
The Potential of Epigallocatechin Gallate (EGCG) in Targeting Autophagy for Cancer Treatment: A Narrative Review




Abstract
:1. Introduction
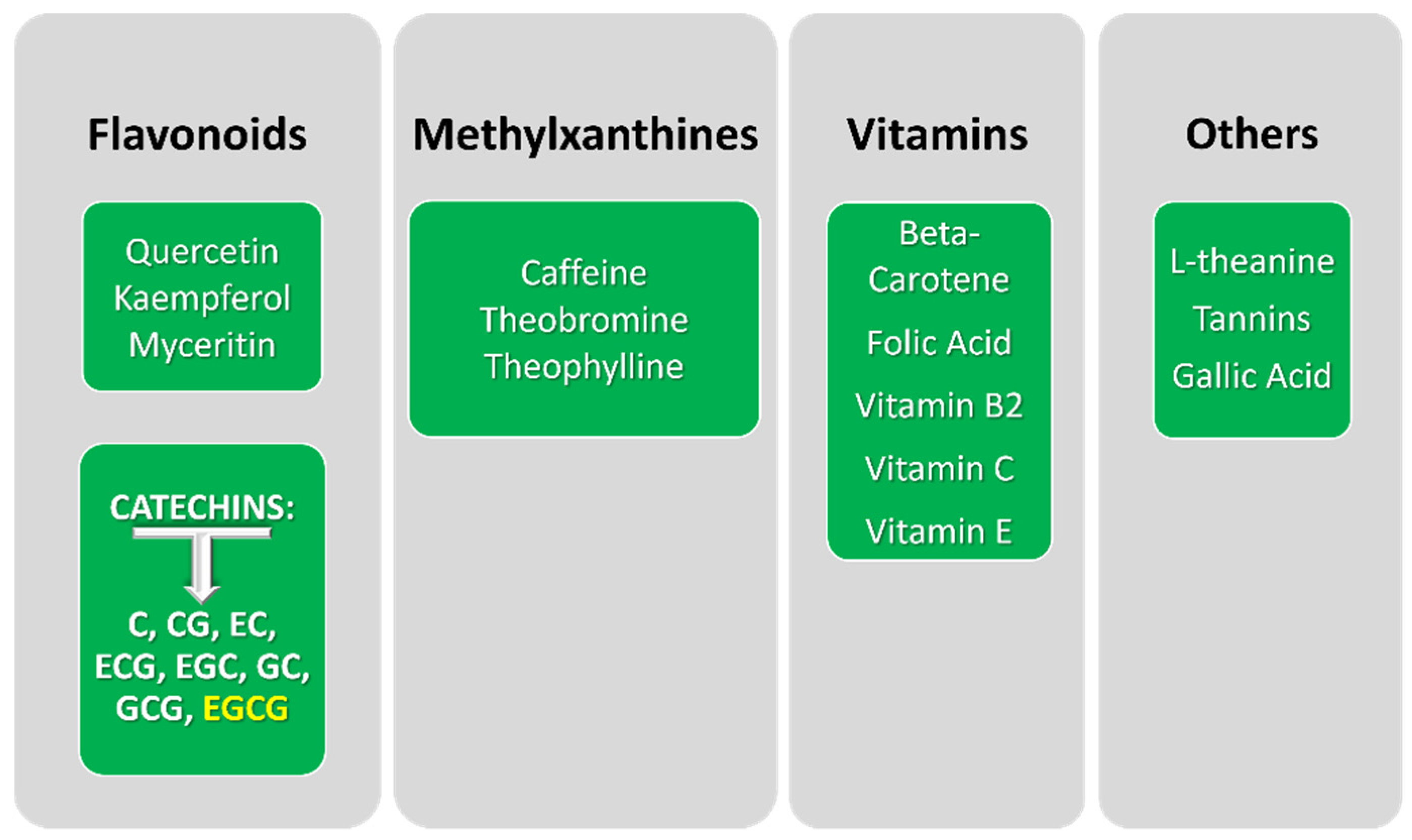
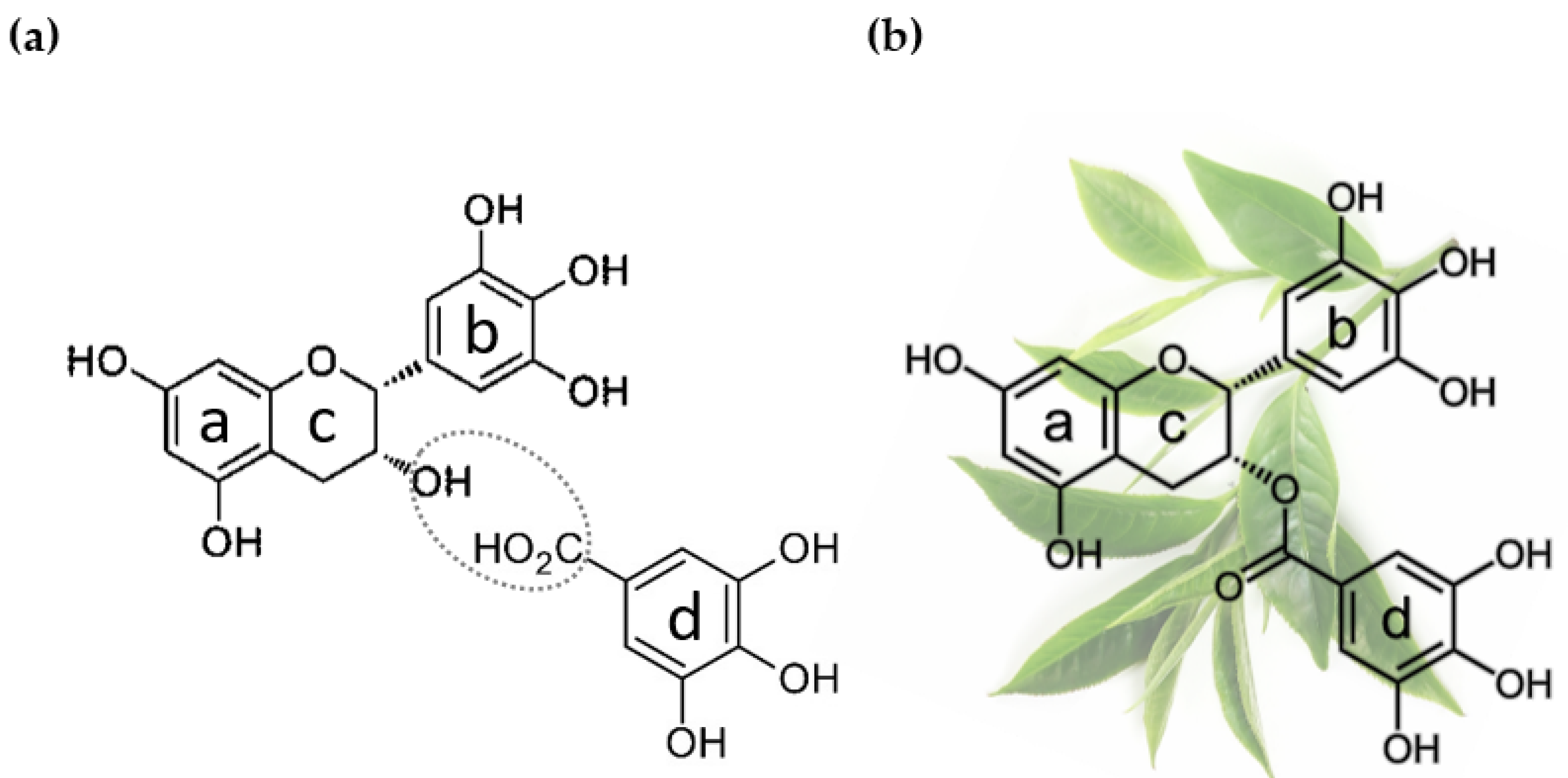
2. Epigallocatechin Gallate: Origin and Application in Cancer Research
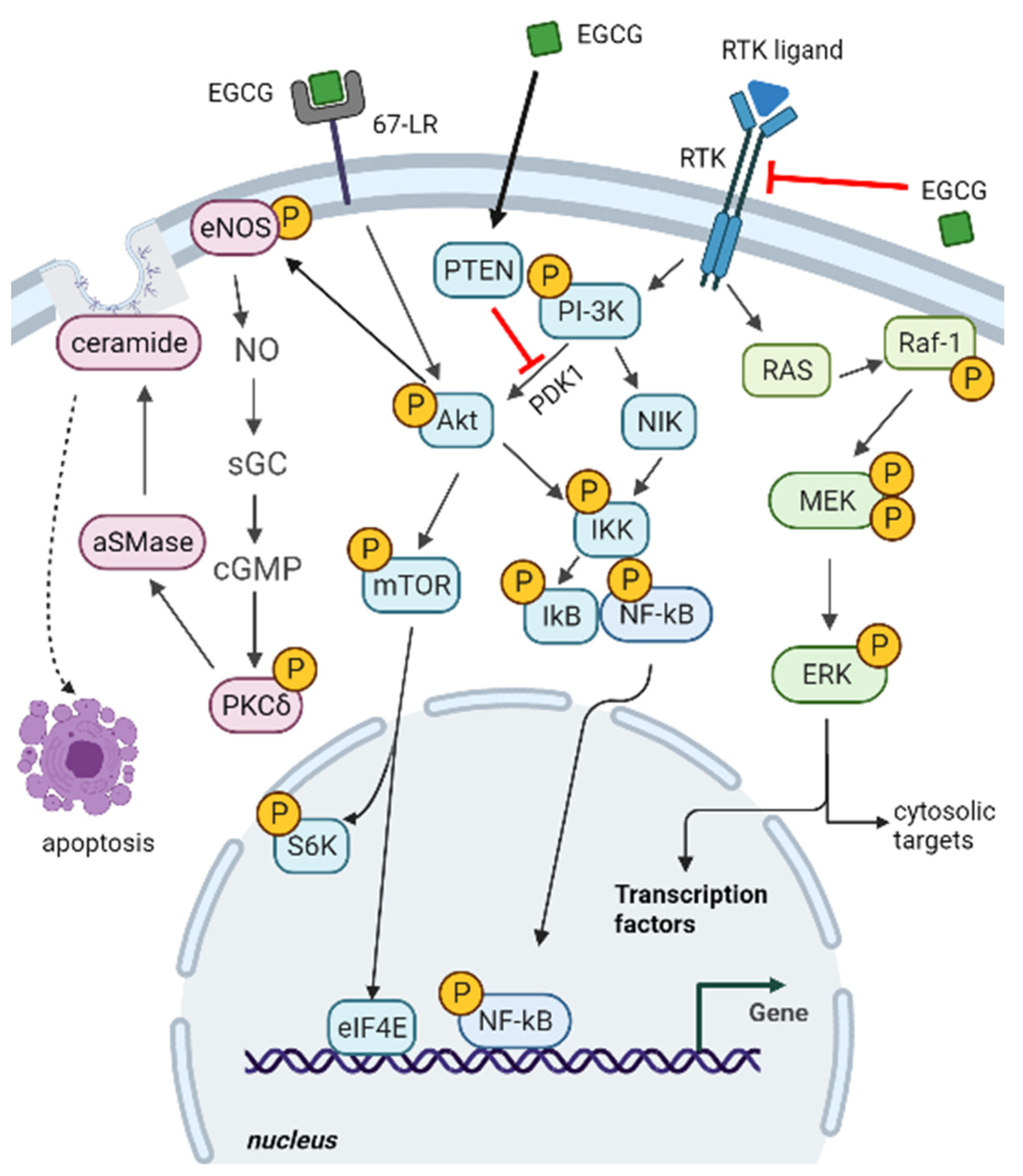
2.1. Epigallocatechin Gallate Impact on Cancer-Related Signalling Pathways
2.1.1. ERK and PI3K-Akt Pathways
2.1.2. 67-LR Pathway
2.1.3. Death Receptors-Dependent Apoptosis and Cell Redox Balance
3. Autophagy Mechanism and Function
Autophagy in Cancer
4. Targeting Autophagy in Cancer by EGCG Treatment
4.1. Study Selection
4.2. Evidence of Autophagy Modulation Combined with Anti-Cancer Effects in Response to EGCG Treatment in Experimental Cancer Models
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Yang, Y.; Klionsky, D.J. Autophagy and disease: Unanswered questions. Cell Death Differ. 2020, 27, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Kocak, M.; Ezazi Erdi, S.; Jorba, G.; Maestro, I.; Farrés, J.; Kirkin, V.; Martinez, A.; Pless, O. Targeting autophagy in disease: Established and new strategies. Autophagy 2022, 18, 473–495. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.W.; Lee, S.H. The Roles of Autophagy in Cancer. Int. J. Mol. Sci. 2018, 19, 3466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, S.F.; Wang, X.L.; Yang, X.Q.; Chen, N. Autophagy-associated targeting pathways of natural products during cancer treatment. Asian Pac. J. Cancer Prev. 2014, 15, 10557–10563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musial, C.; Siedlecka-Kroplewska, K.; Kmiec, Z.; Gorska-Ponikowska, M. Modulation of Autophagy in Cancer Cells by Dietary Polyphenols. Antioxidants 2021, 10, 123. [Google Scholar] [CrossRef]
- Mohammad, R.M.; Muqbil, I.; Lowe, L.; Yedjou, C.; Hsu, H.Y.; Lin, L.T.; Siegelin, M.D.; Fimognari, C.; Kumar, N.B.; Dou, Q.P.; et al. Broad targeting of resistance to apoptosis in cancer. Semin. Cancer Biol. 2015, 35, S78–S103. [Google Scholar] [CrossRef]
- Su, M.; Mei, Y.; Sinha, S. Role of the Crosstalk between Autophagy and Apoptosis in Cancer. J. Oncol. 2013, 2013, 102735. [Google Scholar] [CrossRef] [Green Version]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Akkoc, Y.; Kig, C.; Bayraktar, O.; Gozuacik, D.; Kutlu, O. Autophagy as a molecular target for cancer treatment. Eur. J. Pharm. Sci. 2019, 134, 116–137. [Google Scholar] [CrossRef]
- Sharifi-Rad, M.; Pezzani, R.; Redaelli, M.; Zorzan, M.; Imran, M.; Khalil, A.A.; Salehi, B.; Sharopov, F.; Cho, W.C.; Sharifi-Rad, J. Preclinical Pharmacological Activities of Epigallocatechin-3-gallate in Signaling Pathways: An Update on Cancer. Molecules 2020, 25, 467. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Cao, M.; Fang, F. The Role of Epigallocatechin-3-Gallate in Autophagy and Endoplasmic Reticulum Stress (ERS)-Induced Apoptosis of Human Diseases. Med. Sci. Monit. 2020, 26, e924558. [Google Scholar] [CrossRef] [PubMed]
- Min, K.; Kwon, T.K. Anticancer effects and molecular mechanisms of epigallocatechin-3-gallate. Integr. Med. Res. 2014, 3, 16–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almatrood, S.A.; Almatroudi, A.; Khan, A.A.; Alhumaydh, F.A.; Alsahl, M.A.; Rahmani, A.H. Potential Therapeutic Targets of Epigallocatechin Gallate (EGCG), the Most Abundant Catechin in Green Tea, and Its Role in the Therapy of Various Types of Cancer. Molecules 2020, 25, 3146. [Google Scholar] [CrossRef]
- Rahman, M.A.; Hannan, M.A.; Dash, R.; Rahman, M.H.; Islam, R.; Uddin, M.J.; Sohag, A.A.M.; Rahman, M.H.; Rhim, H. Phytochemicals as a Complement to Cancer Chemotherapy: Pharmacological Modulation of the Autophagy-Apoptosis Pathway. Front. Pharmacol. 2021, 12, 639628. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ho, C.T.; Zhou, J.; Santos, J.S.; Armstrong, L.; Granato, D. Chemistry and Biological Activities of Processed Camellia sinensis Teas: A Comprehensive Review. Compr. Rev. Food Sci. Food Saf. 2019, 18, 1474–1495. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Wang, Y.; Li, D.; Chen, Y.; Qiao, X.; Fardous, R.; Lewandowski, A.; Liu, J.; Chan, T.H.; Dou, Q.P. Perspectives on the recent developments with green tea polyphenols in drug discovery. Expert Opin. Drug Discov. 2018, 13, 643–660. [Google Scholar] [CrossRef]
- Chacko, S.M.; Thambi, P.T.; Kuttan, R.; Nishigaki, I. Beneficial effects of green tea: A literature review. Chin. Med. 2010, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Bansal, S.; Syan, N.; Mathur, P.; Choudhary, S. Pharmacological profile of green tea and its polyphenols: A review. Med. Chem. Res. 2012, 21, 3347–3360. [Google Scholar] [CrossRef]
- Tauber, A.L.; Schweiker, S.S.; Levonis, S.M. From tea to treatment; epigallocatechin gallate and its potential involvement in minimizing the metabolic changes in cancer. Nutr. Res. 2020, 74, 23–36. [Google Scholar] [CrossRef]
- Kochman, J.; Jakubczyk, K.; Antoniewicz, J.; Mruk, H.; Janda, K. Health Benefits and Chemical Composition of Matcha Green Tea: A Review. Molecules 2020, 26, 85. [Google Scholar] [CrossRef]
- Negri, A.; Naponelli, V.; Rizzi, F.; Bettuzzi, S. Molecular targets of epigallocatechin—gallate (EGCG): A special focus on signal transduction and cancer. Nutrients 2018, 10, 1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, R.Y.; Li, H.B.; Sui, Z.Q.; Corke, H. Absorption, metabolism, anti-cancer effect and molecular targets of epigallocatechin gallate (EGCG): An updated review. Crit. Rev. Food Sci. Nutr. 2018, 58, 924–941. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Suthakar, G.; Srivastava, R.K. Epigallocatechin-3-gallate inhibits cell cycle and induces apoptosis in pancreatic cancer. Front. Biosci. 2007, 12, 5039–5051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Han, X.; Ou, D.; Liu, T.; Li, Z.; Jiang, G.; Liu, J.; Zhang, J. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl. Microbiol. Biotechnol. 2020, 104, 575–587. [Google Scholar] [CrossRef]
- Popova, N.V.; Jücker, M. The Role of mTOR Signaling as a Therapeutic Target in Cancer. Int. J. Mol. Sci. 2021, 22, 1743. [Google Scholar] [CrossRef]
- Zughaibi, T.A.; Suhail, M.; Tarique, M.; Tabrez, S. Targeting PI3K/Akt/mTOR Pathway by Different Flavonoids: A Cancer Chemopreventive Approach. Int. J. Mol. Sci. 2021, 22, 2455. [Google Scholar] [CrossRef]
- Owusu-Brackett, N.; Shariati, M.; Meric-Bernstam, F. Role of PI3K/AKT/mTOR in Cancer Signaling. In Predictive Biomarkers in Oncology; Springer: Cham, Switzerland, 2019; pp. 263–270. [Google Scholar] [CrossRef]
- Ocana, A.; Vera-Badillo, F.; Al-Mubarak, M.; Templeton, A.J.; Corrales-Sanchez, V.; Diez-Gonzalez, L.; Cuenca-Lopez, M.D.; Seruga, B.; Pandiella, A.; Amir, E. Activation of the PI3K/mTOR/AKT pathway and survival in solid tumors: Systematic review and meta-analysis. PLoS ONE 2014, 9, e95219. [Google Scholar] [CrossRef]
- Christian, F.; Smith, E.L.; Carmody, R.J. The Regulation of NF-κB Subunits by Phosphorylation. Cells 2016, 5, 12. [Google Scholar] [CrossRef] [Green Version]
- Caporali, A.; Davalli, P.; Astancolle, S.; D’Arca, D.; Brausi, M.; Bettuzzi, S.; Corti, A. The chemopreventive action of catechins in the TRAMP mouse model of prostate carcinogenesis is accompanied by clusterin over-expression. Carcinogenesis 2004, 25, 2217–2224. [Google Scholar] [CrossRef] [Green Version]
- Panico, F.; Casali, C.; Rossi, G.; Rizzi, F.; Morandi, U.; Bettuzzi, S.; Davalli, P.; Corbetta, L.; Storelli, E.S.; Corti, A.; et al. Prognostic role of clusterin in resected adenocarcinomas of the lung. Lung Cancer 2013, 79, 294–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.X.; Shi, Y.L.; Zhang, L.J.; Wang, K.R.; Xiang, L.P.; Cai, Z.Y.; Lu, J.L.; Ye, J.H.; Liang, Y.R.; Zheng, X.Q. Inhibitory Effects of (−)-Epigallocatechin-3-gallate on Esophageal Cancer. Molecules 2019, 24, 954. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Fu, M.; Wang, J.; Huang, F.; Liu, H.; Huangfu, M.; Yu, D.; Liu, H.; Li, X.; Guan, X.; et al. PTEN/AKT/mTOR signaling mediates anticancer effects of epigallocatechin-3-gallate in ovarian cancer. Oncol. Rep. 2020, 43, 1885–1896. [Google Scholar] [CrossRef]
- Montuori, N.; Selleri, C.; Risitano, A.M.A.M.; Raiola, A.M.A.M.; Ragno, P.; Del Vecchio, L.; Rotoli, B.; Rossi, G. Expression of the 67-kDa laminin receptor in acute myeloid leukemia cells mediates adhesion to laminin and is frequently associated with monocytic differentiation. Clin. Cancer Res. 1999, 5, 1465–1472. [Google Scholar] [PubMed]
- Pesapane, A.; Ragno, P.; Selleri, C.; Montuori, N. Recent Advances in the Function of the 67 kDa Laminin Receptor and its Targeting for Personalized Therapy in Cancer. Curr. Pharm. Des. 2017, 23, 4745–4757. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.L.; Xu, J.; Yao, H.J.; Luo, K.L.; Li, J.M.; Wu, T.; Wu, G.Z. Inhibition of human 67-kDa laminin receptor sensitizes multidrug resistance colon cancer cell line SW480 for apoptosis induction. Tumour Biol. 2016, 37, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Casaletto, J.B.; McClatchey, A.I. Spatial regulation of receptor tyrosine kinases in development and cancer. Nat. Rev. Cancer 2012, 12, 387–400. [Google Scholar] [CrossRef]
- Tsukamoto, S.; Hirotsu, K.; Kumazoe, M.; Goto, Y.; Sugihara, K.; Suda, T.; Tsurudome, Y.; Suzuki, T.; Yamashita, S.; Kim, Y.; et al. Green tea polyphenol EGCG induces lipid-raft clustering and apoptotic cell death by activating protein kinase Cδ and acid sphingomyelinase through a 67 kDa laminin receptor in multiple myeloma cells. Biochem. J. 2012, 443, 525–534. [Google Scholar] [CrossRef]
- Huang, Y.; Kumazoe, M.; Bae, J.; Yamada, S.; Takai, M.; Hidaka, S.; Yamashita, S.; Kim, Y.; Won, Y.; Murata, M.; et al. Green tea polyphenol epigallocatechin-O-gallate induces cell death by acid sphingomyelinase activation in chronic myeloid leukemia cells. Oncol. Rep. 2015, 34, 1162–1168. [Google Scholar] [CrossRef] [Green Version]
- Kumazoe, M.; Sugihara, K.; Tsukamoto, S.; Huang, Y.; Tsurudome, Y.; Suzuki, T.; Suemasu, Y.; Ueda, N.; Yamashita, S.; Kim, Y.; et al. 67-kDa laminin receptor increases cGMP to induce cancer-selective apoptosis. J. Clin. Investig. 2013, 123, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int. J. Mol. Sci. 2018, 19, 448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, O.S.; Jung, J.H.; Shin, E.A.; Park, J.E.; Park, W.Y.; Kim, S.H. Epigallocatechin-3-Gallate Induces Apoptosis as a TRAIL Sensitizer via Activation of Caspase 8 and Death Receptor 5 in Human Colon Cancer Cells. Biomedicines 2020, 8, 84. [Google Scholar] [CrossRef] [Green Version]
- Yao, K.; Ye, P.P.; Zhang, L.; Tan, J.; Tang, X.J.; Zhang, Y.D. Epigallocatechin gallate protects against oxidative stress-induced mitochondria-dependent apoptosis in human lens epithelial cells. Mol. Vis. 2008, 14, 217. [Google Scholar] [PubMed]
- Lambert, J.D.; Elias, R.J. The antioxidant and pro-oxidant activities of green tea polyphenols: A role in cancer prevention. Arch. Biochem. Biophys. 2010, 501, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Yan, Z.; Zhong, Y.; Duan, Y.; Chen, Q.; Li, F. Antioxidant mechanism of tea polyphenols and its impact on health benefits. Anim. Nutr. (Zhongguo Xu Mu Shou Yi Xue Hui) 2020, 6, 115–123. [Google Scholar] [CrossRef]
- Chow, H.H.S.; Hakim, I.A.; Vining, D.R.; Crowell, J.A.; Tome, M.E.; Ranger-Moore, J.; Cordova, C.A.; Mikhael, D.M.; Briehl, M.M.; Alberts, D.S. Modulation of human glutathione s-transferases by polyphenon e intervention. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1662–1666. [Google Scholar] [CrossRef] [Green Version]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell. Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef]
- Afzal, M.; Safer, A.M.; Menon, M. Green tea polyphenols and their potential role in health and disease. Inflammopharmacology 2015, 23, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Chikara, S.; Nagaprashantha, L.D.; Singhal, J.; Horne, D.; Awasthi, S.; Singhal, S.S. Oxidative stress and dietary phytochemicals: Role in cancer chemoprevention and treatment. Cancer Lett. 2018, 413, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.; Kim, J. Autophagy: An Essential Degradation Program for Cellular Homeostasis and Life. Cells 2018, 7, 278. [Google Scholar] [CrossRef] [Green Version]
- Towers, C.G.; Wodetzki, D.; Thorburn, A. Autophagy and cancer: Modulation of cell death pathways and cancer cell adaptations. J. Cell Biol. 2020, 219, e201909033. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Song, X.; Yang, Y.; Wan, X.; Alvarez, A.A.; Sastry, N.; Feng, H.; Hu, B.; Cheng, S.Y. Autophagy and Hallmarks of Cancer. Crit. Rev. Oncog. 2018, 23, 247–267. [Google Scholar] [CrossRef]
- Nishimura, T.; Tooze, S.A. Emerging roles of ATG proteins and membrane lipids in autophagosome formation. Cell Discov. 2020, 6, 32. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The multifaceted role of autophagy in cancer and the microenvironment. Med. Res. Rev. 2019, 39, 517–560. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, N. Autophagy Is a Promoter for Aerobic Exercise Performance during High Altitude Training. Oxid. Med. Cell. Longev. 2018, 2018, 3617508. [Google Scholar] [CrossRef] [Green Version]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793. [Google Scholar] [CrossRef] [Green Version]
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Laddha, S.V.; Ganesan, S.; Chan, C.S.; White, E. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Mol. Cancer Res. 2014, 12, 485–490. [Google Scholar] [CrossRef] [Green Version]
- Ionov, Y.; Nowak, N.; Perucho, M.; Markowitz, S.; Cowell, J.K. Manipulation of nonsense mediated decay identifies gene mutations in colon cancer Cells with microsatellite instability. Oncogene 2004, 23, 639–645. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.W.J.Y.; Jeong, E.G.; Soung, Y.H.; Nam, S.W.; Lee, J.W.J.Y.; Yoo, N.J.; Lee, S.H. Decreased expression of tumour suppressor Bax-interacting factor-1 (Bif-1), a Bax activator, in gastric carcinomas. Pathology 2006, 38, 312–315. [Google Scholar] [CrossRef]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front. Oncol. 2020, 10, 8418. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the context-dependent role for autophagy in cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Folkerts, H.; Hilgendorf, S.; Wierenga, A.T.J.; Jaques, J.; Mulder, A.B.; Coffer, P.J.; Schuringa, J.J.; Vellenga, E. Inhibition of autophagy as a treatment strategy for p53 wild-type acute myeloid leukemia. Cell Death Dis. 2017, 8, e2927. [Google Scholar] [CrossRef] [Green Version]
- Zaarour, R.F.; Azakir, B.; Hajam, E.Y.; Nawafleh, H.; Zeinelabdin, N.A.; Engelsen, A.S.T.; Thiery, J.; Jamora, C.; Chouaib, S. Role of Hypoxia-Mediated Autophagy in Tumor Cell Death and Survival. Cancers 2021, 13, 533. [Google Scholar] [CrossRef]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, X.T.; Shi, Y.H.; Zhou, J.; Peng, Y.F.; Liu, W.R.; Shi, G.M.; Gao, Q.; Wang, X.Y.; Song, K.; Fan, J.; et al. MicroRNA-30a suppresses autophagy-mediated anoikis resistance and metastasis in hepatocellular carcinoma. Cancer Lett. 2018, 412, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.W.; Moon, J.H.; Park, S.Y. Activation of autophagic flux by epigallocatechin gallate mitigates TRAIL-induced tumor cell apoptosis via down-regulation of death receptors. Oncotarget 2016, 7, 65660–65668. [Google Scholar] [CrossRef]
- Xie, J.; Yun, J.P.; Yang, Y.N.; Hua, F.; Zhang, X.W.; Lin, H.; Lv, X.X.; Li, K.; Zhang, P.C.; Hu, Z.W. A novel ECG analog 4-(S)-(2,4,6-trimethylthiobenzyl)-epigallocatechin gallate selectively induces apoptosis of B16-F10 melanoma via activation of autophagy and ROS. Sci. Rep. 2017, 7, 42194. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.Y.; Chen, C.Y.; Chiou, Y.H.; Shyu, H.W.; Lin, K.H.; Chou, M.C.; Huang, M.H.; Wang, Y.F. Epigallocatechin-3-Gallate Suppresses Human Herpesvirus 8 Replication and Induces ROS Leading to Apoptosis and Autophagy in Primary Effusion Lymphoma Cells. Int. J. Mol. Sci. 2017, 19, 16. [Google Scholar] [CrossRef] [Green Version]
- Wei, R.; Mao, L.; Xu, P.; Zheng, X.; Hackman, R.M.; MacKenzie, G.G.; Wang, Y. Suppressing glucose metabolism with epigallocatechin-3-gallate (EGCG) reduces breast cancer cell growth in preclinical models. Food Funct. 2018, 9, 5682–5696. [Google Scholar] [CrossRef]
- Enkhbat, T.; Nishi, M.; Yoshikawa, K.; Jun, H.; Tokunaga, T.; Takasu, C.; Kashihara, H.; Ishikawa, D.; Tominaga, M.; Shimada, M. Epigallocatechin-3-gallate Enhances Radiation Sensitivity in Colorectal Cancer Cells through Nrf2 Activation and Autophagy. Anticancer Res. 2018, 38, 6247–6252. [Google Scholar] [CrossRef]
- Hsieh, C.H.; Lu, C.H.; Kuo, Y.Y.; Chen, W.T.; Chao, C.Y. Studies on the non-invasive anticancer remedy of the triple combination of epigallocatechin gallate, pulsed electric field, and ultrasound. PLoS ONE 2018, 13, e0201920. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, S.; Yang, J.; Yi, P.; Xu, P.; Yi, M.; Peng, W. Integrated transcriptomic and metabolomic analyses to characterize the anti-cancer effects of (−)-epigallocatechin-3-gallate in human colon cancer cells. Toxicol. Appl. Pharmacol. 2020, 401, 115100. [Google Scholar] [CrossRef]
- Chen, X.; Liu, B.; Tong, R.; Ding, S.; Wu, J.; Lei, Q.; Fang, W. Improved Stability and Targeted Cytotoxicity of Epigallocatechin-3-Gallate Palmitate for Anticancer Therapy. Langmuir 2021, 37, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Seiler, K.; Mosimann, S.; Rentsch, V.; Sharma, K.; Pandey, A.V.; McKenna, S.L.; Tschan, M.P. Reducing FASN expression sensitizes acute myeloid leukemia cells to differentiation therapy. Cell Death Differ. 2021, 28, 2465–2481. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Li, J.; Kang, L.; Liu, X.; Luo, J.; Zhang, L.; Li, Y.; Cai, J. Epigallocatechin-3-gallate induces autophagy-related apoptosis associated with LC3B II and Beclin expression of bladder cancer cells. J. Food Biochem. 2021, 45, 6. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Chang, C.; Chen, Y.; Bi, F.; Ji, C.; Liu, W. EGCG overcomes gefitinib resistance by inhibiting autophagy and augmenting cell death through targeting ERK phosphorylation in NSCLC. Onco. Targets. Ther. 2019, 12, 6033–6043. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Chen, D.; Zhu, K. SOX2OT variant 7 contributes to the synergistic interaction between EGCG and Doxorubicin to kill osteosarcoma via autophagy and stemness inhibition. J. Exp. Clin. Cancer Res. 2018, 37, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Issinger, O.G.; Guerra, B. Phytochemicals in cancer and their effect on the PI3K/AKT-mediated cellular signalling. Biomed. Pharmacother. 2021, 139, 111650. [Google Scholar] [CrossRef]
- Ouyang, J.; Zhu, K.; Liu, Z.; Huang, J. Prooxidant Effects of Epigallocatechin-3-Gallate in Health Benefits and Potential Adverse Effect. Oxid. Med. Cell. Longev. 2020, 2020, 9723686. [Google Scholar] [CrossRef]
- López-Lázaro, M. Dual role of hydrogen peroxide in cancer: Possible relevance to cancer chemoprevention and therapy. Cancer Lett. 2007, 252, 1–8. [Google Scholar] [CrossRef]
- Faralli, A.; Shekarforoush, E.; Mendes, A.C.; Chronakis, I.S. Enhanced Transepithelial Permeation of Gallic Acid and (−)-Epigallocatechin Gallate across Human Intestinal Caco-2 Cells Using Electrospun Xanthan Nanofibers. Pharmaceutics 2019, 11, 155. [Google Scholar] [CrossRef] [Green Version]
- Sahadevan, R.; Singh, S.; Binoy, A.; Sadhukhan, S. Chemico-biological aspects of (−)- epigallocatechin- 3-gallate (EGCG) to improve its stability, bioavailability and membrane permeability: Current status and future prospects. Crit. Rev. Food Sci. Nutr. 2022, 29, 1–30. [Google Scholar] [CrossRef]
- Cai, Z.Y.; Li, X.M.; Liang, J.P.; Xiang, L.P.; Wang, K.R.; Shi, Y.L.; Yang, R.; Shi, M.; Ye, J.H.; Lu, J.L.; et al. Bioavailability of Tea Catechins and Its Improvement. Molecules 2018, 23, 2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Taylor, L.S.; Ferruzzi, M.G.; Mauer, L.J. Kinetic study of catechin stability: Effects of pH, concentration, and temperature. J. Agric. Food Chem. 2012, 60, 12531–12539. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.Q.; Liang, Y.F.; Ma, S.B.; Li, H.; Gao, W.Y. Stability and stabilization of (−)-gallocatechin gallate under various experimental conditions and analyses of its epimerization, auto-oxidation, and degradation by LC-MS. J. Sci. Food Agric. 2019, 99, 5984–5993. [Google Scholar] [CrossRef] [PubMed]
- Murakami, A. Dose-dependent functionality and toxicity of green tea polyphenols in experimental rodents. Arch. Biochem. Biophys. 2014, 557, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naponelli, V.; Ramazzina, I.; Lenzi, C.; Bettuzzi, S.; Rizzi, F. Green tea catechins for prostate cancer prevention: Present achievements and future challenges. Antioxidants 2017, 6, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurahashi, N.; Sasazuki, S.; Iwasaki, M.; Inoue, M. Green tea consumption and prostate cancer risk in Japanese men: A prospective study. Am. J. Epidemiol. 2008, 167, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Bettuzzi, S.; Brausi, M.; Rizzi, F.; Castagnetti, G.; Peracchia, G.; Corti, A. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: A preliminary report from a one-year proof-of-principle study. Cancer Res. 2006, 66, 1234–1240. [Google Scholar] [CrossRef] [Green Version]
- Tsao, A.S.; Liu, D.; Martin, J.; Tang, X.M.; Lee, J.J.; El-Naggar, A.K.; Wistuba, I.; Culotta, K.S.; Mao, L.; Gillenwater, A.; et al. Phase II randomized, placebo-controlled trial of green tea extract in patients with high-risk oral premalignant lesions. Cancer Prev. Res. 2009, 2, 931–941. [Google Scholar] [CrossRef] [Green Version]
- Page, M.J.; Moher, D.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. PRISMA 2020 explanation and elaboration: Updated guidance and exemplars for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell line/Animal | Principal Techniques | Main Results | Conclusions | Reference |
|---|---|---|---|---|
| HCT116 human colon carcinoma cell line | Crystal violet staining for cell viability, LDH cytotoxicity assay, western blotting (LC3, p62), RNA interference | EGCG and TRAIL co-treatment:
| EGCG can protect against TRAIL-induced cell death by activating autophagy in TRAIL-sensitive cells. Since autophagy activation may prevent cancer cell death, an autophagy inhibitor is recommended in combination with drugs such as TRAIL because of possible autophagic pathway alterations. | [75] |
| B16-F10 mouse melanoma cells, AML-12 mouse hepatocytes, Male C57BL/6 mice | CCK-8 assay for cell viability, flow cytometric analysis of autophagy flux activation (GFP-LC3, Bafilomycin A1 treatment) and apoptosis, flow cytometry with dihydroethidium for measurement of intracellular ROS, B16-F10 xenograft mouse model for in vivo study | In B16 melanoma cells, the EGC analogue 4-(S)-(2,4,6-trimethylthiobenzyl)-epigallocatechin gallate: activated autophagy and reduced cell viability by inducing apoptosis;selectively induced ROS accumulation with consequent cell damage;suppressed tumor growth in vivo, while inducing ROS accumulation.Pharmacological inhibition of ROS by NAC attenuated induced autophagy and apoptosis. | The EGC analogue has autophagy- and ROS-inducing ability. Induced autophagy may act as a downstream sensor of ROS that sequentially induces cell death. | [76] |
| Primary effusion lymphoma (PEL) cells (HHV8-positive) | Trypan blue exclusion assay for cell viability, Caspase 3 activity assay, western blotting (LC3, Beclin 1, MAPKs), acridine orange for acidic vesicular organelle staining | EGCG suppressed viral particle production, and inhibited PEL cell line growth. EGCG induced apoptosis and autophagy through ROS generation; 3-MA autophagy inhibitor and Caspase 3 inhibitor failed to rescue the cytotoxic effect of EGCG. NAC co-treatment reduced ROS level, cytotoxicity, Caspase 3 activation, and autophagy in EGCG-treated PEL cells. | In PEL cells, EGCG induces cell death via a mechanism involving ROS generation, leading to autophagy and apoptosis. | [77] |
| Mouse 4T1 breast cancer cell line, Balb/c mice | CCK-8 assay for cell viability, flow cytometric analysis of cell apoptosis, Caspase activity assay, western blotting (Beclin 1, ATG5, LC3B), glycolysis-related enzyme activity tests | EGCG inhibited the growth of 4T1 cell line by inducing apoptosis and autophagy. EGCG reduced the expression level of HIF1α and GLUT1, and affected the glycolytic pathway by decreasing activity and/or level of HK, PFK and LDH. EGCG reduced breast cancer xenograft growth in mice. | EGCG suppresses glucose metabolism and has antitumor activity through the induction of apoptosis and autophagy. It might be tested as an adjuvant agent against breast cancer. | [78] |
| HCT-116 colon cancer cell line | CCK-8 assay for cell viability, immunofluorescence microscopy (Nfr2 nuclear translocation), qRT-PCR (LC3 and Caspase 9) | EGCG increased cell sensitivity to X-ray irradiation and reduced proliferation.Combination treatment with EGCG and radiation:
| Combination treatment with EGCG and radiation enhanced the expression of proteins and mRNAs related to autophagy and apoptosis. | [79] |
| PANC-1 human pancreatic cancer cell line, HepG2 human hepatocellular carcinoma cell line | MTT proliferation assay, flow cytometry with dihydroethidium for measurement of intracellular ROS, MDC staining for autophagic vacuoles detection, western blotting (LC3, pAkt, Caspase 3 and 9) | Application of low strength pulsed electric field and low energy ultrasound enhanced the growth inhibition effect of EGCG on PANC-1 cells. The triple treatment:
| EGCG combined with non-invasive and mild physical stimulations might be a promising strategy for anticancer treatment. | [80] |
| HT-29 human colorectal adenocarcinoma cell line | MTT proliferation assay, flow cytometry and TUNEL staining for cell apoptosis, MDC staining for autophagic vacuoles detection, western blotting (LC3B, Beclin1, Caspase 3 and 9), transcriptomics, and metabolomics analyses | EGCG inhibited cell proliferation, and induced apoptosis and autophagy in HT-29 cells. EGCG treatment was associated with significant changes in gene-expression and metabolic profile. Differential metabolites of CRC are involved in the metabolism of glutathione, glycerophospholipids, starch, and sucrose, among others. | The anti-proliferative activity of EGCG is closely related to apoptosis and autophagy. Transcriptome and metabolome analyses reveal that the anti-CRC effect of EGCG may depend on its modulation of glycerophospholipids metabolism. | [81] |
| HeLa cell line, HEK293 cell line | MTT proliferation assay, DCFDA ROS assay, flow cytometric analysis of cell apoptosis, mRFP-GFP-LC3 plasmid transfection and confocal microscopy, MDC staining for autophagic vacuoles detection, western blotting (LC3, Beclin 1, Caspase 3 and 9) | EGCG-palmitate remained stable in DMEM medium for a longer time than EGCG. In cancerous cells, EGCG-palmitate induced a lower cell proliferation rate, as compared with normal cells, and promoted apoptosis and autophagy, both resulting from excess of ROS generation. | EGCG-palmitate displayed improved stability and targeted cytotoxicity for cancerous cells. EGCG palmitate expresses its pro-oxidative bioactivity when working as an anticancer drug, and its antioxidant potential in normal cells. | [82] |
| HT93, OCI/AML2, MOLM-13 and NB4 human AML cell lines | Western blotting (FASN, LC3B, p-mTOR), shRNA transfection for FASN knockdown, acridine orange for acidic vesicular organelle staining, immunofluorescence microscopy | FASN is upregulated in tumor-associated myeloid cells and becomes a target for autophagic degradation during all-trans retinoic acid-induced differentiation of APL cells. Co-treatment with EGCG improved the response to all-trans retinoic acid in NB4 cells, and enhanced FASN protein degradation by autophagy. Lowering FASN expression is associated to mTOR pathway inhibition, promoting autophagy. | Differentiation therapy holds great promise for cancer treatment. Co-treatment with EGCG improves the response to all-trans retinoic acid in APL cell lines and significantly re-sensitizes refractory non-APL AML cells. | [83] |
| T24 and 5637 human bladder transitional cell carcinoma cell lines | MTT proliferation assay, flow cytometric analysis of cell apoptosis, western blotting (LC3B, Beclin 1, mTOR/p-mTOR, Caspase 3 and 9), shRNA transfection for ATG5 knockdown | EGCG inhibited proliferation and induced apoptosis in T24 and 5637 cells EGCG regulated apoptosis- and autophagy-related protein expression, and significantly increased autophagosome formation in T24 and 5637 cells. In 5637 cells:
| EGCG treatment inactivates PI3K/Akt/mTOR pathway, resulting in cancer cell growth inhibition. EGCG inhibits bladder cancer cells proliferation by facilitating crosstalk between apoptosis and autophagy. | [84] |
| A549 human lung carcinoma cell line, BALB/C male nude mice | MTS proliferation assay, GFP-LC3 plasmid transfection and confocal microscopy, flow cytometry and TUNEL staining for cell apoptosis, western blotting (LC3, ATG5, pERK, p-MEK) | EGCG and Gef synergized in inhibiting the proliferation of Gef-resistant NSCLC cell; the synergy was confirmed also in A549 mouse xenograft models. EGCG inhibited Gef-induced pro-survival autophagy and ERK phosphorylation in A549 cells, thus promoting cell death by apoptosis. EGCG alleviated Gef resistance by inhibiting Raf/MEK/ ERK pathway. | EGCG overcomes A549 Gef resistance by inhibiting Gef-induced autophagy and induced cell death by targeting ERK pathway. This study presents an effective strategy to overcome acquired Gef resistance in NSCLC. | [85] |
| SaoS2 and U2OS osteosarcoma cell lines | MTT proliferation assay, qRT-PCR (Atg5 and Beclin 1), LC3 immunofluorescence staining, western blotting (LC3), MDC staining for autophagic vacuoles detection, sphere-forming assay | Cell growth inhibition was significantly upregulated when Dox was used in combination with EGCG. EGCG reduced the Dox-induced pro-survival autophagy by decreasing SOX2OT variant 7. EGCG partially inactivated the Notch3/DLL3 signaling cascade, targeting LncRNA SOX2OT variant 7 to reduce the stemness and abate drug-resistance of osteosarcoma cells. | EGCG produced synergistic effects with Dox on osteosarcoma cell growth inhibition by targeting LncRNA SOX2OT variant 7. This study provides a basis for developing anti-tumor treatments targeting osteosarcoma stem cells. | [86] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrari, E.; Bettuzzi, S.; Naponelli, V. The Potential of Epigallocatechin Gallate (EGCG) in Targeting Autophagy for Cancer Treatment: A Narrative Review. Int. J. Mol. Sci. 2022, 23, 6075. https://doi.org/10.3390/ijms23116075
Ferrari E, Bettuzzi S, Naponelli V. The Potential of Epigallocatechin Gallate (EGCG) in Targeting Autophagy for Cancer Treatment: A Narrative Review. International Journal of Molecular Sciences. 2022; 23(11):6075. https://doi.org/10.3390/ijms23116075
Chicago/Turabian StyleFerrari, Elena, Saverio Bettuzzi, and Valeria Naponelli. 2022. "The Potential of Epigallocatechin Gallate (EGCG) in Targeting Autophagy for Cancer Treatment: A Narrative Review" International Journal of Molecular Sciences 23, no. 11: 6075. https://doi.org/10.3390/ijms23116075
APA StyleFerrari, E., Bettuzzi, S., & Naponelli, V. (2022). The Potential of Epigallocatechin Gallate (EGCG) in Targeting Autophagy for Cancer Treatment: A Narrative Review. International Journal of Molecular Sciences, 23(11), 6075. https://doi.org/10.3390/ijms23116075