
Dicarbonyl Stress in Diabetic Vascular Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Clinical Situation

2. Endothelial Function
3. The Observations—Cells, Animals, Humans
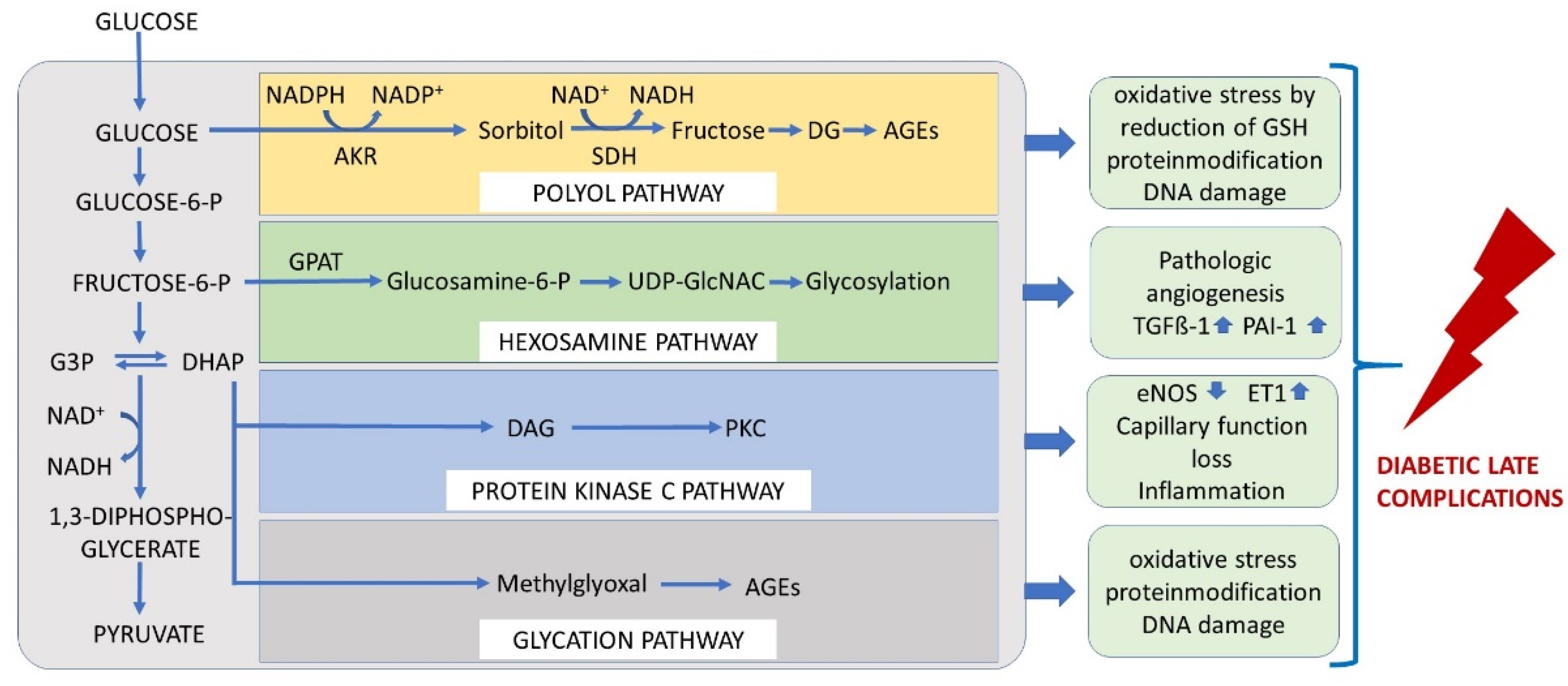
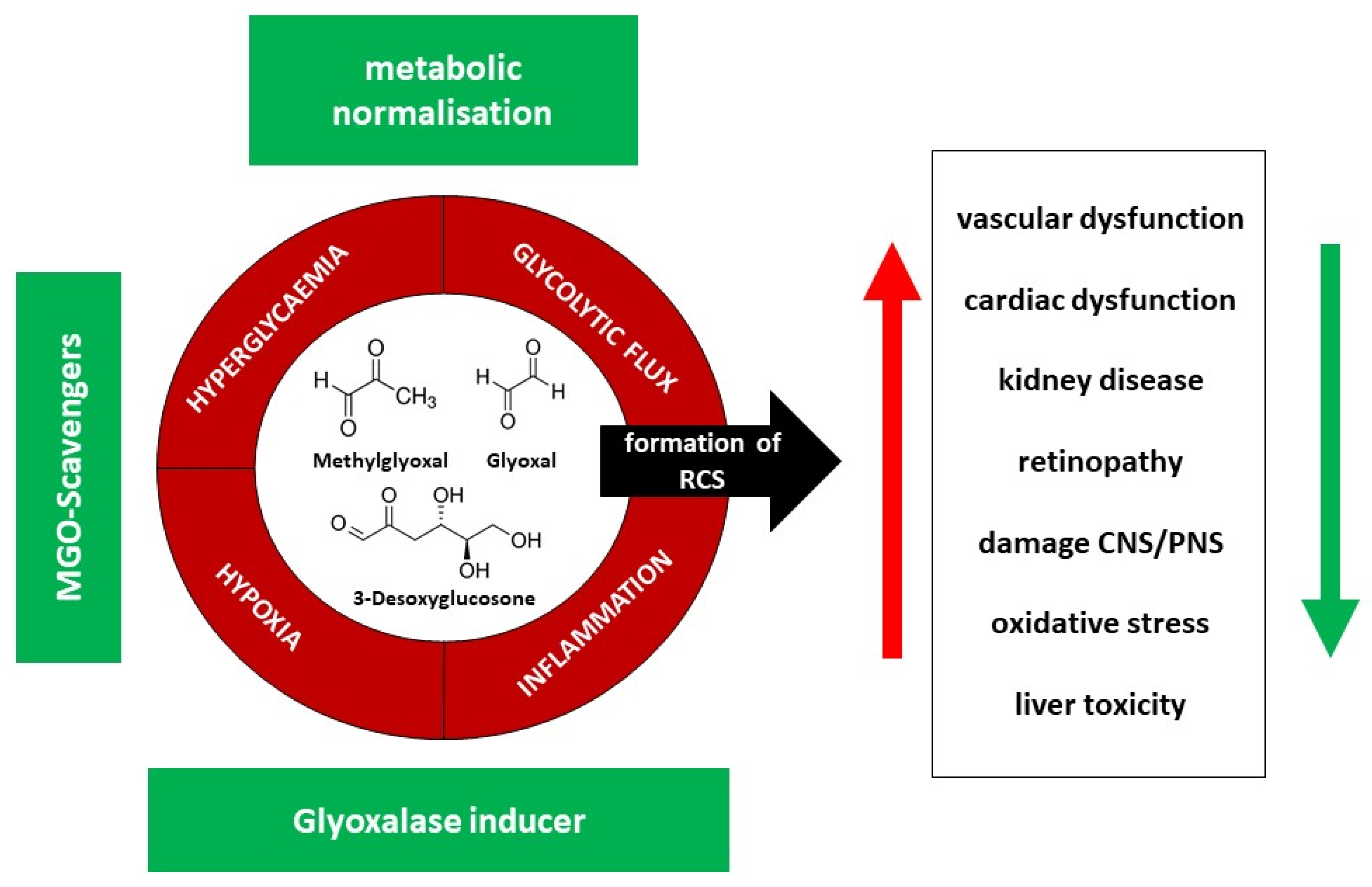
4. The Mechanisms
5. Therapeutic Possibilities
6. Role of MGO in Selected Vascular Diseases
6.1. Microvascular Disease
6.2. Macrovascular Disease
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chalmers, J.; Cooper, M.E. UKPDS and the legacy effect. N. Engl. J. Med. 2008, 359, 1618–1620. [Google Scholar] [CrossRef] [PubMed]
- Tancredi, M.; Rosengren, A.; Svensson, A.M.; Kosiborod, M.; Pivodic, A.; Gudbjornsdottir, S.; Dahlqvist, S.; Wedel, H.; Lind, M. Glycaemic control and excess mortality in patients with type 2 diabetes. Diabetologia 2015, 58, S137. [Google Scholar]
- Rabbani, N.; Thornalley, P.J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophs. Res. Commun. 2015, 458, 221–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Methylglyoxal-induced dicarbonyl stress in aging and disease: First steps towards glyoxalase 1-based treatments. Clin. Sci. 2016, 130, 1677–1696. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Glyoxalase Centennial conference: Introduction, history of research on the glyoxalase system and future prospects. Biochem. Soc. Trans. 2014, 42, 413–418. [Google Scholar] [CrossRef]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Iacobini, C.; Vitale, M.; Pugliese, G.; Menini, S. Normalizing HIF-1alpha Signaling Improves Cellular Glucose Metabolism and Blocks the Pathological Pathways of Hyperglycemic Damage. Biomedicines 2021, 9, 1139. [Google Scholar] [CrossRef]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef]
- Triggle, C.R.; Ding, H.; Marei, I.; Anderson, T.J.; Hollenberg, M.D. Why the endothelium? The endothelium as a target to reduce diabetes-associated vascular disease. Can. J. Physiol. Pharmacol. 2020, 98, 415–430. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Vascular complications in diabetes mellitus: The role of endothelial dysfunction. Clin. Sci. 2005, 109, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Sena, C.M.; Pereira, A.M.; Seica, R. Endothelial dysfunction-A major mediator of diabetic vascular disease. BBA Mol. Basis Dis. 2013, 1832, 2216–2231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stratmann, B.; Engelbrecht, B.; Espelage, B.C.; Klusmeier, N.; Tiemann, J.; Gawlowski, T.; Mattern, Y.; Eisenacher, M.; Meyer, H.E.; Rabbani, N.; et al. Glyoxalase 1-knockdown in human aortic endothelial cells-effect on the proteome and endothelial function estimates. Sci. Rep. 2016, 6, 37737. [Google Scholar] [CrossRef] [PubMed]
- Makinen, V.P.; Civelek, M.; Meng, Q.Y.; Zhang, B.; Zhu, J.; Levian, C.; Huan, T.X.; Segre, A.V.; Ghosh, S.; Vivar, J.; et al. Integrative Genomics Reveals Novel Molecular Pathways and Gene Networks for Coronary Artery Disease. PLoS Genet. 2014, 10, e1004502. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, O.; Niessen, P.M.; Miyata, T.; Ostergaard, J.A.; Flyvbjerg, A.; Peutz-Kootstra, C.J.; Sieber, J.; Mundel, P.H.; Brownlee, M.; Janssen, B.J.; et al. Glyoxalase-1 overexpression reduces endothelial dysfunction and attenuates early renal impairment in a rat model of diabetes. Diabetologia 2014, 57, 224–235. [Google Scholar] [CrossRef] [Green Version]
- Berlanga, J.; Cibrian, D.; Guillen, I.; Freyre, F.; Alba, J.S.; Lopez-Saura, P.; Merino, N.; Aldama, A.; Quintela, A.M.; Triana, M.E.; et al. Methylglyoxal administration induces diabetes-like microvascular changes and perturbs the healing process of cutaneous wounds. Clin. Sci. 2005, 109, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Sena, C.M.; Matafome, P.; Crisostomo, J.; Rodrigues, L.; Fernandes, R.; Pereira, P.; Seica, R.M. Methylglyoxal promotes oxidative stress and endothelial dysfunction. Pharmacol. Res. 2012, 65, 497–506. [Google Scholar] [CrossRef]
- Negrean, M.; Stirban, A.; Stratmann, B.; Gawlowski, T.; Horstmann, T.; Gotting, C.; Kleesiek, K.; Mueller-Roesel, M.; Koschinsky, T.; Uribarri, J.; et al. Effects of low- and high-advanced glycation endproduct meals on macro- and microvascular endothelial function and oxidative stress in patients with type 2 diabetes mellitus. Am. J. Clin. Nutr. 2007, 85, 1236–1243. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, M. The pathobiology of diabetic complications: A unifying mechanism. Diabetes 2005, 54, 1615–1625. [Google Scholar] [CrossRef] [Green Version]
- Vandekeere, S.; Dewerchin, M.; Carmeliet, P. Angiogenesis Revisited: An Overlooked Role of Endothelial Cell Metabolism in Vessel Sprouting. Microcirculation 2015, 22, 509–517. [Google Scholar] [CrossRef]
- Goveia, J.; Stapor, P.; Carmeliet, P. Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol. Med. 2014, 6, 1105–1120. [Google Scholar] [CrossRef] [PubMed]
- Jarisarapurin, W.; Kunchana, K.; Chularojmontri, L.; Wattanapitayakul, S.K. Unripe Carica papaya Protects Methylglyoxal-Invoked Endothelial Cell Inflammation and Apoptosis via the Suppression of Oxidative Stress and Akt/MAPK/NF-kappaB Signals. Antioxidants 2021, 10, 1158. [Google Scholar] [CrossRef] [PubMed]
- Brouwers, O.; Teerlink, T.; van Bezu, J.; Barto, R.; Stehouwer, C.D.; Schalkwijk, C.G. Methylglyoxal and methylglyoxal-arginine adducts do not directly inhibit endothelial nitric oxide synthase. Ann. N. Y. Acad. Sci. 2008, 1126, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Maessen, D.E.; Hanssen, N.M.; Scheijen, J.L.; van der Kallen, C.J.; van Greevenbroek, M.M.; Stehouwer, C.D.; Schalkwijk, C.G. Post-Glucose Load Plasma alpha-Dicarbonyl Concentrations Are Increased in Individuals with Impaired Glucose Metabolism and Type 2 Diabetes: The CODAM Study. Diabetes Care 2015, 38, 913–920. [Google Scholar] [CrossRef] [Green Version]
- Siragusa, M.; Fleming, I. The eNOS signalosome and its link to endothelial dysfunction. Pflugers Arch. 2016, 468, 1125–1137. [Google Scholar] [CrossRef]
- Kietadisorn, R.; Juni, R.P.; Moens, A.L. Tackling endothelial dysfunction by modulating NOS uncoupling: New insights into its pathogenesis and therapeutic possibilities. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E481–E495. [Google Scholar] [CrossRef] [Green Version]
- Mukohda, M.; Morita, T.; Okada, M.; Hara, Y.; Yamawaki, H. Long-term methylglyoxal treatment causes endothelial dysfunction of rat isolated mesenteric artery. J. Vet. Med. Sci. 2013, 75, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Parveen, A.; Do, M.H.; Kang, M.C.; Yumnam, S.; Kim, S.Y. Molecular mechanisms of methylglyoxal-induced aortic endothelial dysfunction in human vascular endothelial cells. Cell Death Dis. 2020, 11, 403. [Google Scholar] [CrossRef]
- Herrera-Zelada, N.; Zuniga-Cuevas, U.; Ramirez-Reyes, A.; Lavandero, S.; Riquelme, J.A. Targeting the Endothelium to Achieve Cardioprotection. Front. Pharmacol. 2021, 12, 636134. [Google Scholar] [CrossRef]
- Maessen, D.E.; Brouwers, O.; Gaens, K.H.; Wouters, K.; Cleutjens, J.P.; Janssen, B.J.; Miyata, T.; Stehouwer, C.D.; Schalkwijk, C.G. Delayed Intervention with Pyridoxamine Improves Metabolic Function and Prevents Adipose Tissue Inflammation and Insulin Resistance in High-Fat Diet-Induced Obese Mice. Diabetes 2016, 65, 956–966. [Google Scholar] [CrossRef] [Green Version]
- Stirban, A.; Negrean, M.; Stratmann, B.; Gawlowski, T.; Horstmann, T.; Gotting, C.; Kleesiek, K.; Mueller-Roesel, M.; Koschinsky, T.; Uribarri, J.; et al. Benfotiamine prevents macro- and microvascular endothelial dysfunction and oxidative stress following a meal rich in advanced glycation end products in individuals with type 2 diabetes. Diabetes Care 2006, 29, 2064–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakumar, P.; Rohilla, A.; Krishan, P.; Solairaj, P.; Thangathirupathi, A. The multifaceted therapeutic potential of benfotiamine. Pharmacol. Res. 2010, 61, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.; Tesfaye, S.; Spallone, V.; Gurieva, I.; Al Kaabi, J.; Mankovsky, B.; Martinka, E.; Radulian, G.; Nguyen, K.T.; Stirban, A.O.; et al. Screening, diagnosis and management of diabetic sensorimotor polyneuropathy in clinical practice: International expert consensus recommendations. Diabetes Res. Clin. Pract. 2022, 186, 109063. [Google Scholar] [CrossRef] [PubMed]
- Nenna, A.; Nappi, F.; Avtaar Singh, S.S.; Sutherland, F.W.; Di Domenico, F.; Chello, M.; Spadaccio, C. Pharmacologic Approaches Against Advanced Glycation End Products (AGEs) in Diabetic Cardiovascular Disease. Res. Cardiovasc. Med. 2015, 4, e26949. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.Y.; Lu, C.H.; Wu, C.H.; Li, K.J.; Kuo, Y.M.; Hsieh, S.C.; Yu, C.L. The Development of Maillard Reaction, and Advanced Glycation End Product (AGE)-Receptor for AGE (RAGE) Signaling Inhibitors as Novel Therapeutic Strategies for Patients with AGE-Related Diseases. Molecules 2020, 25, 5591. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef] [Green Version]
- Yau, J.W.; Rogers, S.L.; Kawasaki, R.; Lamoureux, E.L.; Kowalski, J.W.; Bek, T.; Chen, S.J.; Dekker, J.M.; Fletcher, A.; Grauslund, J.; et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care 2012, 35, 556–564. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.; Mitchell, P.; Wong, T.Y. Diabetic retinopathy. Lancet 2010, 376, 124–136. [Google Scholar] [CrossRef]
- Lee, R.; Wong, T.Y.; Sabanayagam, C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis. 2015, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Fosmark, D.S.; Berg, J.P.; Jensen, A.B.; Sandvik, L.; Agardh, E.; Agardh, C.D.; Hanssen, K.F. Increased retinopathy occurrence in type 1 diabetes patients with increased serum levels of the advanced glycation endproduct hydroimidazolone. Acta Ophthalmol. 2009, 87, 498–500. [Google Scholar] [CrossRef]
- Fosmark, D.S.; Torjesen, P.A.; Kilhovd, B.K.; Berg, T.J.; Sandvik, L.; Hanssen, K.F.; Agardh, C.D.; Agardh, E. Increased serum levels of the specific advanced glycation end product methylglyoxal-derived hydroimidazolone are associated with retinopathy in patients with type 2 diabetes mellitus. Metabolism 2006, 55, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karachalias, N.; Babaei-Jadidi, R.; Ahmed, N.; Thornalley, P.J. Accumulation of fructosyl-lysine and advanced glycation end products in the kidney, retina and peripheral nerve of streptozotocin-induced diabetic rats. Biochem. Soc. Trans. 2003, 31, 1423–1425. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.G.; Smith, D.G.; Bhat, M.; Nagaraj, R.H. Glyoxalase I is critical for human retinal capillary pericyte survival under hyperglycemic conditions. J. Biol. Chem. 2006, 281, 11864–11871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, N.; Thornalley, P.J.; Dawczynski, J.; Franke, S.; Strobel, J.; Stein, G.; Haik, G.M. Methylglyoxal-derived hydroimidazolone advanced glycation end-products of human lens proteins. Investig. Ophthalmol. Vis. Sci. 2003, 44, 5287–5292. [Google Scholar] [CrossRef]
- Gilbertson, D.T.; Liu, J.; Xue, J.L.; Louis, T.A.; Solid, C.A.; Ebben, J.P.; Collins, A.J. Projecting the number of patients with end-stage renal disease in the United States to the year 2015. J. Am. Soc. Nephrol. 2005, 16, 3736–3741. [Google Scholar] [CrossRef]
- Satchell, S.C.; Tooke, J.E. What is the mechanism of microalbuminuria in diabetes: A role for the glomerular endothelium? Diabetologia 2008, 51, 714–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berhane, A.M.; Weil, E.J.; Knowler, W.C.; Nelson, R.G.; Hanson, R.L. Albuminuria and estimated glomerular filtration rate as predictors of diabetic end-stage renal disease and death. Clin. J. Am. Soc. Nephrol. 2011, 6, 2444–2451. [Google Scholar] [CrossRef] [Green Version]
- Hanssen, N.M.J.; Scheijen, J.; Jorsal, A.; Parving, H.H.; Tarnow, L.; Rossing, P.; Stehouwer, C.D.A.; Schalkwijk, C.G. Higher Plasma Methylglyoxal Levels Are Associated with Incident Cardiovascular Disease in Individuals with Type 1 Diabetes: A 12-Year Follow-up Study. Diabetes 2017, 66, 2278–2283. [Google Scholar] [CrossRef] [Green Version]
- Hanssen, N.M.J.; Westerink, J.; Scheijen, J.; van der Graaf, Y.; Stehouwer, C.D.A.; Schalkwijk, C.G.; Group, S.S. Higher Plasma Methylglyoxal Levels Are Associated with Incident Cardiovascular Disease and Mortality in Individuals with Type 2 Diabetes. Diabetes Care 2018, 41, 1689–1695. [Google Scholar] [CrossRef] [Green Version]
- Malik, R.A. The pathology of human diabetic neuropathy. Diabetes 1997, 46 (Suppl. 2), S50–S53. [Google Scholar] [CrossRef]
- Andersen, S.T.; Witte, D.R.; Dalsgaard, E.M.; Andersen, H.; Nawroth, P.; Fleming, T.; Jensen, T.M.; Finnerup, N.B.; Jensen, T.S.; Lauritzen, T.; et al. Risk Factors for Incident Diabetic Polyneuropathy in a Cohort with Screen-Detected Type 2 Diabetes Followed for 13 Years: ADDITION-Denmark. Diabetes Care 2018, 41, 1068–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bierhaus, A.; Fleming, T.; Stoyanov, S.; Leffler, A.; Babes, A.; Neacsu, C.; Sauer, S.K.; Eberhardt, M.; Schnolzer, M.; Lasitschka, F.; et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 2012, 18, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Genuth, S.; Sun, W.; Cleary, P.; Gao, X.; Sell, D.R.; Lachin, J.; Group, D.E.R.; Monnier, V.M. Skin advanced glycation end products glucosepane and methylglyoxal hydroimidazolone are independently associated with long-term microvascular complication progression of type 1 diabetes. Diabetes 2015, 64, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Fukunaga, M.; Miyata, S.; Liu, B.F.; Miyazaki, H.; Hirota, Y.; Higo, S.; Hamada, Y.; Ueyama, S.; Kasuga, M. Methylglyoxal induces apoptosis through activation of p38 MAPK in rat Schwann cells. Biochem. Biophys. Res. Commun. 2004, 320, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, M.J.; Filipovic, M.R.; Leffler, A.; de la Roche, J.; Kistner, K.; Fischer, M.J.; Fleming, T.; Zimmermann, K.; Ivanovic-Burmazovic, I.; Nawroth, P.P.; et al. Methylglyoxal activates nociceptors through transient receptor potential channel A1 (TRPA1): A possible mechanism of metabolic neuropathies. J. Biol. Chem. 2012, 287, 28291–28306. [Google Scholar] [CrossRef] [Green Version]
- Jack, M.; Wright, D. Role of advanced glycation endproducts and glyoxalase I in diabetic peripheral sensory neuropathy. Transl. Res. 2012, 159, 355–365. [Google Scholar] [CrossRef] [Green Version]
- Ussher, J.R.; Greenwell, A.A.; Nguyen, M.A.; Mulvihill, E.E. Cardiovascular Effects of Incretin-Based Therapies: Integrating Mechanisms with Cardiovascular Outcome Trials. Diabetes 2022, 71, 173–183. [Google Scholar] [CrossRef]
- Giugliano, D.; Longo, M.; Scappaticcio, L.; Bellastella, G.; Maiorino, M.I.; Esposito, K. SGLT-2 inhibitors and cardiorenal outcomes in patients with or without type 2 diabetes: A meta-analysis of 11 CVOTs. Cardiovasc. Diabetol. 2021, 20, 236. [Google Scholar] [CrossRef]
- Rabbani, N.; Chittari, M.V.; Bodmer, C.W.; Zehnder, D.; Ceriello, A.; Thornalley, P.J. Increased glycation and oxidative damage to apolipoprotein B100 of LDL cholesterol in patients with type 2 diabetes and effect of metformin. Diabetes 2010, 59, 1038–1045. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, N.; Godfrey, L.; Xue, M.; Shaheen, F.; Geoffrion, M.; Milne, R.; Thornalley, P.J. Glycation of LDL by methylglyoxal increases arterial atherogenicity: A possible contributor to increased risk of cardiovascular disease in diabetes. Diabetes 2011, 60, 1973–1980. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, L.; Yamada-Fowler, N.; Smith, J.; Thornalley, P.J.; Rabbani, N. Arginine-directed glycation and decreased HDL plasma concentration and functionality. Nutr. Diabetes 2014, 4, e134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nigro, C.; Leone, A.; Fiory, F.; Prevenzano, I.; Nicolo, A.; Mirra, P.; Beguinot, F.; Miele, C. Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging. Cells 2019, 8, 749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, T.; Matafome, P.; Sereno, J.; Almeida, J.; Castelhano, J.; Gamas, L.; Neves, C.; Goncalves, S.; Carvalho, C.; Arslanagic, A.; et al. Methylglyoxal-induced glycation changes adipose tissue vascular architecture, flow and expansion, leading to insulin resistance. Sci. Rep. 2017, 7, 1698. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.M.; Wu, L.Y. Accumulation of endogenous methylglyoxal impaired insulin signaling in adipose tissue of fructose-fed rats. Mol. Cell. Biochem. 2007, 306, 133–139. [Google Scholar] [CrossRef]
- Dhar, A.; Dhar, I.; Jiang, B.; Desai, K.M.; Wu, L.Y. Chronic Methylglyoxal Infusion by Minipump Causes Pancreatic beta-Cell Dysfunction and Induces Type 2 Diabetes in Sprague-Dawley Rats. Diabetes 2011, 60, 899–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelbrecht, B.; Mattern, Y.; Scheibler, S.; Tschoepe, D.; Gawlowski, T.; Stratmann, B. Methylglyoxal impairs GLUT4 trafficking and leads to increased glucose uptake in L6 myoblasts. Horm. Metab. Res. 2014, 46, 77–84. [Google Scholar] [CrossRef]
- Engelbrecht, B.; Stratmann, B.; Hess, C.; Tschoepe, D.; Gawlowski, T. Impact of GLO1 knock down on GLUT4 trafficking and glucose uptake in L6 myoblasts. PLoS ONE 2013, 8, e65195. [Google Scholar] [CrossRef] [Green Version]
- Hanssen, N.M.; Wouters, K.; Huijberts, M.S.; Gijbels, M.J.; Sluimer, J.C.; Scheijen, J.L.; Heeneman, S.; Biessen, E.A.; Daemen, M.J.; Brownlee, M.; et al. Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a rupture-prone phenotype. Eur. Heart J. 2014, 35, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Hartog, J.W.; Voors, A.A.; Schalkwijk, C.G.; Scheijen, J.; Smilde, T.D.; Damman, K.; Bakker, S.J.; Smit, A.J.; van Veldhuisen, D.J. Clinical and prognostic value of advanced glycation end-products in chronic heart failure. Eur. Heart J. 2007, 28, 2879–2885. [Google Scholar] [CrossRef]
- Mulder, D.J.; van Haelst, P.L.; Graaff, R.; Gans, R.O.; Zijlstra, F.; Smit, A.J. Skin autofluorescence is elevated in acute myocardial infarction and is associated with the one-year incidence of major adverse cardiac events. Neth. Heart J. 2009, 17, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Kalra, B.S.; Roy, V. Efficacy of metabolic modulators in ischemic heart disease: An overview. J. Clin. Pharmacol. 2012, 52, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Masoud, W.G.; Ussher, J.R.; Wang, W.; Jaswal, J.S.; Wagg, C.S.; Dyck, J.R.; Lygate, C.A.; Neubauer, S.; Clanachan, A.S.; Lopaschuk, G.D. Failing mouse hearts utilize energy inefficiently and benefit from improved coupling of glycolysis and glucose oxidation. Cardiovasc. Res. 2014, 101, 30–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blackburn, N.J.R.; Vulesevic, B.; McNeill, B.; Cimenci, C.E.; Ahmadi, A.; Gonzalez-Gomez, M.; Ostojic, A.; Zhong, Z.; Brownlee, M.; Beisswenger, P.J.; et al. Methylglyoxal-derived advanced glycation end products contribute to negative cardiac remodeling and dysfunction post-myocardial infarction. Basic Res. Cardiol. 2017, 112, 57. [Google Scholar] [CrossRef] [PubMed]
- Heber, S.; Haller, P.M.; Kiss, A.; Jager, B.; Huber, K.; Fischer, M.J.M. Association of Plasma Methylglyoxal Increase after Myocardial Infarction and the Left Ventricular Ejection Fraction. Biomedicines 2022, 10, 605. [Google Scholar] [CrossRef] [PubMed]
- Kannel, W.B.; Hjortland, M.; Castelli, W.P. Role of diabetes in congestive heart failure: The Framingham study. Am. J. Cardiol. 1974, 34, 29–34. [Google Scholar] [CrossRef]
- Bugger, H.; Abel, E.D. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia 2014, 57, 660–671. [Google Scholar] [CrossRef] [Green Version]
- Vulesevic, B.; McNeill, B.; Giacco, F.; Maeda, K.; Blackburn, N.J.; Brownlee, M.; Milne, R.W.; Suuronen, E.J. Methylglyoxal-Induced Endothelial Cell Loss and Inflammation Contribute to the Development of Diabetic Cardiomyopathy. Diabetes 2016, 65, 1699–1713. [Google Scholar] [CrossRef] [Green Version]
- Papadaki, M.; Kampaengsri, T.; Barrick, S.K.; Campbell, S.G.; von Lewinski, D.; Rainer, P.P.; Harris, S.P.; Greenberg, M.J.; Kirk, J.A. Myofilament glycation in diabetes reduces contractility by inhibiting tropomyosin movement, is rescued by cMyBPC domains. J. Mol. Cell. Cardiol. 2022, 162, 1–9. [Google Scholar] [CrossRef]
- Gawlowski, T.; Stratmann, B.; Stork, I.; Engelbrecht, B.; Brodehl, A.; Niehaus, K.; Korfer, R.; Tschoepe, D.; Milting, H. Heat shock protein 27 modification is increased in the human diabetic failing heart. Horm. Metab. Res. 2009, 41, 594–599. [Google Scholar] [CrossRef]
- Papadaki, M.; Holewinski, R.J.; Previs, S.B.; Martin, T.G.; Stachowski, M.J.; Li, A.; Blair, C.A.; Moravec, C.S.; Van Eyk, J.E.; Campbell, K.S.; et al. Diabetes with heart failure increases methylglyoxal modifications in the sarcomere, which inhibit function. JCI Insight 2018, 3, e121264. [Google Scholar] [CrossRef] [Green Version]
- Folsom, A.R.; Rasmussen, M.L.; Chambless, L.E.; Howard, G.; Cooper, L.S.; Schmidt, M.I.; Heiss, G. Prospective associations of fasting insulin, body fat distribution, and diabetes with risk of ischemic stroke. The Atherosclerosis Risk in Communities (ARIC) Study Investigators. Diabetes Care 1999, 22, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, L.B.; Adams, R.; Becker, K.; Furberg, C.D.; Gorelick, P.B.; Hademenos, G.; Hill, M.; Howard, G.; Howard, V.J.; Jacobs, B.; et al. Primary prevention of ischemic stroke: A statement for healthcare professionals from the Stroke Council of the American Heart Association. Stroke 2001, 32, 280–299. [Google Scholar] [CrossRef] [PubMed]
- Himmelmann, A.; Hansson, L.; Svensson, A.; Harmsen, P.; Holmgren, C.; Svanborg, A. Predictors of stroke in the elderly. Acta Med. Scand. 1988, 224, 439–443. [Google Scholar] [CrossRef]
- Kuusisto, J.; Mykkanen, L.; Pyorala, K.; Laakso, M. Non-insulin-dependent diabetes and its metabolic control are important predictors of stroke in elderly subjects. Stroke 1994, 25, 1157–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, R.X.; McNeil, J.J.; OMalley, H.M.; Davis, S.M.; Thrift, A.G.; Donnan, G.A. Risk factors for stroke due to cerebral infarction in young adults. Stroke 1997, 28, 1913–1918. [Google Scholar] [CrossRef]
- Rohr, J.; Kittner, S.; Feeser, B.; Hebel, J.R.; Whyte, M.G.; Weinstein, A.; Kanarak, N.; Buchholz, D.; Earley, C.; Johnson, C.; et al. Traditional risk factors and ischemic stroke in young adults: The Baltimore-Washington Cooperative Young Stroke Study. Arch. Neurol. 1996, 53, 603–607. [Google Scholar] [CrossRef]
- Gao, M.; Sun, L.; Liu, Y.L.; Xie, J.W.; Qin, L.; Xue, J.; Wang, Y.T.; Guo, K.M.; Ma, M.M.; Li, X.Y. Reduction of glyoxalase 1 (GLO1) aggravates cerebrovascular remodeling via promoting the proliferation of basilar smooth muscle cells in hypertension. Biochem. Biophys. Res. Commun. 2019, 518, 278–285. [Google Scholar] [CrossRef]
- Alomar, F.; Singh, J.; Jang, H.S.; Rozanzki, G.J.; Shao, C.H.; Padanilam, B.J.; Mayhan, W.G.; Bidasee, K.R. Smooth muscle-generated methylglyoxal impairs endothelial cell-mediated vasodilatation of cerebral microvessels in type 1 diabetic rats. Br. J. Pharmacol. 2016, 173, 3307–3326. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Xu, H.; Hu, Y.; He, P.; Ni, Z.; Xu, H.; Zhang, Z.; Dai, H. Edaravone protected human brain microvascular endothelial cells from methylglyoxal-induced injury by inhibiting AGEs/RAGE/oxidative stress. PLoS ONE 2013, 8, e76025. [Google Scholar] [CrossRef] [Green Version]
- Toth, A.E.; Toth, A.; Walter, F.R.; Kiss, L.; Veszelka, S.; Ozsvari, B.; Puskas, L.G.; Heimesaat, M.M.; Dohgu, S.; Kataoka, Y.; et al. Compounds blocking methylglyoxal-induced protein modification and brain endothelial injury. Arch. Med. Res. 2014, 45, 753–764. [Google Scholar] [CrossRef]
- Wu, F.; Feng, J.Z.; Qiu, Y.H.; Yu, F.B.; Zhang, J.Z.; Zhou, W.; Yu, F.; Wang, G.K.; An, L.N.; Ni, F.H.; et al. Activation of receptor for advanced glycation end products contributes to aortic remodeling and endothelial dysfunction in sinoaortic denervated rats. Atherosclerosis 2013, 229, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Birukov, A.; Cuadrat, R.; Polemiti, E.; Eichelmann, F.; Schulze, M.B. Advanced glycation end-products, measured as skin autofluorescence, associate with vascular stiffness in diabetic, pre-diabetic and normoglycemic individuals: A cross-sectional study. Cardiovasc. Diabetol. 2021, 20, 110. [Google Scholar] [CrossRef] [PubMed]
- de Vos, L.C.; Noordzij, M.J.; Mulder, D.J.; Smit, A.J.; Lutgers, H.L.; Dullaart, R.P.; Kamphuisen, P.W.; Zeebregts, C.J.; Lefrandt, J.D. Skin autofluorescence as a measure of advanced glycation end products deposition is elevated in peripheral artery disease. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 131–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bezold, V.; Rosenstock, P.; Scheffler, J.; Geyer, H.; Horstkorte, R.; Bork, K. Glycation of macrophages induces expression of pro-inflammatory cytokines and reduces phagocytic efficiency. Aging 2019, 11, 5258–5275. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stratmann, B. Dicarbonyl Stress in Diabetic Vascular Disease. Int. J. Mol. Sci. 2022, 23, 6186. https://doi.org/10.3390/ijms23116186
Stratmann B. Dicarbonyl Stress in Diabetic Vascular Disease. International Journal of Molecular Sciences. 2022; 23(11):6186. https://doi.org/10.3390/ijms23116186
Chicago/Turabian StyleStratmann, Bernd. 2022. "Dicarbonyl Stress in Diabetic Vascular Disease" International Journal of Molecular Sciences 23, no. 11: 6186. https://doi.org/10.3390/ijms23116186
APA StyleStratmann, B. (2022). Dicarbonyl Stress in Diabetic Vascular Disease. International Journal of Molecular Sciences, 23(11), 6186. https://doi.org/10.3390/ijms23116186