modelling in Drug Design. Biomolecules 2019, 9, 216. [Google Scholar] [CrossRef] [Green Version]
- Zhavoronkov, A.; Ivanenkov, Y.A.; Aliper, A.; Veselov, M.S.; Aladinskiy, V.A.; Aladinskaya, A.V.; Terentiev, V.A.; Polykovskiy, D.A.; Kuznetsov, M.D.; Asadulaev, A.; et al. Deep Learning Enables Rapid Identification of Potent DDR1 Kinase Inhibitors. Nat. Biotechnol. 2019, 37, 1038–1040. [Google Scholar] [CrossRef] [PubMed]
- Walters, W.P. Virtual Chemical Libraries: Miniperspective. J. Med. Chem. 2019, 62, 1116–1124. [Google Scholar] [CrossRef] [PubMed]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular Docking and High-Throughput Screening for Novel Inhibitors of Protein Tyrosine Phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Braga, R.C.; Alves, V.M.; Silva, A.C.; Nascimento, M.N.; Silva, F.C.; Liao, L.M.; Andrade, C.H. Virtual Screening Strategies in Medicinal Chemistry: The State of the Art and Current Challenges. Curr. Top. Med. Chem. 2014, 14, 1899–1912. [Google Scholar] [CrossRef] [PubMed]
- Mayr, L.M.; Fuerst, P. The Future of High-Throughput Screening. J. Biomol. Screen. 2008, 13, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Reichman, M.; Simpson, P.B. Open Innovation in Early Drug Discovery: Roadmaps and Roadblocks. Drug Discov. Today 2016, 21, 779–788. [Google Scholar] [CrossRef]
- Gimeno, A.; Ojeda-Montes, M.; Tomás-Hernández, S.; Cereto-Massagué, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. The Light and Dark Sides of Virtual Screening: What Is There to Know? Int. J. Mol. Sci. 2019, 20, 1375. [Google Scholar] [CrossRef] [Green Version]
- Subramaniam, S.; Mehrotra, M.; Gupta, D. Virtual High Throughput Screening (VHTS)—A Perspective. Bioinformation 2008, 3, 14–17. [Google Scholar] [CrossRef] [Green Version]
- McInnes, C. Virtual Screening Strategies in Drug Discovery. Curr. Opin. Chem. Biol. 2007, 11, 494–502. [Google Scholar] [CrossRef]
- Chen, Z.; Li, H.; Zhang, Q.; Bao, X.; Yu, K.; Luo, X.; Zhu, W.; Jiang, H. Pharmacophore-Based Virtual Screening versus Docking-Based Virtual Screening: A Benchmark Comparison against Eight Targets. Acta Pharmacol. Sin. 2009, 30, 1694–1708. [Google Scholar] [CrossRef] [Green Version]
- Patrick, G. An Introduction to Medicinal Chemistry, 6th ed.; Oxford University Press: New York, NY, USA, 2017; ISBN 978-0-19-874969-1. [Google Scholar]
- Seidel, T.; Wieder, O.; Garon, A.; Langer, T. Applications of the Pharmacophore Concept in Natural Product Inspired Drug Design. Mol. Inform. 2020, 39, 2000059. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Docking, Virtual High Throughput Screening and in Silico Fragment-Based Drug Design. J. Cell. Mol. Med. 2009, 13, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Sousa, S.F.; Fernandes, P.A.; Ramos, M.J. Protein-Ligand Docking: Current Status and Future Challenges. Proteins Struct. Funct. Bioinform. 2006, 65, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking Covalent Inhibitors: A Parameter Free Approach to Pose Prediction and Scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- Furlan, V.; Konc, J.; Bren, U. Inverse Molecular Docking as a Novel Approach to Study Anticarcinogenic and Anti-Neuroinflammatory Effects of Curcumin. Molecules 2018, 23, 3351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jukič, M.; Janežič, D.; Bren, U. Ensemble Docking Coupled to Linear Interaction Energy Calculations for Identification of Coronavirus Main Protease (3CLpro) Non-Covalent Small-Molecule Inhibitors. Molecules 2020, 25, 5808. [Google Scholar] [CrossRef] [PubMed]
- Bakken, G.A.; Bell, A.S.; Boehm, M.; Everett, J.R.; Gonzales, R.; Hepworth, D.; Klug-McLeod, J.L.; Lanfear, J.; Loesel, J.; Mathias, J.; et al. Shaping a Screening File for Maximal Lead Discovery Efficiency and Effectiveness: Elimination of Molecular Redundancy. J. Chem. Inf. Model. 2012, 52, 2937–2949. [Google Scholar] [CrossRef]
- Njoroge, M.; Njuguna, N.M.; Mutai, P.; Ongarora, D.S.B.; Smith, P.W.; Chibale, K. Recent Approaches to Chemical Discovery and Development against Malaria and the Neglected Tropical Diseases Human African Trypanosomiasis and Schistosomiasis. Chem. Rev. 2014, 114, 11138–11163. [Google Scholar] [CrossRef] [PubMed]
- Morgan, P.; Brown, D.G.; Lennard, S.; Anderton, M.J.; Barrett, J.C.; Eriksson, U.; Fidock, M.; Hamrén, B.; Johnson, A.; March, R.E.; et al. Impact of a Five-Dimensional Framework on R&D Productivity at AstraZeneca. Nat. Rev. Drug Discov. 2018, 17, 167–181. [Google Scholar] [CrossRef]
- Blay, V.; Tolani, B.; Ho, S.P.; Arkin, M.R. High-Throughput Screening: Today’s Biochemical and Cell-Based Approaches. Drug Discov. Today 2020, 25, 1807–1821. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Galloway, W.R.J.D.; Spring, D.R. A Question of Library Design. Nature 2011, 470, 42–43. [Google Scholar] [CrossRef]
- Peakman, M.-C.; Troutman, M.; Gonzales, R.; Schmidt, A. Experimental Screening Strategies to Reduce Attrition Risk. In Attrition in the Pharmaceutical Industry; Alex, A., Harris, C.J., Smith, D.A., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2015; pp. 180–214. ISBN 978-1-118-81958-6. [Google Scholar]
- Capdeville, R.; Buchdunger, E.; Zimmermann, J.; Matter, A. Glivec (STI571, Imatinib), a Rationally Developed, Targeted Anticancer Drug. Nat. Rev. Drug Discov. 2002, 1, 493–502. [Google Scholar] [CrossRef]
- Talele, T.T.; Khedkar, S.A.; Rigby, A.C. Successful Applications of Computer Aided Drug Discovery: Moving Drugs from Concept to the Clinic. Curr. Top. Med. Chem. 2010, 10, 127–141. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Peng, Z.; Sutton, S.C.; Na, J.; Kostrowicki, J.; Yang, B.; Thacher, T.; Kong, X.; Mattaparti, S.; Zhou, J.Z.; et al. Pfizer Global Virtual Library (PGVL): A Chemistry Design Tool Powered by Experimentally Validated Parallel Synthesis Information. ACS Comb. Sci. 2012, 14, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Peng, Z.; Kostrowicki, J.; Kuki, A. LEAP into the Pfizer Global Virtual Library (PGVL) Space: Creation of Readily Synthesizable Design Ideas Automatically. Methods Mol. Biol. 2011, 685, 253–276. [Google Scholar] [CrossRef]
- Nicolaou, C.A.; Watson, I.A.; Hu, H.; Wang, J. The Proximal Lilly Collection: Mapping, Exploring and Exploiting Feasible Chemical Space. J. Chem. Inf. Model. 2016, 56, 1253–1266. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Berdini, V.; Hartshorn, M.J.; Mooij, W.T.M.; Murray, C.W.; Taylor, R.D.; Watson, P. Virtual Screening Using Protein−Ligand Docking: Avoiding Artificial Enrichment. J. Chem. Inf. Comput. Sci. 2004, 44, 793–806. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Müller, A.T.; Huisman, B.J.H.; Fuchs, J.A.; Schneider, P.; Schneider, G. Generative Recurrent Networks for De Novo Drug Design. Mol. Inform. 2018, 37, 1700111. [Google Scholar] [CrossRef] [Green Version]
- Colby, S.M.; Nuñez, J.R.; Hodas, N.O.; Corley, C.D.; Renslow, R.R. Deep Learning to Generate in Silico Chemical Property Libraries and Candidate Molecules for Small Molecule Identification in Complex Samples. Anal. Chem. 2020, 92, 1720–1729. [Google Scholar] [CrossRef] [Green Version]
- Rigoni, D.; Navarin, N.; Sperduti, A. A Systematic Assessment of Deep Learning Models for Molecule Generation. arXiv 2020, arXiv:2008.09168. [Google Scholar]
- Xue, D.; Gong, Y.; Yang, Z.; Chuai, G.; Qu, S.; Shen, A.; Yu, J.; Liu, Q. Advances and Challenges in Deep Generative Models for de Novo Molecule Generation. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2019, 9, e1395. [Google Scholar] [CrossRef]
- Walters, W.P.; Barzilay, R. Applications of Deep Learning in Molecule Generation and Molecular Property Prediction. Acc. Chem. Res. 2021, 54, 263–270. [Google Scholar] [CrossRef]
- Leach, A. The in Silico World of Virtual Libraries. Drug Discov. Today 2000, 5, 326–336. [Google Scholar] [CrossRef]
- Bernardo, P.H.; Tong, J.C. In Silico Design of Small Molecules. In Chemical Genomics and Proteomics; Zanders, E.D., Ed.; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2012; Volume 800, pp. 25–31. ISBN 978-1-61779-348-6. [Google Scholar]
- Śledź, P.; Caflisch, A. Protein Structure-Based Drug Design: From Docking to Molecular Dynamics. Curr. Opin. Struct. Biol. 2018, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Dobson, P.D.; Patel, Y.; Kell, D.B. ‘Metabolite-Likeness’ as a Criterion in the Design and Selection of Pharmaceutical Drug Libraries. Drug Discov. Today 2009, 14, 31–40. [Google Scholar] [CrossRef] [PubMed]
- T Garcia-Sosa, A.; Maran, U.; Hetenyi, C. Molecular Property Filters Describing Pharmacokinetics and Drug Binding. Curr. Med. Chem. 2012, 19, 1646–1662. [Google Scholar] [CrossRef]
- Olah, M.M.; Bologa, C.G.; Oprea, T.I. Strategies for Compound Selection. Curr. Drug Discov. Technol. 2004, 1, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Irwin, J.J.; Duan, D.; Torosyan, H.; Doak, A.K.; Ziebart, K.T.; Sterling, T.; Tumanian, G.; Shoichet, B.K. An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 2015, 58, 7076–7087. [Google Scholar] [CrossRef] [Green Version]
- Dalby, A.; Nourse, J.G.; Hounshell, W.D.; Gushurst, A.K.I.; Grier, D.L.; Leland, B.A.; Laufer, J. Description of Several Chemical Structure File Formats Used by Computer Programs Developed at Molecular Design Limited. J. Chem. Inf. Comput. Sci. 1992, 32, 244–255. [Google Scholar] [CrossRef]
- Martin, Y.C. Let’s Not Forget Tautomers. J. Comput. Aided Mol. Des. 2009, 23, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Oellien, F.; Cramer, J.; Beyer, C.; Ihlenfeldt, W.-D.; Selzer, P.M. The Impact of Tautomer Forms on Pharmacophore-Based Virtual Screening. J. Chem. Inf. Model. 2006, 46, 2342–2354. [Google Scholar] [CrossRef]
- Brooks, W.H.; Guida, W.C.; Daniel, K.G. The Significance of Chirality in Drug Design and Development. Curr. Top. Med. Chem. 2011, 11, 760–770. [Google Scholar] [CrossRef]
- Brooks, W.H.; McCloskey, D.E.; Daniel, K.G.; Ealick, S.E.; Secrist, J.A.; Waud, W.R.; Pegg, A.E.; Guida, W.C. In Silico Chemical Library Screening and Experimental Validation of a Novel 9-Aminoacridine Based Lead-Inhibitor of Human S-Adenosylmethionine Decarboxylase. J. Chem. Inf. Model. 2007, 47, 1897–1905. [Google Scholar] [CrossRef]
- Meng, E.C.; Gschwend, D.A.; Blaney, J.M.; Kuntz, I.D. Orientational Sampling and Rigid-Body Minimization in Molecular Docking. Proteins Struct. Funct. Genet. 1993, 17, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Price, D.A.; Blagg, J.; Jones, L.; Greene, N.; Wager, T. Physicochemical Drug Properties Associated with in Vivo Toxicological Outcomes: A Review. Expert Opin. Drug Metab. Toxicol. 2009, 5, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Lenci, E.; Trabocchi, A. Peptidomimetic Toolbox for Drug Discovery. Chem. Soc. Rev. 2020, 49, 3262–3277. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Cao, S.; Su, P.-C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit Identification and Optimization in Virtual Screening: Practical Recommendations Based on a Critical Literature Analysis: Miniperspective. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef] [Green Version]
- Downs, G.M.; Barnard, J.M. Clustering Methods and Their Uses in Computational Chemistry. In Reviews in Computational Chemistry; Lipkowitz, K.B., Boyd, D.B., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2002; Volume 18, pp. 1–40. ISBN 978-0-471-21576-9. [Google Scholar]
- Böcker, A.; Derksen, S.; Schmidt, E.; Teckentrup, A.; Schneider, G. A Hierarchical Clustering Approach for Large Compound Libraries. J. Chem. Inf. Model. 2005, 45, 807–815. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Walters, W.P.; Murcko, M.A. Library Filtering Systems and Prediction of Drug-Like Properties. In Methods and Principles in Medicinal Chemistry; Böhm, H., Schneider, G., Eds.; Wiley: Hoboken, NJ, USA, 2000; pp. 15–32. ISBN 978-3-527-30153-9. [Google Scholar]
- Walters, W.P.; Murcko, M.A. Prediction of “Drug-Likeness”. Adv. Drug Deliv. Rev. 2002, 54, 255–271. [Google Scholar] [CrossRef]
- Bruns, R.F.; Watson, I.A. Rules for Identifying Potentially Reactive or Promiscuous Compounds. J. Med. Chem. 2012, 55, 9763–9772. [Google Scholar] [CrossRef] [PubMed]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A “rule of Three” for Fragment-Based Lead Discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Morelli, X.; Bourgeas, R.; Roche, P. Chemical and Structural Lessons from Recent Successes in Protein–Protein Interaction Inhibition (2P2I). Curr. Opin. Chem. Biol. 2011, 15, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I.; Davis, A.M.; Teague, S.J.; Leeson, P.D. Is There a Difference between Leads and Drugs? A Historical Perspective. J. Chem. Inf. Comput. Sci. 2001, 41, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Lee, M.L.; Schneider, G. Scaffold Architecture and Pharmacophoric Properties of Natural Products and Trade Drugs: Application in the Design of Natural Product-Based Combinatorial Libraries. J. Comb. Chem. 2001, 3, 284–289. [Google Scholar] [CrossRef]
- Van De Waterbeemd, H.; Camenisch, G.; Folkers, G.; Raevsky, O.A. Estimation of Caco-2 Cell Permeability Using Calculated Molecular Descriptors. Quant. Struct.-Act. Relatsh. 1996, 15, 480–490. [Google Scholar] [CrossRef]
- Morin-Allory, L.; Mozziconacci, J.C.; Arnoult, E.; Baurin, N.; Marot, C. Preparation of a Molecular Database from a Set of 2 Million Compounds for Virtual Screening Applications: Gathering, Structural Analysis and Filtering; Institut de Chimie Organique et Analytique, Universite d’Orleans: Orléans, France, 2003. [Google Scholar]
- Fichert, T.; Yazdanian, M.; Proudfoot, J.R. A Structure-Permeability Study of Small Drug-like Molecules. Bioorg. Med. Chem. Lett. 2003, 13, 719–722. [Google Scholar] [CrossRef]
- Muegge, I. Pharmacophore Features of Potential Drugs. Chem. Weinh. Bergstr. Ger. 2002, 8, 1976–1981. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Ajay; Bemis, G.W.; Murcko, M.A. Designing Libraries with CNS Activity. J. Med. Chem. 1999, 42, 4942–4951. [Google Scholar] [CrossRef]
- Oprea, T.I. Property Distribution of Drug-Related Chemical Databases. J. Comput. Aided Mol. Des. 2000, 14, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.; Ou, C.; He, J.; Liu, L.; Shan, H.; Lei, C.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 Main Protease as Drug Target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Jukič, M.; Janežič, D.; Bren, U. Potential Novel Thioether-Amide or Guanidine-Linker Class of SARS-CoV-2 Virus RNA-Dependent RNA Polymerase Inhibitors Identified by High-Throughput Virtual Screening Coupled to Free-Energy Calculations. Int. J. Mol. Sci. 2021, 22, 11143. [Google Scholar] [CrossRef]
- Jukič, M.; Škrlj, B.; Tomšič, G.; Pleško, S.; Podlipnik, Č.; Bren, U. Prioritisation of Compounds for 3CLpro Inhibitor Development on SARS-CoV-2 Variants. Molecules 2021, 26, 3003. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Huang, B.; Tang, J.; Liu, S.; Liu, M.; Ye, Y.; Liu, Z.; Xiong, Y.; Zhu, W.; Cao, D.; et al. The Complex Structure of GRL0617 and SARS-CoV-2 PLpro Reveals a Hot Spot for Antiviral Drug Discovery. Nat. Commun. 2021, 12, 488. [Google Scholar] [CrossRef]
- Gorgulla, C.; Boeszoermenyi, A.; Wang, Z.-F.; Fischer, P.D.; Coote, P.W.; Padmanabha Das, K.M.; Malets, Y.S.; Radchenko, D.S.; Moroz, Y.S.; Scott, D.A.; et al. An Open-Source Drug Discovery Platform Enables Ultra-Large Virtual Screens. Nature 2020, 580, 663–668. [Google Scholar] [CrossRef]
- Turk, B. Targeting Proteases: Successes, Failures and Future Prospects. Nat. Rev. Drug Discov. 2006, 5, 785–799. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Bond, J.S. Proteases: Multifunctional Enzymes in Life and Disease. J. Biol. Chem. 2008, 283, 30433–30437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, S.; Hamilton, A. Protein-Protein Interaction Inhibitors: Small Molecules from Screening Techniques. Curr. Top. Med. Chem. 2007, 7, 922–927. [Google Scholar] [CrossRef]
- Toogood, P.L. Inhibition of Protein−Protein Association by Small Molecules: Approaches and Progress. J. Med. Chem. 2002, 45, 1543–1558. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Features/Cutoffs | Developer/ Reference |
|---|---|---|
| PAINS | Removal of frequent hitters (promiscuous compounds) in HTS assays | Cancer Therapeutics-CRC P/L [64] |
| REOS | Set of rules and of functional group filters for the removal of problematic structures dubbed REOS (Rapid elimination of swill) Maintaining compounds using the following cutoffs: H-bond donor ≤ 5, H-bond acceptors ≤ 10, −2 ≤ formal charge ≤ +2, number of rotatable bonds ≤ 8, 200 ≤ Molecular weight ≤ 500, 20 ≤ number of heavy atoms ≤ 50, −2 ≤ logP ≤ 5 | Vertex [65,66] |
| Aggregators | Tanimoto coefficient similarity search to a database of known aggregators | Irwin et al. [52] |
| Ely Lilly rules | Hybrid method of physiochemical and functional group filters for identification of promiscuous compounds | Bruns at Ely Lilly [67] |
| Lipinski (Rule of 5) | A set of rules for drug-likeness: Molecular Weight ≤ 500, logP ≤ 5, H-bond donors ≤ 5, H-bond acceptors ≤ 10 | Lipinski at Pfizer [46] |
| Rule of 3 | A set of cutoff rules for lead-like discovery: Molecular Weight ≤ 300, logP ≤ 3, H-bond donor ≤ 3, H-bond acceptors ≤ 3 | Congreve et al. [68] |
| Rule of 4 | A set of rules derived from protein–protein interaction inhibitors: Molecular Weight ≥ 400, logP ≥ 4, number of rings ≥ 4, H-bond acceptors ≥ 4 | Morelli [69] |
| Oprea Lead-like and drug-like | A set of rules based on the lead-like vs. drug-like comparison: Molecular Weight < 450, −3.5 ≤ logP < 4.5, −4 ≤ logD ≤ 4, number of rings ≤ 4, nonterminal single bonds ≤ 10, H-bond donor ≤ 5, H-bond acceptor ≤ 8 | Oprea group [70] |
| Ghose | A set of rules for drug-likeness with cutoffs: 180 ≤ Molecular Weight ≤ 480, 40 ≤ molecular refractivity ≤ 130, −0.4 ≤ logP ≤ 5.6, 20 ≤ number of atoms ≤ 70 | Ghose et al. [71] |
| Veber | Two rules to meet the criteria for drug-likeness: rotatable bonds ≤ 10, Polar Surface Area ≤ 140 Å2 | Veber et al [72] |
| Lee | Physiochemical properties of highly-drug like molecules: Mean Molecular Weight = 356 Mean logP = 2.1 | Lee at Hoffman-La Roche [73] |
| van de Waterbeemd | Physiochemical properties for permeability and blood brain barrier permeability: Molecular Weight ≤ 450, Polar Surface Area ≤ 90 Å2 | van de Waterbeemd [74] |
| Mozzicconacci | Set of rules to filter for drug-like molecules: Rotatable bonds ≤ 15, rings ≤ 6, oxygen atoms ≥ 1, nitrogen atoms ≥ 1, halogen atoms ≤ 7 | Mozziconacci [75] |
| Fichert | Rules for permeability based on a small drug set: Molecular Weight ≤ 500, 0 ≤ logD ≤ 3 | Fichert et al. [76] |
| Muegge method | Bioavailability prediction rules dubbed Muegge method: 200 ≤ MW ≤ 600, −2 ≤ logP ≤ 5, PSA ≤ 150 Å2, number of rings ≤ 7, number of carbons ≥ 4, number of heteroatoms > 1, Number of rotatable bonds ≤ 15, H-bond acceptors ≤ 10, H-bond donors ≤ 5 | Muegge [77] |
| Egan | Set of rules designed to predict bioavailability: logP ≤ 5.88, PSA ≤ 131.6 Å2 | Egan et al. [78] |
| Murcko filter | Set of rules derived from the rule of 5 coupled with 1D and 2D descriptor analysis to determine central nervous system activity. MW 200–400, 0 ≤ logP ≤ 5.2, H-bond acceptors ≤ 4, H-bond donor ≤ 3, rotatable bonds ≤ 7 | Ajay et al. [79] |
| Database Name | No. of Compounds | MW | TPSA | SlogP | HBA | HBD | No. of Rings | No. of Rotatable Bonds |
|---|---|---|---|---|---|---|---|---|
| Chembridge | 16,777 | 391.5 ± 62 | 80 ± 26 | 3.2 ± 1.5 | 4.9 ± 1.7 | 1.4 ± 1.0 | 4.3 ± 0.8 | 3.9 ± 1.4 |
| Enamine | 16,800 | 362 ± 61 | 79.3 ± 21 | 2.7 ± 1.2 | 4.5 ± 1.4 | 1.58 ± 0.8 | 3.1 ± 0.8 | 5.1 ± 1.8 |
| LifeChemical | 7311 | 404 ± 75 | 84.8 ± 23 | 3.1 ± 1.4 | 5.8 ± 1.8 | 1.4 ± 0.9 | 3.6 ± 0.9 | 5.6 ± 1.9 |
| Otava | 9018 | 383 ± 56 | 77.3 ± 20 | 3.7 ± 1.0 | 5.2 ± 1.5 | 1 ± 0.8 | 3.9 ± 0.8 | 4.0 ± 1.5 |
| TargetMol | 2448 | 460 ± 211 | 110 ± 151 | 2.6 ± 4.6 | 6.6 ± 4.0 | 2.2 ± 3.2 | 3.6 ± 1.5 | 7.5 ± 4.8 |
| Joined SARS-CoV-2-Targeted Library | 52,354 | 385 ± 79 | 81 ± 40 | 3.1 ± 1.7 | 5.0 ± 1.9 | 1.4 ± 1.1 | 3.7 ± 1.0 | 4.7 ± 2.1 |
| Chembridge | Enamine | LifeChemicals | Otava | TargetMol | |
|---|---|---|---|---|---|
| Unfiltered | 16,777 | 16,800 | 7311 | 9018 | 2448 |
| Isolated filter: | Number of filtered out compounds (%) | ||||
| REOS | 1160 (7%) | 4565 (27%) | 1414 (19%) | 1486 (17%) | 858 (35%) |
| PAINS | 454 (3%) | 267 (2%) | 11 (~0%) | 430 (5%) | 248 (10%) |
| Aggregators | 9053 (54%) | 6702 (40%) | 4002 (55%) | 6784 (75%) | 1445 (60%) |
| Lipinski Ro5 | 258 (2%) | 193 (1%) | 380 (5%) | 5 (~0%) | 522 (21%) |
| All filters | 10,000 (60%) | 9412 (56%) | 4937 (68%) | 7202 (80%) | 1887 (77%) |
| Database Name | No. of Compounds | MW | TPSA | SlogP | HBA | HBD | No. of Rings | No. of Rotatable Bonds |
|---|---|---|---|---|---|---|---|---|
| ApexBio | 824 | 348 ± 181 | 96 ± 59 | 1.9 ± 2.8 | 5.0 ± 3.2 | 2.5 ± 2.0 | 2.5 ± 1.7 | 5.2 ± 4.5 |
| Asinex | 6640 | 383 ± 34 | 79 ± 18 | 2.9 ± 1.0 | 5.3 ± 1.3 | 0.9 ± 0.7 | 3.7 ± 0.6 | 4.7 ± 1.5 |
| Chemdiv | 41,801 | 406 ± 63 | 74 ± 20 | 3.6 ± 1.2 | 5.0 ± 1.6 | 1.1 ± 0.7 | 3.7 ± 0.8 | 5.4 ± 1.8 |
| Enamine | 117 | 336 ± 167 | 90 ± 58 | 2.2 ± 2 | 4.4 ± 2.6 | 2.0 ± 1.8 | 2.5 ± 1.5 | 4.5 ± 4.4 |
| LifeChemicals | 25,535 | 390 ± 70 | 81 ± 22 | 3.0 ± 1.4 | 5.3 ± 1.7 | 1.0 ± 0.7 | 3.3 ± 0.9 | 4.9 ± 1.9 |
| Otava | 8034 | 352 ± 71 | 79 ± 23 | 3.0 ± 1.2 | 4.6 ± 1.6 | 1.4 ± 0.9 | 3.2 ± 1.1 | 4.5 ± 1.8 |
| SelleckChem | 227 | 409 ± 168 | 106 ± 52 | 2.4 ± 2 | 5.45 ± 2.6 | 2.4 ± 1.7 | 2.9 ± 1.7 | 6.2 ± 4.4 |
| TargetMol | 295 | 410 ± 183 | 107 ± 60 | 2.4 ± 2.2 | 5.6 ± 3.1 | 2.5 ± 2.0 | 3.0 ± 1.8 | 5.9 ± 4.5 |
| Joined Protease Inhibitor Databases | 83,473 | 394 ± 70 | 77 ± 23 | 3.3 ± 1.3 | 5.0 ± 1.7 | 1.1 ± 0.8 | 3.5 ± 0.9 | 5.1 ± 1.9 |
| ApexBio | Asinex | Chemdiv | Enamine | LifeChemicals | Otava | SelleckChem | TargetMol | |
|---|---|---|---|---|---|---|---|---|
| Unfiltered | 824 | 6640 | 41,801 | 117 | 25,535 | 8034 | 227 | 295 |
| Isolated filter: | Number of filtered out compounds (%) | |||||||
| REOS | 397(48%) | 273(4%) | 2151(5%) | 54(46%) | 3583(14%) | 1025(13%) | 110(48%) | 146(49%) |
| PAINS | 58(7%) | 129(2%) | 1060(3%) | 7(6%) | 430(2%) | 307(4%) | 7(3%) | 16(5%) |
| Aggregators | 294(36%) | 3254(49%) | 29,313(70%) | 36(31%) | 14,059(55%) | 4216(52%) | 88(39%) | 118(40%) |
| Lipinski Ro5 | 113(14%) | 0(0%) | 1045(2%) | 11(9%) | 409(2%) | 2(~0%) | 37(16%) | 55(19%) |
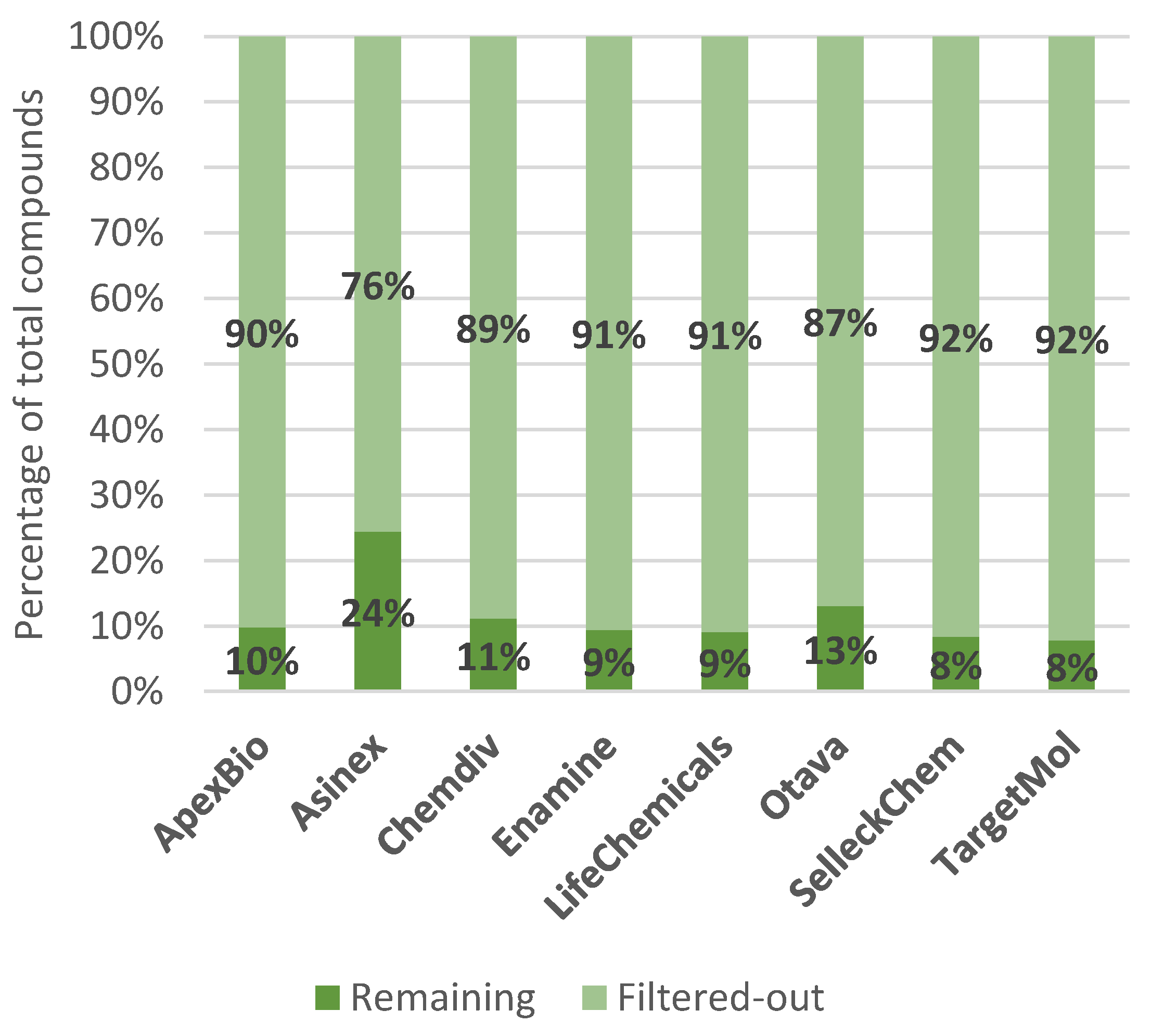
| All filters | 743(90%) | 5019(76%) | 37,137(89%) | 106(91%) | 23,215(91%) | 6987(87%) | 208(92%) | 272(92%) |
| Database Name | No. of Compounds | MW | TPSA | SlogP | HBA | HBD | No. of Rings | No. of Rotatable Bonds |
|---|---|---|---|---|---|---|---|---|
| Asinex | 11,439 | 386 ± 53 | 70 ± 19 | 3.1 ± 1.2 | 4.9 ± 1.3 | 0.8 ± 0.6 | 3.7 ± 0.7 | 4.9 ± 1.5 |
| Chemdiv | 212,906 | 408 ± 61 | 72 ± 20 | 3.5 ± 1.3 | 5.0 ± 1.6 | 0.8 ± 0.7 | 3.9 ± 0.8 | 5.1 ± 1.9 |
| Enamine | 40,640 | 357 ± 49 | 70 ± 17 | 2.8 ± 0.9 | 4.7 ± 1.3 | 1 ± 0.7 | 3.5 ± 0.7 | 4.3 ± 1.6 |
| LifeChemicals | 36,426 | 393 ± 81 | 77 ± 22 | 3.3 ± 1.1 | 5.4 ± 1.8 | 1.0 ± 0.7 | 3.6 ± 1.0 | 5.3 ± 2.2 |
| Otava | 3849 | 437 ± 71 | 86 ± 25 | 3.8 ± 1.6 | 5.4 ± 1.6 | 1.4 ± 1.0 | 3.9 ± 1.1 | 6.4 ± 2.4 |
| SelleckChem | 188 | 472 ± 183 | 101 ± 66 | 3.6 ± 2.3 | 6.3 ± 3.7 | 2.1 ± 1.7 | 3.8 ± 1.5 | 6.2 ± 3.9 |
| Joined PPI databases | 305,448 | 400 ± 65 | 73 ± 20 | 3.3 ± 1.2 | 5.0 ± 1.6 | 0.9 ± 0.7 | 3.8 ± 0.8 | 5.0 ± 1.9 |
| Asinex | Chemdiv | Enamine | LifeChemicals | Otava | SelleckChem | |
|---|---|---|---|---|---|---|
| Unfiltered | 11,439 | 212,906 | 40,640 | 36,426 | 3849 | 188 |
| Isolated filter: | Number of filtered out compounds (%) | |||||
| REOS | 148(1%) | 12,546(6%) | 970(2%) | 3191(9%) | 686(18%) | 87(47%) |
| PAINS | 18(~0%) | 3891(2%) | 239(1%) | 547(2%) | 164(4%) | 22(12%) |
| Aggregators | 6579(58%) | 132,015(62%) | 17,499(43%) | 22,721(38%) | 2682(70%) | 126(67%) |
| Lipinski Ro5 | 26(~0%) | 5936(3%) | 16(~0%) | 814(89%) | 424(11%) | 47(25%) |
| All filters | 8137(71%) | 174,713(82%) | 28,386(70%) | 31,886(88%) | 3581(93%) | 177(94%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kralj, S.; Jukič, M.; Bren, U. Commercial SARS-CoV-2 Targeted, Protease Inhibitor Focused and Protein–Protein Interaction Inhibitor Focused Molecular Libraries for Virtual Screening and Drug Design. Int. J. Mol. Sci. 2022, 23, 393. https://doi.org/10.3390/ijms23010393
Kralj S, Jukič M, Bren U. Commercial SARS-CoV-2 Targeted, Protease Inhibitor Focused and Protein–Protein Interaction Inhibitor Focused Molecular Libraries for Virtual Screening and Drug Design. International Journal of Molecular Sciences. 2022; 23(1):393. https://doi.org/10.3390/ijms23010393
Chicago/Turabian StyleKralj, Sebastjan, Marko Jukič, and Urban Bren. 2022. "Commercial SARS-CoV-2 Targeted, Protease Inhibitor Focused and Protein–Protein Interaction Inhibitor Focused Molecular Libraries for Virtual Screening and Drug Design" International Journal of Molecular Sciences 23, no. 1: 393. https://doi.org/10.3390/ijms23010393
APA StyleKralj, S., Jukič, M., & Bren, U. (2022). Commercial SARS-CoV-2 Targeted, Protease Inhibitor Focused and Protein–Protein Interaction Inhibitor Focused Molecular Libraries for Virtual Screening and Drug Design. International Journal of Molecular Sciences, 23(1), 393. https://doi.org/10.3390/ijms23010393