
Impact of Uremic Toxins on Endothelial Dysfunction in Chronic Kidney Disease: A Systematic Review


Abstract
:1. Introduction
2. Materials and Methods
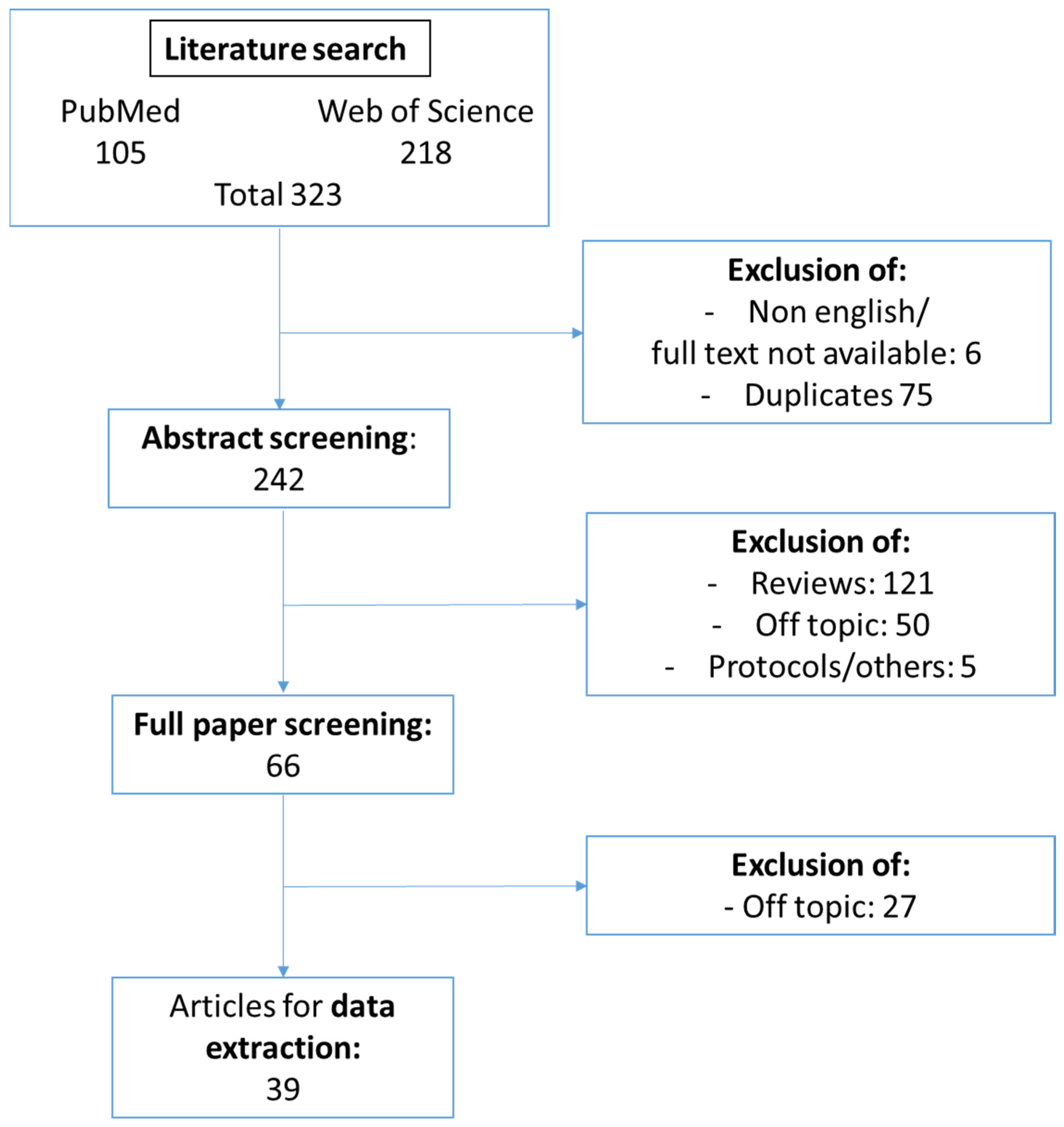
2.1. Search Strategy
2.2. Study Selection Criteria
2.3. Data Extraction
3. Results and Discussion
3.1. Study Selection
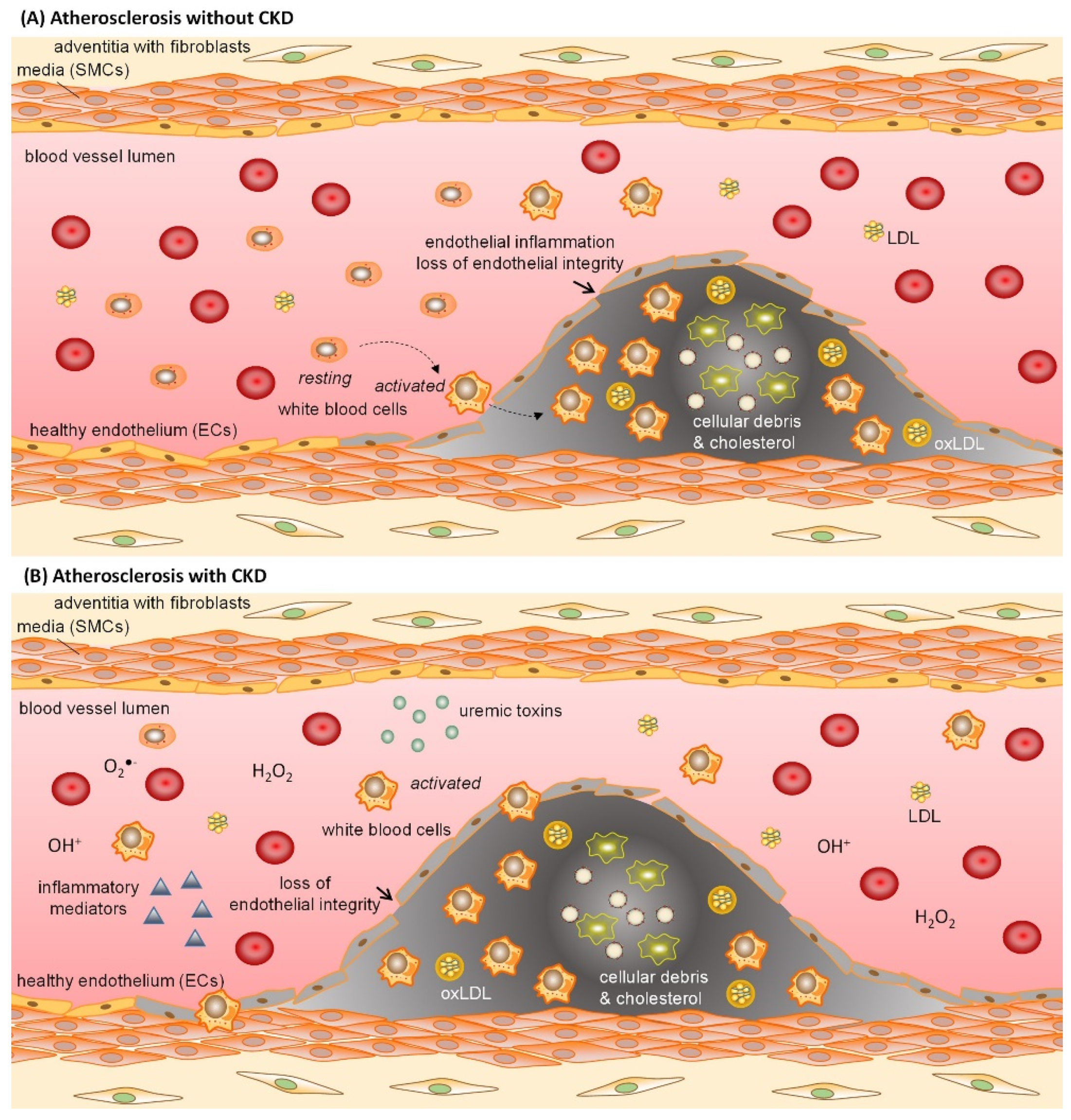
3.2. Pathophysiological Effect of Uremic Toxins on Endothelial Cells
3.3. Identification of Molecular Mechanisms Underlying Uremic Toxin-Induced Endothelial Dysfunction
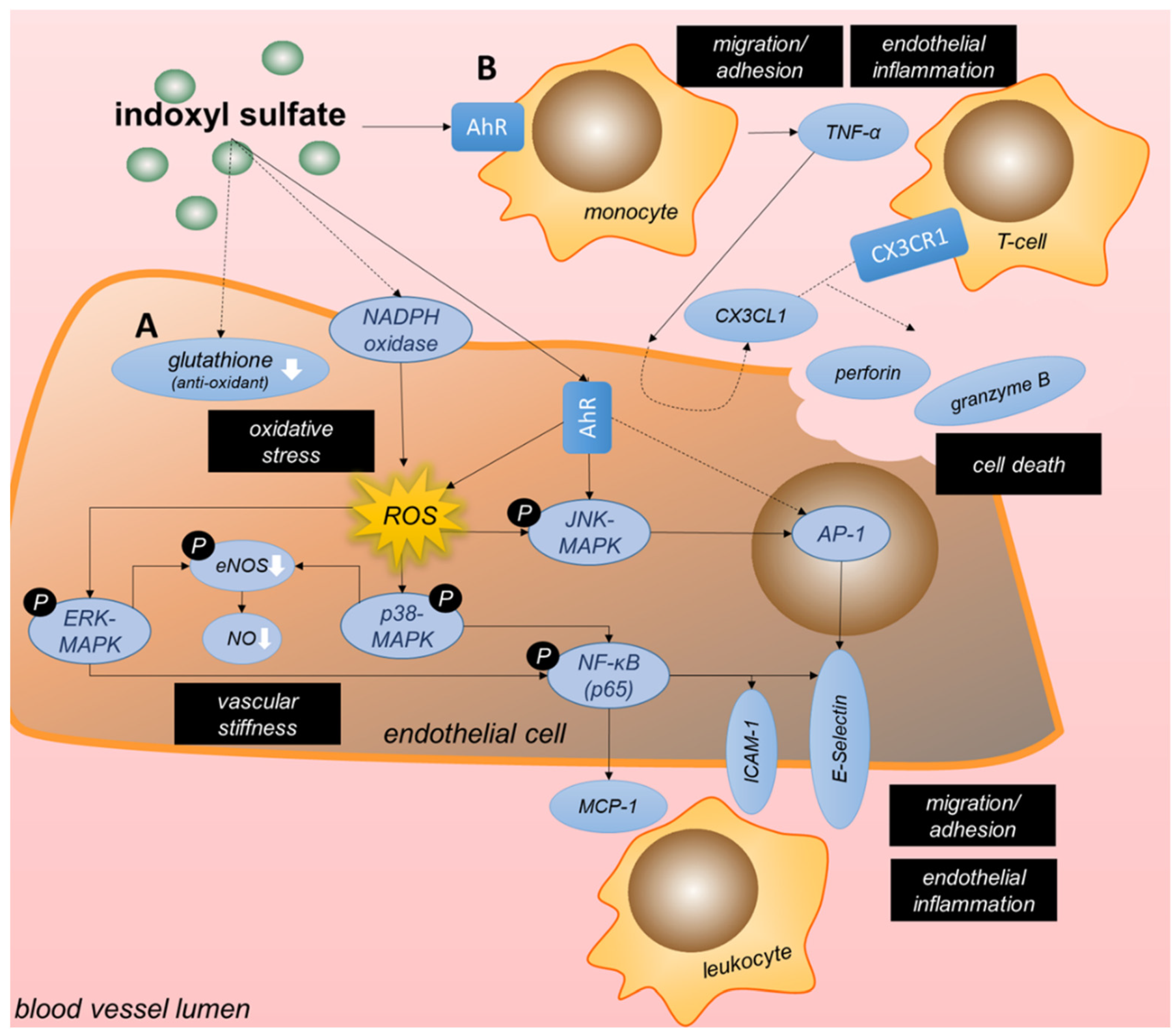
3.3.1. Tryptophan-Derived Toxins Trigger Inflammation and Oxidative Stress in Endothelial Cells
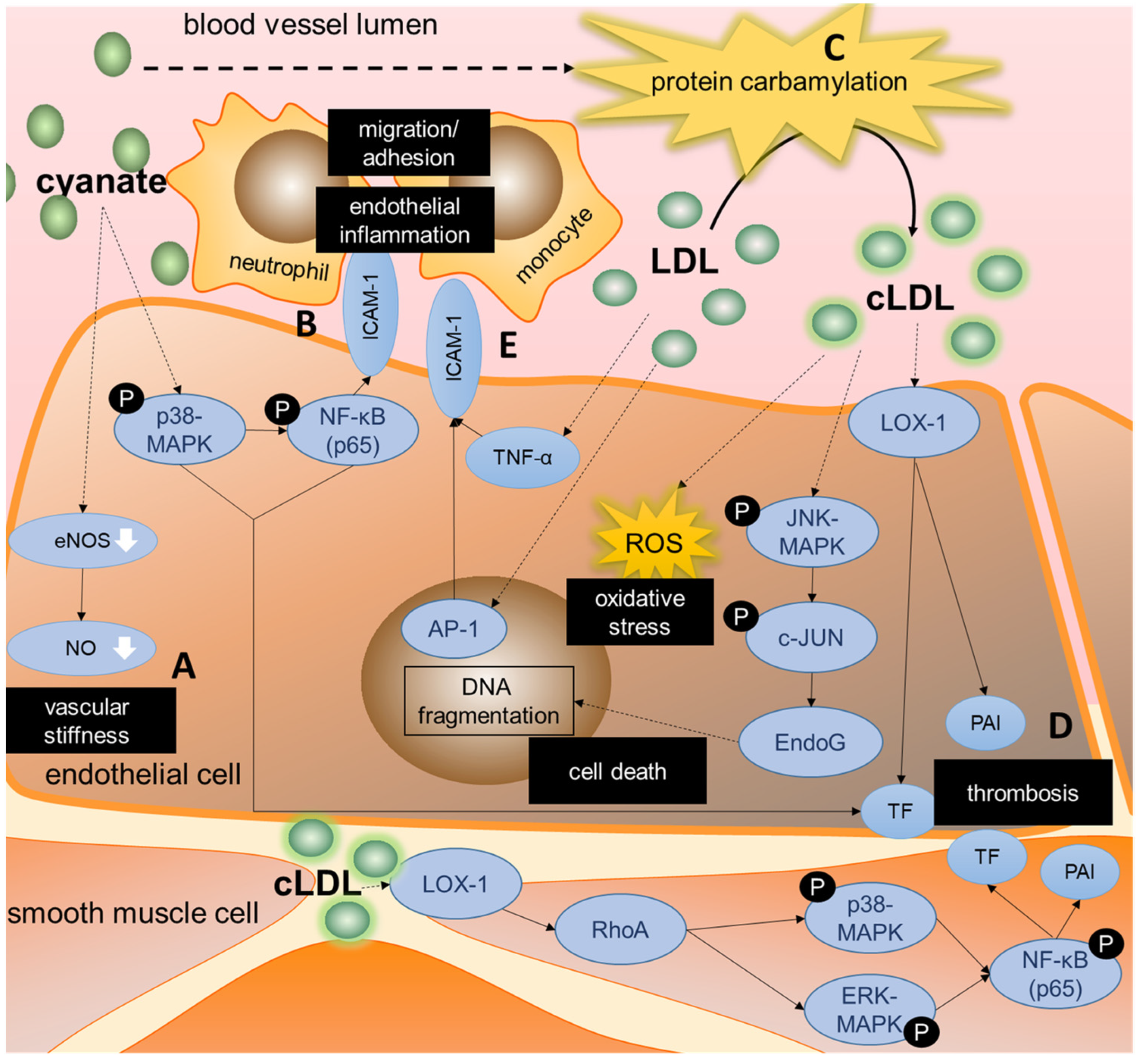
3.3.2. Cyanate Triggers Inflammatory Signaling and Protein Carbamylation
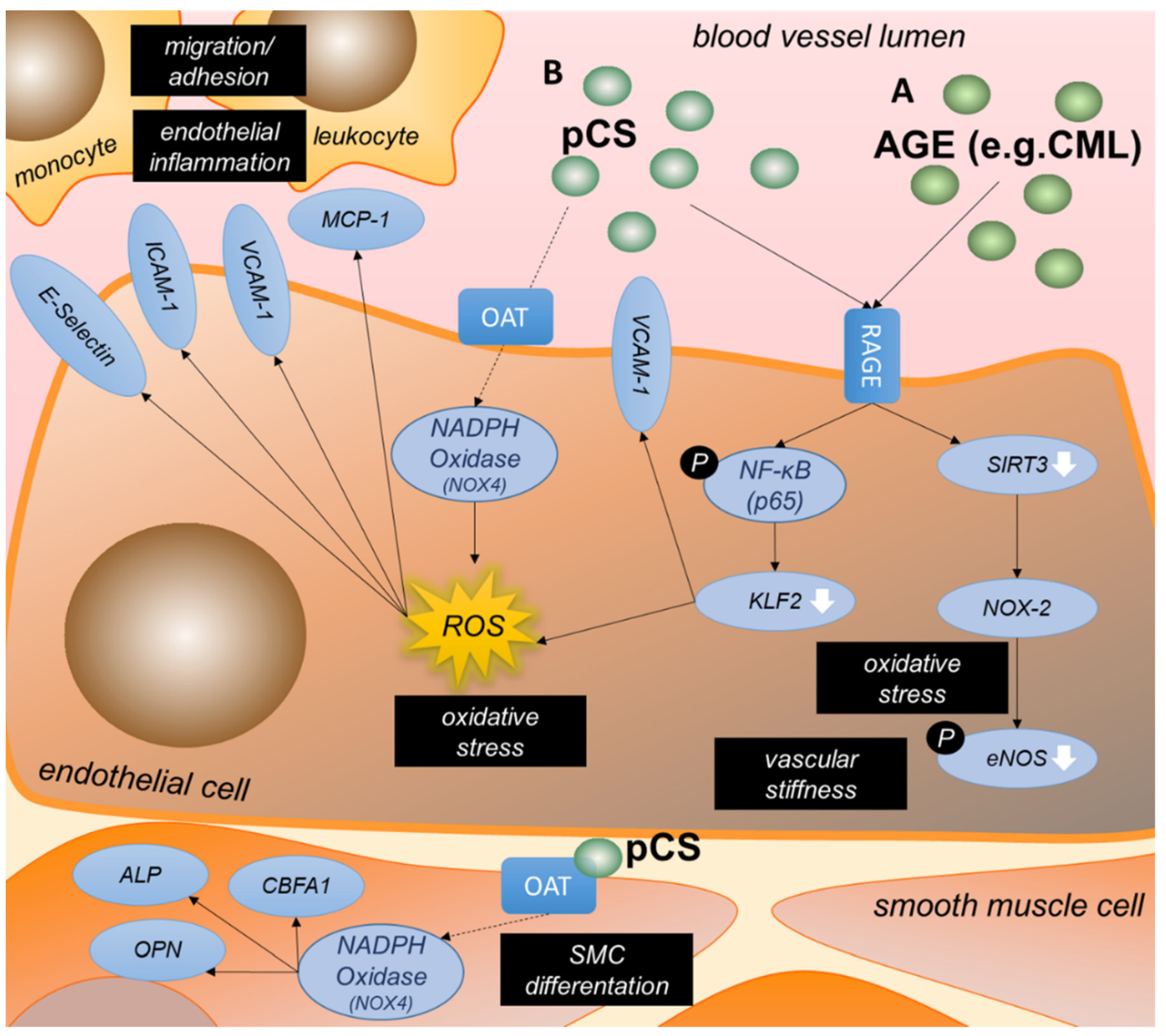
3.3.3. AGEs Are an Important Group of Uremic Toxins Inducing Endothelial Dysfunction
3.3.4. P-Cresyl Sulfate Contributes to Oxidative Stress and Inflammation in Endothelial Cells
3.3.5. Phosphate Reduces NO Production and Triggers Inflammation in Vascular Cells
3.3.6. High ADMA Levels as in CKD Lead to Increased Endothelial Cell Death
3.3.7. Uric Acid Increases Oxidative and ER Stress as well as Endothelial Cell Apoptosis
3.4. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stevens, P.; O’Donoghue, D.; de Lusignan, S.; Van Vlymen, J.; Klebe, B.; Middleton, R.; Hague, N.; New, J.; Farmer, C. Chronic kidney disease management in the United Kingdom: NEOERICA project results. Kidney Int. 2007, 72, 92–99. [Google Scholar] [CrossRef] [Green Version]
- Drey, N.; Roderick, P.; Mullee, M.; Rogerson, M. A population-based study of the incidence and outcomes of diagnosed chronic kidney disease. Am. J. Kidney Dis. 2003, 42, 677–684. [Google Scholar] [CrossRef]
- Thompson, S.; James, M.; Wiebe, N.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M. Cause of Death in Patients with Reduced Kidney Function. J. Am. Soc. Nephrol. 2015, 26, 2504–2511. [Google Scholar] [CrossRef]
- Masuda, C.; Dohi, K.; Sakurai, Y.; Bessho, Y.; Fukuda, H.; Fujii, S.; Sugimoto, T.; Tanabe, M.; Onishi, K.; Shiraki, K.; et al. Impact of Chronic Kidney Disease on the Presence and Severity of Aortic Stenosis in Patients at High Risk for Coronary Artery Disease. Cardiovasc. Ultrasound 2011, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Valdivielso, J.M.; Rodríguez-Puyol, D.; Pascual, J.; Barrios, C.; Bermúdez-López, M.; Sánchez-Niño, M.D.; Pérez-Fernández, M.; Ortiz, A. Atherosclerosis in Chronic Kidney Disease: More, Less, or Just Different? Arter. Thromb. Vasc. Biol. 2019, 39, 1938–1966. [Google Scholar] [CrossRef]
- Briet, M.; Bozec, E.; Laurent, S.; Fassot, C.; London, G.; Jacquot, C.; Froissart, M.; Houillier, P.; Boutouyrie, P. Arterial stiffness and enlargement in mild-to-moderate chronic kidney disease. Kidney Int. 2006, 69, 350–357. [Google Scholar] [CrossRef] [Green Version]
- Temmar, M.; Liabeuf, S.; Renard, C.; Czernichow, S.; El Esper, N.; Shahapuni, I.; Presne, C.; Makdassi, R.; Andrejak, M.; Tribouilloy, C.; et al. Pulse wave velocity and vascular calcification at different stages of chronic kidney disease. J. Hypertens. 2010, 28, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.D.; Hryniewicz, K.; Hriljac, I.; Balidemaj, K.; Dimayuga, C.; Hudaihed, A.; Yasskiy, A. Vascular Endothelial Dysfunction and Mortality Risk in Patients With Chronic Heart Failure. Circulation 2005, 111, 310–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schächinger, V.; Britten, M.B.; Zeiher, A.M. Prognostic Impact of Coronary Vasodilator Dysfunction on Adverse Long-Term Outcome of Coronary Heart Disease. Circulation 2000, 101, 1899–1906. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Soppert, J.; Lehrke, M.; Marx, N.; Jankowski, J.; Noels, H. Lipoproteins and lipids in cardiovascular disease: From mechanistic insights to therapeutic targeting. Adv. Drug Deliv. Rev. 2020, 159, 4–33. [Google Scholar] [CrossRef]
- Cai, H.; Harrison, D.G. Endothelial Dysfunction in Cardiovascular Diseases: The Role of Oxidant Stress. Circ. Res. 2000, 87, 840–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yau, J.W.; Teoh, H.; Verma, S. Endothelial cell control of thrombosis. BMC Cardiovasc. Disord. 2015, 15, 130. [Google Scholar] [CrossRef] [Green Version]
- Hutter, R.; Carrick, F.E.; Valdiviezo, C.; Wolinsky, C.; Rudge, J.S.; Wiegand, S.J.; Fuster, V.; Badimon, J.J.; Sauter, B.V. Vascular Endothelial Growth Factor Regulates Reendothelialization and Neointima Formation in a Mouse Model of Arterial Injury. Circulation 2004, 110, 2430–2435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noels, H.; Zhou, B.; Tilstam, P.V.; Theelen, W.; Li, X.; Pawig, L.; Schmitz, C.; Akhtar, S.; Simsekyilmaz, S.; Shagdarsuren, E.; et al. Deficiency of Endothelial Cxcr4 Reduces Reendothelialization and Enhances Neointimal Hyperplasia After Vascular Injury in Atherosclerosis-Prone Mice. Arter. Thromb. Vasc. Biol. 2014, 34, 1209–1220. [Google Scholar] [CrossRef] [Green Version]
- Vanholder, R.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; Dedeyn, P.; Deppisch, R.; Descamps-Latscha, B.; Henle, T.; et al. Uremic Toxicity: Present State of the Art. Int. J. Artif. Organs 2001, 24, 695–725. [Google Scholar] [CrossRef]
- Nowak, K.L.; Jovanovich, A.; Farmer-Bailey, H.; Bispham, N.; Struemph, T.; Malaczewski, M.; Wang, W.; Chonchol, M. Vascular Dysfunction, Oxidative Stress, and Inflammation in Chronic Kidney Disease. Kidney360 2020, 1, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Oberg, B.P.; McMenamin, E.; Lucas, F.L.; McMonagle, E.; Morrow, J.; Ikizler, T.A.; Himmelfarb, J. Increased prevalence of oxidant stress and inflammation in patients with moderate to severe chronic kidney disease. Kidney Int. 2004, 65, 1009–1016. [Google Scholar] [CrossRef] [Green Version]
- Cachofeiro, V.; Goicochea, M.; de Vinuesa, S.G.; Oubiña, P.; Lahera, V.; Luño, J. Oxidative stress and inflammation, a link between chronic kidney disease and cardiovascular disease. Kidney Int. 2008, 74, S4–S9. [Google Scholar] [CrossRef] [Green Version]
- Annuk, M.; Zilmer, M.; Lind, L.; Linde, T.; Fellström, B. Oxidative Stress and Endothelial Function in Chronic Renal Failure. J. Am. Soc. Nephrol. 2001, 12, 2747–2752. [Google Scholar] [CrossRef]
- Noels, H.; Lehrke, M.; Vanholder, R.; Jankowski, J. Lipoproteins and fatty acids in chronic kidney disease: Molecular and metabolic alterations. Nat. Rev. Nephrol. 2021, 17, 528–542. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [Green Version]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; on behalf of the European Uremic Toxin Work Group. Normal and Pathologic Concentrations of Uremic Toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmar, J.; De La Puente-Secades, S.; Floege, J.; Noels, H.; Jankowski, J.; Orth-Alampour, S. Uremic Toxins Affecting Cardiovascular Calcification: A Systematic Review. Cells 2020, 9, 2428. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Bertrand, E.; Cerini, C.; Faure, V.; Sampol, J.; Vanholder, R.; Berland, Y.; Brunet, P. The uremic solutes p-cresol and indoxyl sulfate inhibit endothelial proliferation and wound repair. Kidney Int. 2004, 65, 442–451. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; The PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA Statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerotic, D.; Suvakov, S.; Matic, M.; Alqudah, A.; Grieve, D.J.; Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Damjanovic, T.; Dimkovic, N.; McClements, L.; et al. GSTM1 Modulates Expression of Endothelial Adhesion Molecules in Uremic Milieu. Oxidative Med. Cell. Longev. 2021, 2021, 6678924. [Google Scholar] [CrossRef]
- Saum, K.; Campos, B.; Celdran-Bonafonte, D.; Nayak, L.; Sangwung, P.; Thakar, C.; Roy-Chaudhury, P.; OwensIII, A.P. Uremic Advanced Glycation End Products and Protein-Bound Solutes Induce Endothelial Dysfunction through Suppression of Krüppel-Like Factor 2. J. Am. Heart Assoc. 2018, 7, e007566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Jérez, A.; Luengo, A.; Carracedo, J.; Ramírez-Chamond, R.; Rodriguez-Puyol, D.; Rodriguez-Puyol, M.; Calleros, L. Effect of uraemia on endothelial cell damage is mediated by the integrin linked kinase pathway. J. Physiol. 2014, 593, 601–618. [Google Scholar] [CrossRef] [Green Version]
- Eloueyk, A.; Osta, B.; Alameldinne, R.; Awad, D. Uremic Serum Induces Inflammation in Cultured Human Endothelial Cells and Triggers Vascular Repair Mechanisms. Inflammation 2019, 42, 2003–2010. [Google Scholar] [CrossRef]
- Nilsson, L.; Lundquist, P.; Kågedal, B.; Larsson, R. Plasma cyanate concentrations in chronic renal failure. Clin. Chem. 1996, 42, 482–483. [Google Scholar] [CrossRef]
- The European Uremic Toxins (EUTox) Database. 2021. Available online: www.uremic-toxins.org (accessed on 1 June 2021).
- Speer, T.; Owala, F.O.; Holy, E.W.; Zewinger, S.; Frenzel, F.L.; Stähli, B.E.; Razavi, M.; Triem, S.; Cvija, H.; Rohrer, L.; et al. Carbamylated low-density lipoprotein induces endothelial dysfunction. Eur. Heart J. 2014, 35, 3021–3032. [Google Scholar] [CrossRef] [Green Version]
- Moore, L.W.; Nolte, J.V.; Gaber, A.O.; Suki, W.N. Association of dietary phosphate and serum phosphorus concentration by levels of kidney function. Am. J. Clin. Nutr. 2015, 102, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, S.; Osaka, M.; Edamatsu, T.; Itoh, Y.; Yoshida, M. Crucial Role of the Aryl Hydrocarbon Receptor (AhR) in Indoxyl Sulfate-Induced Vascular Inflammation. J. Atheroscler. Thromb. 2016, 23, 960–975. [Google Scholar] [CrossRef] [Green Version]
- Masai, N.; Tatebe, J.; Yoshino, G.; Morita, T. Indoxyl sulfate stimulates monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells by inducing oxidative stress through activation of the NADPH oxidase-nuclear factor-κB pathway. Circ. J. 2010, 74, 2216–2224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl Sulfate Upregulates Expression of ICAM-1 and MCP-1 by Oxidative Stress-Induced NF-kappa B Activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Yoo, T.-H.; Hwang, Y.; Lee, G.H.; Kim, B.; Jang, J.; Yu, H.T.; Kim, M.C.; Cho, J.Y.; Lee, C.J.; et al. Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD). Sci. Rep. 2017, 7, 3057. [Google Scholar] [CrossRef] [Green Version]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Yang, K.; Nie, L.; Huang, Y.; Zhang, J.; Xiao, T.; Guan, X.; Zhao, J. Amelioration of uremic toxin indoxyl sulfate-induced endothelial cell dysfunction by Klotho protein. Toxicol. Lett. 2012, 215, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xie, Y.; Yang, B.; Huang, S.; Zhang, Y.; Jia, Z.; Ding, G.; Zhang, A. MicroRNA-214 targets COX-2 to antagonize indoxyl sulfate (IS)-induced endothelial cell apoptosis. Apoptosis 2019, 25, 92–104. [Google Scholar] [CrossRef] [PubMed]
- El-Gamal, D.; Holzer, M.; Gauster, M.; Schicho, R.; Binder, V.; Konya, V.; Wadsack, C.; Schuligoi, R.; Heinemann, A.; Marsche, G. Cyanate Is a Novel Inducer of Endothelial ICAM-1 Expression. Antioxid. Redox Signal. 2012, 16, 129–137. [Google Scholar] [CrossRef] [Green Version]
- El-Gamal, D.; Rao, S.P.; Holzer, M.; Hallström, S.; Haybaeck, J.; Gauster, M.; Wadsack, C.; Kozina, A.; Frank, S.; Schicho, R.; et al. The urea decomposition product cyanate promotes endothelial dysfunction. Kidney Int. 2014, 86, 923–931. [Google Scholar] [CrossRef] [Green Version]
- Ambrosch, A.; Müller, R.; Freytag, C.; Borgmann, S.; Kraus, J.; Dierkes, J.; Neumann, K.H.; König, W. Small-sized low-density lipoproteins of subclass B from patients with end-stage renal disease effectively augment tumor necrosis factor-α-induced adhesive properties in human endothelial cells. Am. J. Kidney Dis. 2002, 39, 972–984. [Google Scholar] [CrossRef]
- Holy, E.W.; Akhmedov, A.; Speer, T.; Camici, G.G.; Zewinger, S.; Bonetti, N.; Beer, J.H.; Lüscher, T.F.; Tanner, F.C. Carbamylated Low-Density Lipoproteins Induce a Prothrombotic State via LOX-1 Impact on Arterial Thrombus Formation In Vivo. J. Am. Coll. Cardiol. 2016, 68, 1664–1676. [Google Scholar] [CrossRef]
- Apostolov, E.O.; Ray, D.; Alobuia, W.M.; Mikhailova, M.V.; Wang, X.; Basnakian, A.G.; Shah, S.V. Endonuclease G mediates endothelial cell death induced by carbamylated LDL. Am. J. Physiol. Circ. Physiol. 2011, 300, H1997–H2004. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-C.; Lee, A.-S.; Liu, S.-H.; Chang, K.-C.; Shen, M.-Y.; Chang, C.-T. Spironolactone ameliorates endothelial dysfunction through inhibition of the AGE/RAGE axis in a chronic renal failure rat model. BMC Nephrol. 2019, 20, 351. [Google Scholar] [CrossRef] [Green Version]
- Linden, E.; Cai, W.; He, J.C.; Xue, C.; Li, Z.; Winston, J.; Vlassara, H.; Uribarri, J. Endothelial Dysfunction in Patients with Chronic Kidney Disease Results from Advanced Glycation End Products (AGE)-Mediated Inhibition of Endothelial Nitric Oxide Synthase through RAGE Activation. Clin. J. Am. Soc. Nephrol. 2008, 3, 691–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Yang, K.; Jing, Y.; Du, R.; Zhu, Z.; Lu, L.; Zhang, R. The effects of low-dose Nepsilon-(carboxymethyl)lysine (CML) and Nepsilon-(carboxyethyl)lysine (CEL), two main glycation free adducts considered as potential uremic toxins, on endothelial progenitor cell function. Cardiovasc. Diabetol. 2012, 11, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Y.J.; Ni, J.W.; Ding, F.H.; Fang, Y.H.; Wang, X.Q.; Wang, H.B.; Chen, X.N.; Chen, N.; Zhan, W.W.; Lu, L.; et al. p-Cresyl sulfate is associated with carotid arteriosclerosis in hemodialysis patients and promotes atherogenesis in apoE–/– mice. Kidney Int. 2016, 89, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Miyamoto, Y.; Enoki, Y.; Ishima, Y.; Kadowaki, D.; Kotani, S.; Nakajima, M.; Tanaka, M.; Matsushita, K.; Mori, Y.; et al. p-Cresyl sulfate, a uremic toxin, causes vascular endothelial and smooth muscle cell damages by inducing oxidative stress. Pharmacol. Res. Perspect. 2014, 3, e00092. [Google Scholar] [CrossRef]
- Meijers, B.K.I.; Van Kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The Uremic Retention Solute p-Cresyl Sulfate and Markers of Endothelial Damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef]
- Peng, A.; Wu, T.; Zeng, C.; Rakheja, D.; Zhu, J.; Ye, T.; Hutcheson, J.; Vaziri, N.D.; Liu, Z.; Mohan, C.; et al. Adverse Effects of Simulated Hyper- and Hypo-Phosphatemia on Endothelial Cell Function and Viability. PLoS ONE 2011, 6, e23268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.-J.; Hsu, S.-C.; Huang, S.-M.; Lee, H.-S.; Lin, S.-H.; Tsai, C.-S.; Shih, C.-C.; Lin, C.-Y. Hyperphosphatemia induces protective autophagy in endothelial cells through the inhibition of Akt/mTOR signaling. J. Vasc. Surg. 2014, 62, 210–221.e2. [Google Scholar] [CrossRef] [Green Version]
- Abbasian, N.; Burton, J.; Herbert, K.; Tregunna, B.-E.; Brown, J.R.; Ghaderi-Najafabadi, M.; Brunskill, N.J.; Goodall, A.; Bevington, A. Hyperphosphatemia, Phosphoprotein Phosphatases, and Microparticle Release in Vascular Endothelial Cells. J. Am. Soc. Nephrol. 2015, 26, 2152–2162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Diao, Z.; Liu, W. Asymmetric dimethylarginine downregulates sarco/endoplasmic reticulum calcium-ATPase 3 and induces endoplasmic reticulum stress in human umbilical vein endothelial cells. Mol. Med. Rep. 2017, 16, 7541–7547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Hong, Q.; Huang, Z.; Xue, P.; Lv, Y.; Fu, B.; Chen, X.; Wu, D. ALDR Enhanced Endothelial Injury in Hyperuricemia Screened using SILAC. Cell. Physiol. Biochem. 2014, 33, 479–490. [Google Scholar] [CrossRef]
- Li, P.; Zhang, L.; Zhang, M.; Zhou, C.; Lin, N. Uric acid enhances PKC-dependent eNOS phosphorylation and mediates cellular ER stress: A mechanism for uric acid-induced endothelial dysfunction. Int. J. Mol. Med. 2016, 37, 989–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, H.; Yamada, K.; Tamai, I. Hyperuricemia enhances intracellular urate accumulation via down-regulation of cell-surface BCRP/ABCG2 expression in vascular endothelial cells. Biochim. Biophys. Acta (BBA)-Biomembr. 2018, 1860, 973–980. [Google Scholar] [CrossRef]
- Pawlak, K.; Kowalewska, A.; Mysliwiec, M.; Pawlak, D. 3-hydroxyanthranilic acid is independently associated with monocyte chemoattractant protein-1 (CCL2) and macrophage inflammatory protein-1 beta (CCL4) in patients with chronic kidney disease. Clin. Biochem. 2010, 43, 1101–1106. [Google Scholar] [CrossRef]
- Frericks, M.; Meissner, M.; Esser, C. Microarray analysis of the AHR system: Tissue-specific flexibility in signal and target genes. Toxicol. Appl. Pharmacol. 2007, 220, 320–332. [Google Scholar] [CrossRef]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The Aryl Hydrocarbon Receptor: Multitasking in the Immune System. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.F.A.; Sciullo, E.; Matsumura, F. Activation of Inflammatory Mediators and Potential Role of Ah-Receptor Ligands in Foam Cell Formation. Cardiovasc. Toxicol. 2004, 4, 363–374. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Sciullo, E.; Wong, P.; Kuzmicky, P.; Kado, N.; Matsumura, F. Induction of proinflammatory cytokines and C-reactive protein in human macrophage cell line U937 exposed to air pollution particulates. Environ. Health Perspect. 2005, 113, 1536–1541. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Nishimura, N.; Kuo, V.; Fiehn, O.; Shahbaz, S.; Van Winkle, L.; Matsumura, F.; Vogel, C.F.A. Activation of aryl hydrocarbon receptor induces vascular inflammation and promotes atherosclerosis in apolipoprotein E–/– mice. Arter. Thromb. Vasc. Biol. 2011, 31, 1260–1267. [Google Scholar] [CrossRef] [Green Version]
- Wakamatsu, T.; Yamamoto, S.; Ito, T.; Sato, Y.; Matsuo, K.; Takahashi, Y.; Kaneko, Y.; Goto, S.; Kazama, J.J.; Gejyo, F.; et al. Indoxyl Sulfate Promotes Macrophage IL-1β Production by Activating Aryl Hydrocarbon Receptor/NF-κ/MAPK Cascades, but the NLRP3 inflammasome Was Not Activated. Toxins 2018, 10, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.Y.; Yoo, T.H.; Cho, J.Y.; Kim, H.C.; Lee, W.W. Indoxyl sulfate-induced TNF-alpha is regulated by crosstalk between the aryl hydrocarbon receptor, NF-kappa B, and SOCS2 in human macrophages. FASEB J. 2019, 33, 10844–10858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The Cardiovascular Effect of the Uremic Solute Indole-3 Acetic Acid. J. Am. Soc. Nephrol. 2014, 26, 876–887. [Google Scholar] [CrossRef]
- Brito, J.S.D.; Borges, N.A.; Anjos, J.S.D.; Nakao, L.S.; Stockler-Pinto, M.B.; Paiva, B.R.; Cardoso-Weide, L.D.C.; Cardozo, L.F.M.D.F.; Mafra, D. Aryl Hydrocarbon Receptor and Uremic Toxins from the Gut Microbiota in Chronic Kidney Disease Patients: Is There a Relationship between Them? Biochemistry 2019, 58, 2054–2060. [Google Scholar] [CrossRef]
- Shah, S.V.; Shukla, A.M.; Bose, C.; Basnakian, A.G.; Rajapurkar, M. Recent Advances in Understanding the Pathogenesis of Atherosclerosis in CKD Patients. J. Ren. Nutr. 2015, 25, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Hsu, S.-C.; Lee, H.-S.; Lin, S.-H.; Tsai, C.-S.; Huang, S.-M.; Shih, C.-C.; Hsu, Y.-J. Enhanced expression of glucose transporter-1 in vascular smooth muscle cells via the Akt/tuberous sclerosis complex subunit 2 (TSC2)/mammalian target of rapamycin (mTOR)/ribosomal S6 protein kinase (S6K) pathway in experimental renal failure. J. Vasc. Surg. 2013, 57, 475–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, J.; Lee, J.; Lee, H.; Sadler, P.; Wilkie, D.; Woodham, R. Nuclear magnetic resonance studies of blood plasma and urine from subjects with chronic renal failure: Identification of trimethylamine-N-oxide. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 1991, 1096, 101–107. [Google Scholar] [CrossRef]
- Blackmore, D.J.; Elder, W.J.; Bowden, C.H. Urea distribution in renal failure. J. Clin. Pathol. 1963, 16, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Nicholls, S.; Rodriguez, E.R.; Kummu, O.; Hörkkö, S.; Barnard, J.W.; Reynolds, W.F.; Topol, E.; DiDonato, J.A.; Hazen, S.L. Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat. Med. 2007, 13, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Arlandson, M.; Decker, T.; Roongta, V.A.; Bonilla, L.; Mayo, K.H.; MacPherson, J.C.; Hazen, S.L.; Slungaard, A. Eosinophil peroxidase oxidation of thiocyanate. Characterization of major reaction products and a potential sulfhydryl-targeted cytotoxicity system. J. Biol. Chem. 2001, 276, 215–224. [Google Scholar] [CrossRef] [Green Version]
- Baldus, S.; Eiserich, J.P.; Mani, A.; Castro, L.; Figueroa, M.; Chumley, P.; Ma, W.; Tousson, A.; White, C.R.; Bullard, D.C.; et al. Endothelial transcytosis of myeloperoxidase confers specificity to vascular ECM proteins as targets of tyrosine nitration. J. Clin. Investig. 2001, 108, 1759–1770. [Google Scholar] [CrossRef] [PubMed]
- Kalim, S.; Karumanchi, S.A.; Thadhani, R.I.; Berg, A.H. Protein carbamylation in kidney disease: Pathogenesis and clinical implications. Am. J. Kidney Dis. Off. J. Natl. Kidney Found. 2014, 64, 793–803. [Google Scholar] [CrossRef] [Green Version]
- Apostolov, E.O.; Ray, D.; Savenka, A.V.; Shah, S.V.; Basnakian, A.G. Chronic Uremia Stimulates LDL Carbamylation and Atherosclerosis. J. Am. Soc. Nephrol. 2010, 21, 1852–1857. [Google Scholar] [CrossRef] [Green Version]
- Apostolov, E.O.; Shah, S.V.; Ray, D.; Basnakian, A.G. Scavenger Receptors of Endothelial Cells Mediate the Uptake and Cellular Proatherogenic Effects of Carbamylated LDL. Arter. Thromb. Vasc. Biol. 2009, 29, 1622–1630. [Google Scholar] [CrossRef] [Green Version]
- Basnakian, A.G.; Shah, S.V.; Ok, E.; Altunel, E.; Apostolov, E.O. Carbamylated LDL. Adv. Clin. Chem. 2010, 51, 25–52. [Google Scholar] [PubMed]
- Gonen, B.; Goldberg, A.P.; Harter, H.R.; Schonfeld, G. Abnormal cell-interactive properties of low-density lipoproteins isolated from patients with chronic renal failure. Metabolism 1985, 34, 10–14. [Google Scholar] [CrossRef]
- Hörkkö, S.; Huttunen, K.; Kervinen, K.; Kesäniemi, Y.A. Decreased clearance of uraemic and mildly carbamylated low-density lipoprotein. Eur. J. Clin. Investig. 1994, 24, 105–113. [Google Scholar] [CrossRef]
- Ok, E.; Basnakian, A.G.; Apostolov, E.O.; Barri, Y.M.; Shah, S.V. Carbamylated low-density lipoprotein induces death of endothelial cells: A link to atherosclerosis in patients with kidney disease. Kidney Int. 2005, 68, 173–178. [Google Scholar] [CrossRef] [Green Version]
- Chu, M.; Wang, A.Y.M.; Chan, I.H.S.; Chui, S.H.; Lam, C.W.K. Serum small-dense LDL abnormalities in chronic renal disease patients. Br. J. Biomed. Sci. 2012, 69, 99–102. [Google Scholar] [CrossRef]
- Tao, J.-L.; Ruan, X.-Z.; Li, H.; Li, X.-M.; Moorhead, J.F.; Varghese, Z. Endoplasmic reticulum stress is involved in acetylated low-density lipoprotein induced apoptosis in THP-1 differentiated macrophages. Chin. Med. J. 2009, 122, 1794–1799. [Google Scholar]
- Miyata, T.; Strihou, C.V.Y.D.; Kurokawa, K.; Baynes, J.W. Alterations in nonenzymatic biochemistry in uremia: Origin and significance of “carbonyl stress” in long-term uremic complications. Kidney Int. 1999, 55, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Peppa, M.; Uribarri, J.; Cai, W.; Lu, M.; Vlassara, H. Glycoxidation and inflammation in renal failure patients. Am. J. Kidney Dis. 2004, 43, 690–695. [Google Scholar] [CrossRef]
- Martinez, A.W.; Recht, N.S.; Hostetter, T.H.; Meyer, T.W. Removal of P-Cresol Sulfate by Hemodialysis. J. Am. Soc. Nephrol. 2005, 16, 3430–3436. [Google Scholar] [CrossRef] [Green Version]
- Meijers, B.; Bammens, B.; De Moor, B.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. Free p-cresol is associated with cardiovascular disease in hemodialysis patients. Kidney Int. 2008, 73, 1174–1180. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Sakaguchi, Y.; Sugimoto, R.; Kaneko, K.-I.; Iwata, H.; Kotani, S.; Nakajima, M.; Ishima, Y.; Otagiri, M.; Maruyama, T. Human organic anion transporters function as a high-capacity transporter for p-cresyl sulfate, a uremic toxin. Clin. Exp. Nephrol. 2013, 18, 814–820. [Google Scholar] [CrossRef]
- Giachelli, C.M.; Speer, M.Y.; Li, X.; Rajachar, R.M.; Yang, H. Regulation of vascular calcification: Roles of phosphate and osteopontin. Circ. Res. 2005, 96, 717–722. [Google Scholar] [CrossRef] [Green Version]
- Mizobuchi, M.; Towler, D.; Slatopolsky, E. Vascular Calcification: The Killer of Patients with Chronic Kidney Disease. J. Am. Soc. Nephrol. 2009, 20, 1453–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Moreno, J.M.; Muñoz-Castañeda, J.R.; Herencia, C.; De Oca, A.M.; Estepa, J.C.; Canalejo, R.; Rodríguez-Ortiz, M.E.; Martínez, P.P.; Aguilera-Tejero, E.; Canalejo, A.; et al. In vascular smooth muscle cells paricalcitol prevents phosphate-induced Wnt/β-catenin activation. Am. J. Physiol. Physiol. 2012, 303, F1136–F1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Moreno, J.M.; Herencia, C.; de Oca, A.M.; Díaz-Tocados, J.M.; Vergara, N.; Gómez-Luna, M.J.; López-Argüello, S.D.; Camargo, A.; Peralbo-Santaella, E.; Rodríguez-Ortiz, M.E.; et al. High phosphate induces a pro-inflammatory response by vascular smooth muscle cells and modulation by vitamin D derivatives. Clin. Sci. 2017, 131, 1449–1463. [Google Scholar] [CrossRef]
- Konya, H.; Miuchi, M.; Satani, K.; Matsutani, S.; Yano, Y.; Tsunoda, T.; Ikawa, T.; Matsuo, T.; Ochi, F.; Kusunoki, Y.; et al. Asymmetric dimethylarginine, a biomarker of cardiovascular complications in diabetes mellitus. World J. Exp. Med. 2015, 5, 110–119. [Google Scholar] [CrossRef]
- Böger, R.H.; Maas, R.; Schulze, F.; Schwedhelm, E. Asymmetric dimethylarginine (ADMA) as a prospective marker of cardiovascular disease and mortality—An update on patient populations with a wide range of cardiovascular risk. Pharmacol. Res. 2009, 60, 481–487. [Google Scholar] [CrossRef]
- Woehlbier, U.; Hetz, C. Modulating stress responses by the UPRosome: A matter of life and death. Trends Biochem. Sci. 2011, 36, 329–337. [Google Scholar] [CrossRef]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Snauwaert, E.; Holvoet, E.; Van Biesen, W.; Raes, A.; Glorieux, G.; Walle, J.V.; Roels, S.; Vanholder, R.; Askiti, V.; Azukaitis, K.; et al. Uremic Toxin Concentrations are Related to Residual Kidney Function in the Pediatric Hemodialysis Population. Toxins 2019, 11, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanbay, M.; Segal, M.; Afsar, B.; Kang, D.-H.; Rodriguez-Iturbe, B.; Johnson, R.J. The role of uric acid in the pathogenesis of human cardiovascular disease. Heart 2013, 99, 759–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, E.-S.; Kim, M.J.; Shin, H.-S.; Jang, Y.-H.; Choi, H.S.; Jo, I.; Johnson, R.J.; Kang, D.-H. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am. J. Physiol. Ren. Physiol. 2013, 304, F471–F480. [Google Scholar] [CrossRef] [PubMed]
- Sarnak, M.J.; Levey, A.S.; Schoolwerth, A.C.; Coresh, J.; Culleton, B.; Hamm, L.L.; McCullough, P.A.; Kasiske, B.L.; Kelepouris, E.; Klag, M.J.; et al. Kidney Disease as a Risk Factor for Development of Cardiovascular Disease. Circulation 2003, 108, 2154–2169. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, P.A.; Covic, A.; Fliser, D.; Fouque, D.; Goldsmith, D.; Kanbay, M.; Mallamaci, F.; Massy, Z.A.; Rossignol, P.; Vanholder, R.; et al. Epidemiology, contributors to, and clinical trials of mortality risk in chronic kidney failure. Lancet 2014, 383, 1831–1843. [Google Scholar] [CrossRef]
- Sallée, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The Aryl Hydrocarbon Receptor-Activating Effect of Uremic Toxins from Tryptophan Metabolism: A New Concept to Understand Cardiovascular Complications of Chronic Kidney Disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef]
- Malatino, L.S.; Stancanelli, B.; Cataliotti, A.; Bellanuova, I.; Fatuzzo, P.M.; Rapisarda, F.A.; Leonardis, D.; Tripepi, G.; Mallamaci, F.; Zoccali, C. Circulating E-selectin as a risk marker in patients with end-stage renal disease. J. Intern. Med. 2007, 262, 479–487. [Google Scholar] [CrossRef]
- Yi, T.; Wang, J.; Zhu, K.; Tang, Y.; Huang, S.; Shui, X.; Ding, Y.; Chen, C.; Lei, W. Aryl Hydrocarbon Receptor: A New Player of Pathogenesis and Therapy in Cardiovascular Diseases. BioMed Res. Int. 2018, 2018, 6058784. [Google Scholar] [CrossRef]
- Zhu, K.; Meng, Q.; Zhang, Z.; Yi, T.; He, Y.; Zheng, J.; Lei, W. Aryl hydrocarbon receptor pathway: Role, regulation and intervention in atherosclerosis therapy (Review). Mol. Med. Rep. 2019, 20, 4763–4773. [Google Scholar] [CrossRef] [Green Version]
- Reichert, S.; Triebert, U.; Santos, A.N.; Hofmann, B.; Schaller, H.-G.; Schlitt, A.; Schulz, S. Soluble form of receptor for advanced glycation end products and incidence of new cardiovascular events among patients with cardiovascular disease. Atherosclerosis 2017, 266, 234–239. [Google Scholar] [CrossRef]
- Uekita, H.; Ishibashi, T.; Shiomi, M.; Koyama, H.; Ohtsuka, S.; Yamamoto, H.; Yamagishi, S.; Inoue, H.; Itabe, H.; Sugimoto, K.; et al. Integral role of receptor for advanced glycation end products (RAGE) in nondiabetic atherosclerosis. Fukushima J. Med. Sci. 2019, 65, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Jandeleit-Dahm, K.; Cooper, M.E. The role of AGEs in cardiovascular disease. Curr. Pharm. Des. 2008, 14, 979–986. [Google Scholar] [CrossRef]
- Baaten, C.C.; Sternkopf, M.; Henning, T.; Marx, N.; Jankowski, J.; Noels, H. Platelet Function in CKD: A Systematic Review and Meta-Analysis. J. Am. Soc. Nephrol. 2021, 32, 1583–1598. [Google Scholar] [CrossRef]
- Silswal, N.; Touchberry, C.D.; Daniel, D.R.; McCarthy, D.L.; Zhang, S.; Andresen, J.; Stubbs, J.R.; Wacker, M.J. FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am. J. Physiol. Metab. 2014, 307, E426–E436. [Google Scholar] [CrossRef] [Green Version]
- Six, I.; Okazaki, H.; Gross, P.; Cagnard, J.; Boudot, C.; Maizel, J.; Drueke, T.B.; Massy, Z.A. Direct, Acute Effects of Klotho and FGF23 on Vascular Smooth Muscle and Endothelium. PLoS ONE 2014, 9, e93423. [Google Scholar]
- Saito, Y.; Yamagishi, T.; Nakamuraa, T.; Ohyamaa, Y.; Aizawaa, H.; Sugaa, T.; Matsumuraab, Y.; Masudaab, H.; Kurabayashia, M.; Kuro-Ob, M.; et al. Klotho Protein Protects against Endothelial Dysfunction. Biochem. Biophys. Res. Commun. 1998, 248, 324–329. [Google Scholar] [CrossRef]
- Nagai, R.; Saito, Y.; Ohyama, Y.; Aizawa, H.; Suga, T.; Nakamura, T.; Kurabayashi, M.; Kuro-o, M. Endothelial dysfunction in the klotho mouse and downregulation of klotho gene expression in various animal models of vascular and metabolic diseases. Cell. Mol. Life Sci. 2000, 57, 738–746. [Google Scholar] [CrossRef]
- Shimada, T.; Takeshita, Y.; Murohara, T.; Sasaki, K.-I.; Egami, K.; Shintani, S.; Katsuda, Y.; Ikeda, H.; Nabeshima, Y.-I.; Imaizumi, T. Angiogenesis and Vasculogenesis Are Impaired in the Precocious-Aging klotho Mouse. Circulation 2004, 110, 1148–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Nakamura, T.; Ohyama, Y.; Suzuki, T.; Iida, A.; Shiraki-Iida, T.; Kuro-o, M.; Nabeshima, Y.-I.; Kurabayashi, M.; Nagai, R. In Vivo klotho Gene Delivery Protects against Endothelial Dysfunction in Multiple Risk Factor Syndrome. Biochem. Biophys. Res. Commun. 2000, 276, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, Y.; Ishikawa, K.; Yasuda, O.; Oguro, R.; Hanasaki, H.; Kida, I.; Takemura, Y.; Ohishi, M.; Katsuya, T.; Rakugi, H. Klotho suppresses TNF-alpha-induced expression of adhesion molecules in the endothelium and attenuates NF-kappaB activation. Endocrine 2009, 35, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wu, L.; Xie, C.; Zhao, X.; Mao, H.; Xing, C. The role of AMP-activated protein kinase α1-mediated endoplasmic reticulum stress in alleviating the toxic effect of uremic toxin indoxyl sulfate on vascular endothelial cells by Klotho. J. Appl. Toxicol. 2021, 41, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Kusaba, T.; Okigaki, M.; Matui, A.; Murakami, M.; Ishikawa, K.; Kimura, T.; Sonomura, K.; Adachi, Y.; Shibuya, M.; Shirayama, T.; et al. Klotho is associated with VEGF receptor-2 and the transient receptor potential canonical-1 Ca2+ channel to maintain endothelial integrity. Proc. Natl. Acad. Sci. USA 2010, 107, 19308–19313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.S.; Son, S.S.; Lee, J.-H.; Lee, S.W.; Jeong, A.R.; Lee, E.S.; Cha, S.-K.; Chung, C.H. Protective effects of klotho on palmitate-induced podocyte injury in diabetic nephropathy. PLoS ONE 2021, 16, e0250666. [Google Scholar]
- Chen, J.; Zhang, X.; Zhang, H.; Liu, T.; Zhang, H.; Teng, J.; Ji, J.; Ding, X. Indoxyl Sulfate Enhance the Hypermethylation of Klotho and Promote the Process of Vascular Calcification in Chronic Kidney Disease. Int. J. Biol. Sci. 2016, 12, 1236–1246. [Google Scholar] [CrossRef] [Green Version]
- Gajjala, P.R.; Fliser, D.; Speer, T.; Jankowski, V.; Jankowski, J. Emerging role of post-translational modifications in chronic kidney disease and cardiovascular disease. Nephrol. Dial. Transplant. 2015, 30, 1814–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saar-Kovrov, V.; Zidek, W.; Orth-Alampour, S.; Fliser, D.; Jankowski, V.; Biessen, E.A.L. Reduction of protein-bound uraemic toxins in plasma of chronic renal failure patients: A systematic review. J. Intern. Med. 2021, 290, 499–526. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin | Inflammation | Oxidative Stress | Cell Death | Migration/ Adhesion | Proliferation | Thrombosis | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Indoxyl sulfate | + | [37,38,39,40] | + | [31,37,38,39,41,42] | + | [31,40,42,43] | + | [37,39,40] | - | [31] | ||
| Cyanate | + | [44,45] | + | [44] | + | [45] | ||||||
| cLDL | + | [46,47] | + | [47,48] | + | [46] | ||||||
| AGE | + | [49] | + | [30,49,50] | + | [30] | + | [30,51] | - | [51] | ||
| p-Cresol/ p-cresyl sulfate | + | [52] | + | [30,31,52,53] | + | [30,31,54] | + | [30,52] | - | [31] | ||
| Phosphate | + | [55] | + | [55,56] | + | [57] | ||||||
| ADMA | + | [58] | ||||||||||
| Uric acid | + | [59,60,61] | + | [60,61] | ||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harlacher, E.; Wollenhaupt, J.; Baaten, C.C.F.M.J.; Noels, H. Impact of Uremic Toxins on Endothelial Dysfunction in Chronic Kidney Disease: A Systematic Review. Int. J. Mol. Sci. 2022, 23, 531. https://doi.org/10.3390/ijms23010531
Harlacher E, Wollenhaupt J, Baaten CCFMJ, Noels H. Impact of Uremic Toxins on Endothelial Dysfunction in Chronic Kidney Disease: A Systematic Review. International Journal of Molecular Sciences. 2022; 23(1):531. https://doi.org/10.3390/ijms23010531
Chicago/Turabian StyleHarlacher, Eva, Julia Wollenhaupt, Constance C. F. M. J. Baaten, and Heidi Noels. 2022. "Impact of Uremic Toxins on Endothelial Dysfunction in Chronic Kidney Disease: A Systematic Review" International Journal of Molecular Sciences 23, no. 1: 531. https://doi.org/10.3390/ijms23010531