
Transcriptome Profiling of the Dorsomedial Prefrontal Cortex in Suicide Victims

 , and
, and
Abstract
:1. Introduction
2. Results
2.1. Transcriptome Sequencing in the DMPFC of the Suicide Brains
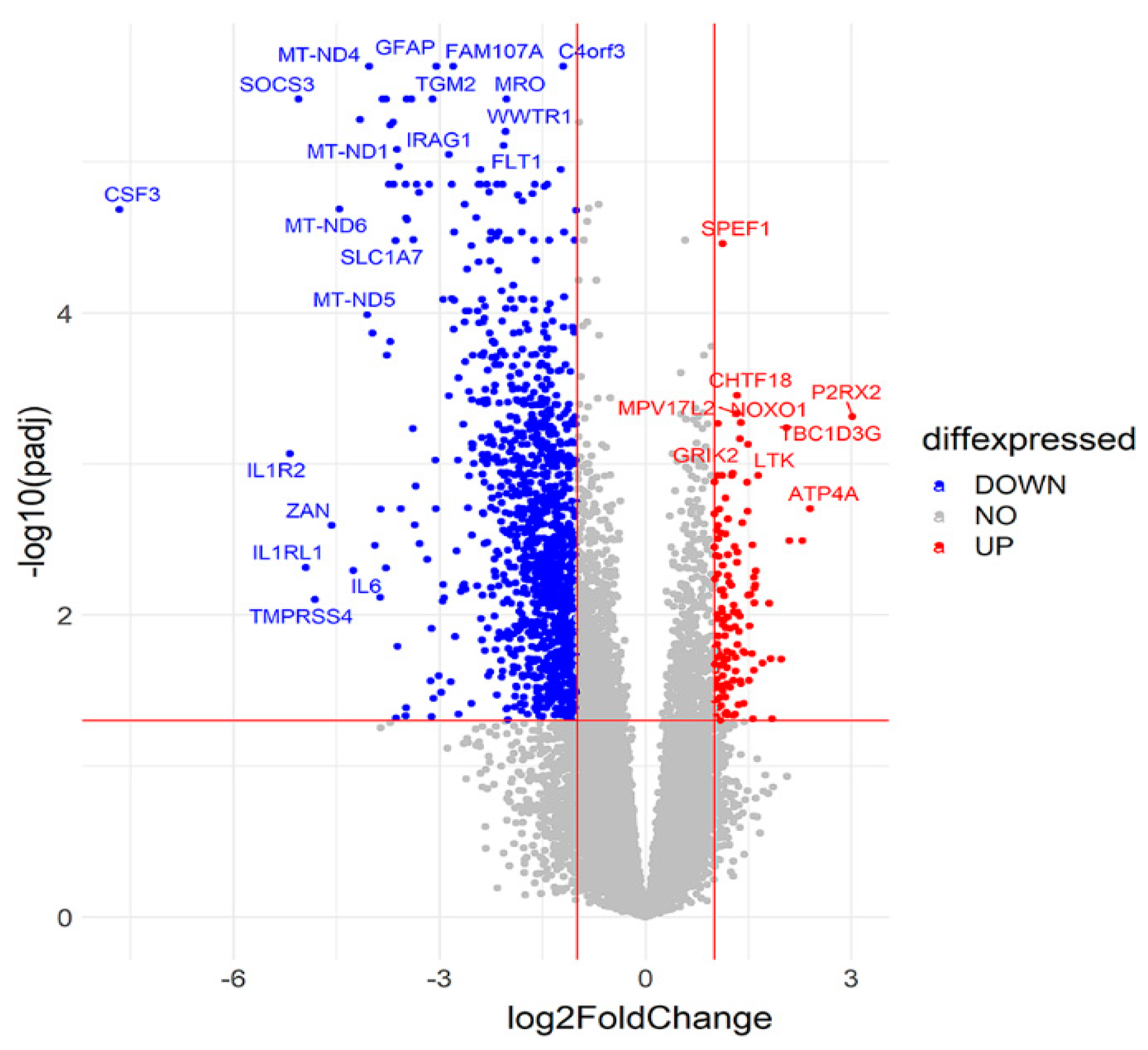
2.2. Comparison of Suicide and Control Samples Based on Differentially Expressed Genes (DEGs)
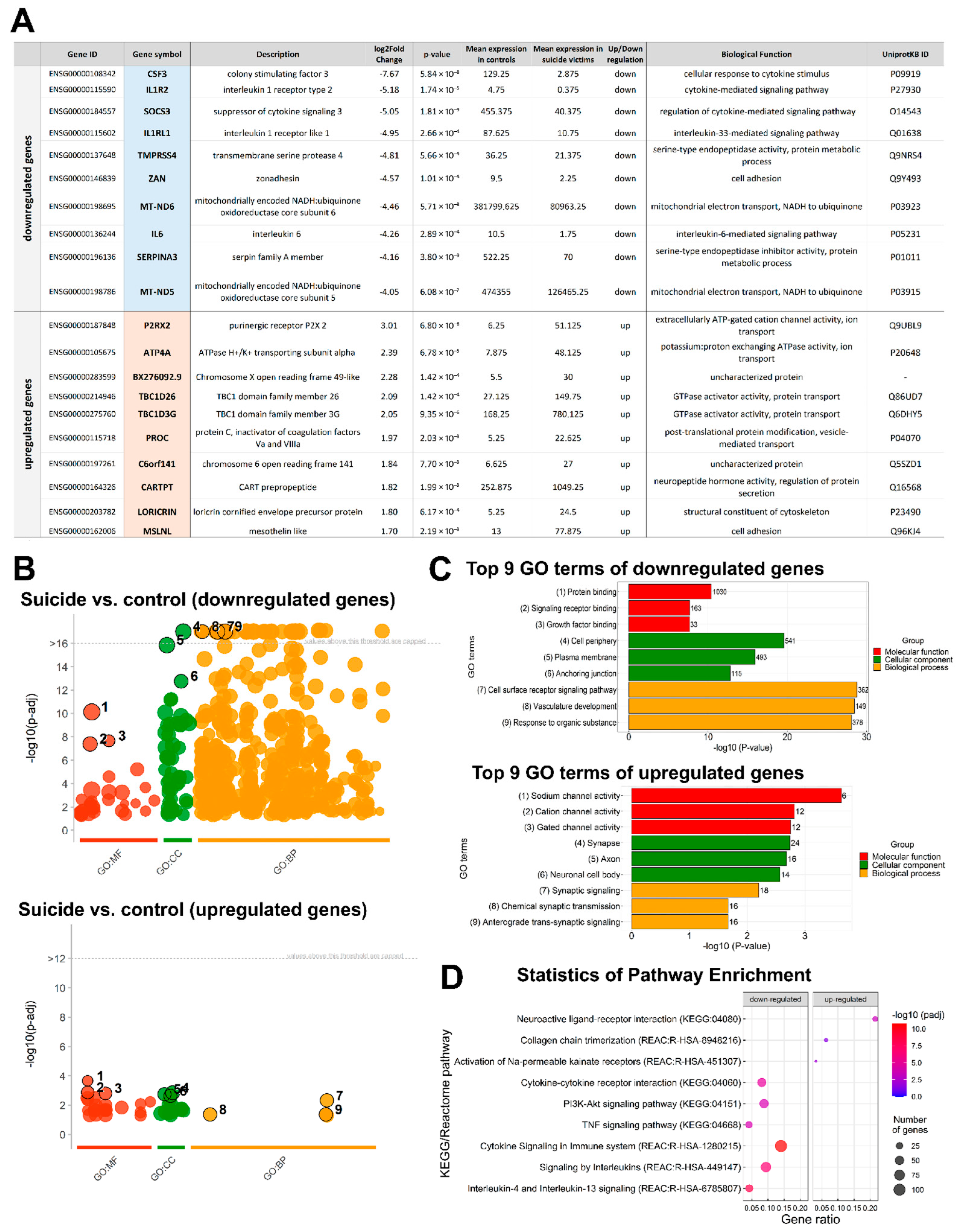
2.3. Functional Annotation and Classification of the DEGs
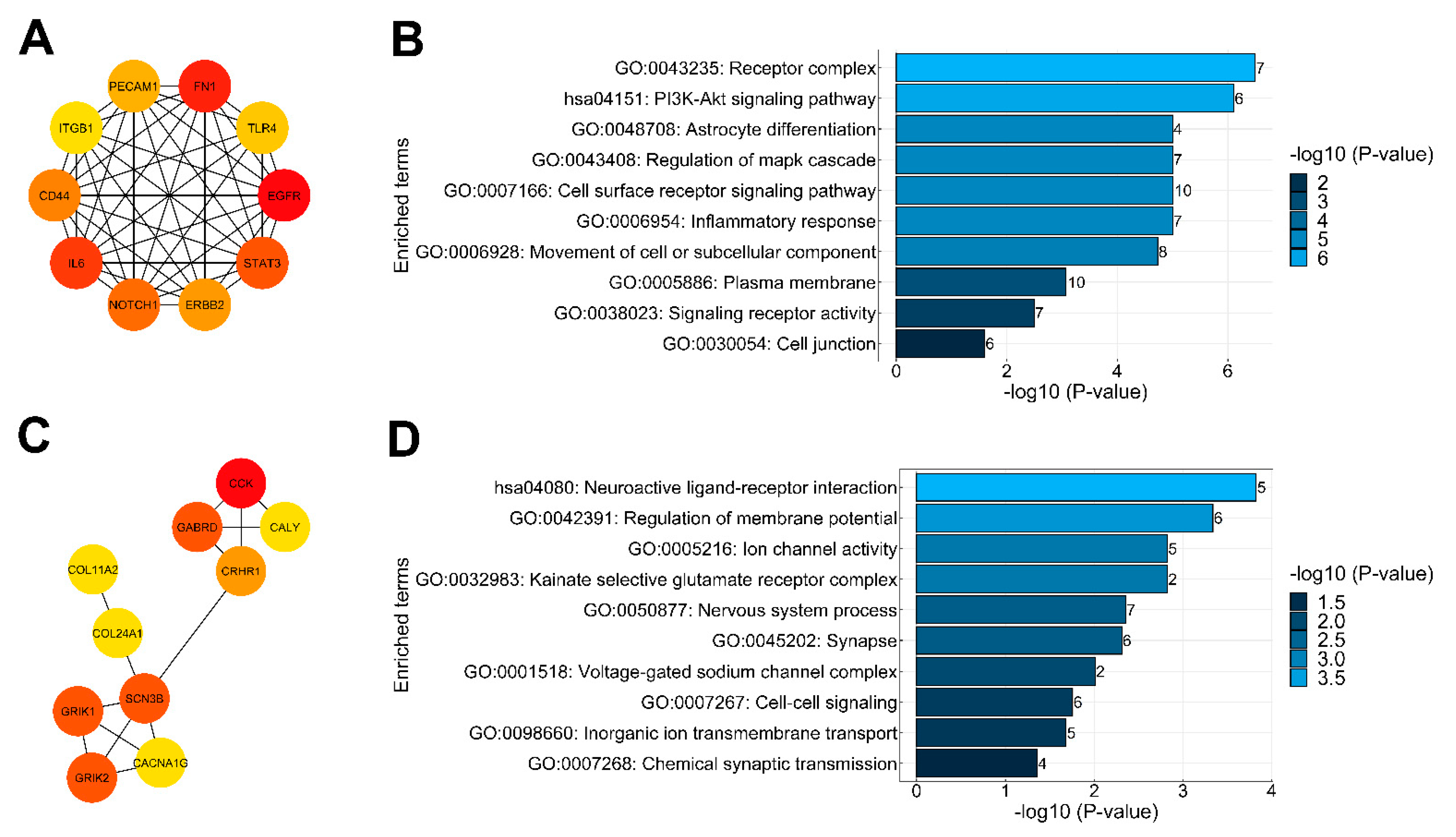
2.4. Protein–Protein Interaction Analysis of DEGs and Identification of Key Genes
2.5. Validation of RNA-Seq Data
2.6. Depression-Focused Gene Set Enrichment
2.7. Co-Expression Network Analysis and Hub Gene Screening in the DMPFC of Suicidal Individuals
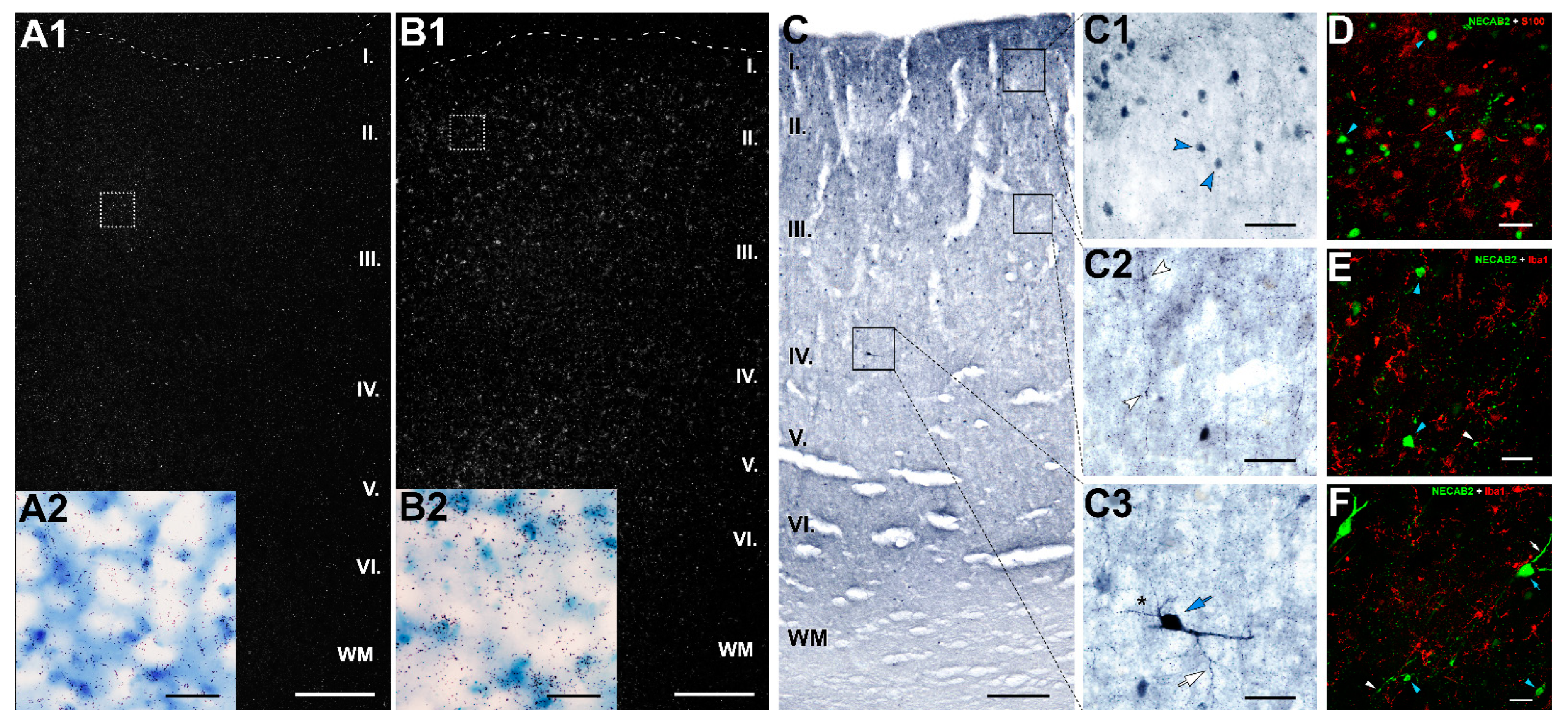
2.8. Distribution of NECAB2 in the DMPFC
3. Discussion
3.1. Conclusions Based on Individual DEGs
3.2. Conclusion Based on the Distribution of NECAB2
3.3. Functional Implications Based on Pathway Analyses
3.4. Functional Cluster Analysis of Gene Expressions in the DMPFC
3.5. Functions Supported by Known PPIs of DEGs
3.6. Genes Associated with Depression and Comorbidities
4. Materials and Methods
4.1. Human Brain Tissue Samples
4.2. Tissue Preparation
4.3. RNA Sequencing Analysis and Data
4.4. Gene Ontology and Pathway Enrichment Analysis of DEGs
4.5. Compare RNA-Seq Results with the Allen Human Brain Atlas Database
4.6. Protein–Protein Interaction Network Construction
4.7. Disease-Associated Gene Sets
4.8. Co-Expression Network Construction and Functional Annotation
4.9. Validation of Expression Changes by qRT-PCR
4.10. Preparation of In Situ Hybridization Probes
4.11. In Situ Hybridization Histochemistry
4.12. Tissue Collection for Immunolabeling
4.13. DAB Immunolabeling
4.14. Double Labeling of NECAB2
4.15. Microscopy and Photography
4.16. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cauda, F.; D’Agata, F.; Sacco, K.; Duca, S.; Geminiani, G.; Vercelli, A. Functional connectivity of the insula in the resting brain. Neuroimage 2011, 55, 8–23. [Google Scholar] [CrossRef]
- Seeley, W.W.; Menon, V.; Schatzberg, A.F.; Keller, J.; Glover, G.H.; Kenna, H.; Reiss, A.L.; Greicius, M.D. Dissociable intrinsic connectivity networks for salience processing and executive control. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 2349–2356. [Google Scholar] [CrossRef]
- Isaacs, B.R.; Forstmann, B.U.; Temel, Y.; Keuken, M.C. The Connectivity Fingerprint of the Human Frontal Cortex, Subthalamic Nucleus, and Striatum. Front. Neuroanat. 2018, 12, 60. [Google Scholar] [CrossRef]
- Cacciola, A.; Calamuneri, A.; Milardi, D.; Mormina, E.; Chillemi, G.; Marino, S.; Naro, A.; Rizzo, G.; Anastasi, G.; Quartarone, A. A Connectomic Analysis of the Human Basal Ganglia Network. Front. Neuroanat. 2017, 11, 85. [Google Scholar] [CrossRef] [Green Version]
- Kotz, S. Differential Input of the Supplementary Motor Area to a Dedicated Temporal Processing Network: Functional and Clinical Implications. Front. Integr. Neurosci. 2011, 5, 86. [Google Scholar] [CrossRef] [Green Version]
- Buckner, R.L.; Andrews-Hanna, J.R.; Schacter, D.L. The brain’s default network: Anatomy, function, and relevance to disease. Ann. New York Acad. Sci. 2008, 1124, 1–38. [Google Scholar] [CrossRef] [Green Version]
- Cavanna, A.E.; Trimble, M.R. The precuneus: A review of its functional anatomy and behavioural correlates. Brain 2006, 129, 564–583. [Google Scholar] [CrossRef] [Green Version]
- Báez-Mendoza, R.; Vázquez, Y.; Mastrobattista, E.P.; Williams, Z.M. Neuronal Circuits for Social Decision-Making and Their Clinical Implications. Front. Neurosci. 2021, 15, 720294. [Google Scholar] [CrossRef]
- Anticevic, A.; Cole, M.W.; Murray, J.D.; Corlett, P.R.; Wang, X.J.; Krystal, J.H. The role of default network deactivation in cognition and disease. Trends Cogn. Sci. 2012, 16, 584–592. [Google Scholar] [CrossRef] [Green Version]
- Kolling, N.; O’Reilly, J.X. State-change decisions and dorsomedial prefrontal cortex: The importance of time. Curr. Opin. Behav. Sci. 2018, 22, 152–160. [Google Scholar] [CrossRef]
- Lieberman, M.D.; Straccia, M.A.; Meyer, M.L.; Du, M.; Tan, K.M. Social, self, (situational), and affective processes in medial prefrontal cortex (MPFC): Causal, multivariate, and reverse inference evidence. Neurosci. Biobehav. Rev. 2019, 99, 311–328. [Google Scholar] [CrossRef]
- Raschle, N.M.; Menks, W.M.; Fehlbaum, L.V.; Tshomba, E.; Stadler, C. Structural and Functional Alterations in Right Dorsomedial Prefrontal and Left Insular Cortex Co-Localize in Adolescents with Aggressive Behaviour: An ALE Meta-Analysis. PLoS ONE 2015, 10, e0136553. [Google Scholar] [CrossRef]
- Solbakk, A.K.; Lovstad, M. Effects of focal prefrontal cortex lesions on electrophysiological indices of executive attention and action control. Scand. J. Psychol. 2014, 55, 233–243. [Google Scholar] [CrossRef]
- Kalisch, R.; Gerlicher, A.M. Making a mountain out of a molehill: On the role of the rostral dorsal anterior cingulate and dorsomedial prefrontal cortex in conscious threat appraisal, catastrophizing, and worrying. Neurosci. Biobehav. Rev. 2014, 42, 1–8. [Google Scholar] [CrossRef]
- Northoff, G.; Heinzel, A.; De Greck, M.; Bermpohl, F.; Dobrowolny, H.; Panksepp, J. Self-referential processing in our brain—A meta-analysis of imaging studies on the self. Neuroimage 2006, 31, 440–457. [Google Scholar] [CrossRef]
- Figley, T.D.; Bhullar, N.; Courtney, S.M.; Figley, C.R. Probabilistic atlases of default mode, executive control and salience network white matter tracts: An fMRI-guided diffusion tensor imaging and tractography study. Front. Hum. Neurosci. 2015, 9, 585. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Rao, B.; Li, S.; Gao, L.; Xie, Y.; Dai, X.; Fu, K.; Peng, X.Z.; Xu, H. Altered Effective Connectivity Measured by Resting-State Functional Magnetic Resonance Imaging in Posterior Parietal-Frontal-Striatum Circuit in Patients With Disorder of Consciousness. Front. Neurosci. 2021, 15, 766633. [Google Scholar] [CrossRef]
- Utevsky, A.V.; Smith, D.V.; Huettel, S.A. Precuneus is a functional core of the default-mode network. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 932–940. [Google Scholar] [CrossRef] [Green Version]
- Bruner, E.; Pereira-Pedro, A.S.; Chen, X.; Rilling, J.K. Precuneus proportions and cortical folding: A morphometric evaluation on a racially diverse human sample. Ann. Anat. Anat. Anz. 2017, 211, 120–128. [Google Scholar] [CrossRef]
- Bruner, E.; Rangel de Lázaro, G.; de la Cuétara, J.M.; Martín-Loeches, M.; Colom, R.; Jacobs, H.I. Midsagittal brain variation and MRI shape analysis of the precuneus in adult individuals. J. Anat. 2014, 224, 367–376. [Google Scholar] [CrossRef] [Green Version]
- Bruton, O.J. Is there a “g-neuron”? Establishing a systematic link between general intelligence (g) and the von Economo neuron. Intelligence 2021, 86, 101540. [Google Scholar] [CrossRef]
- Cavanna, A.E. The Precuneus and Consciousness. CNS Spectr. 2007, 12, 545–552. [Google Scholar] [CrossRef]
- Margulies, D.S.; Vincent, J.L.; Kelly, C.; Lohmann, G.; Uddin, L.Q.; Biswal, B.B.; Villringer, A.; Castellanos, F.X.; Milham, M.P.; Petrides, M. Precuneus shares intrinsic functional architecture in humans and monkeys. Proc. Natl. Acad. Sci. USA 2009, 106, 20069–20074. [Google Scholar] [CrossRef] [Green Version]
- Beaty, R.E.; Seli, P.; Schacter, D.L. Network neuroscience of creative cognition: Mapping cognitive mechanisms and individual differences in the creative brain. Curr. Opin. Behav. Sci. 2019, 27, 22–30. [Google Scholar] [CrossRef]
- Fuentealba-Villarroel, F.J.; Renner, J.; Hilbig, A.; Bruton, O.J.; Rasia-Filho, A.A. Spindle-Shaped Neurons in the Human Posteromedial (Precuneus) Cortex. Front. Synaptic Neurosci. 2021, 13, 769228. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, D.; Wang, Y.; Wang, Y.; Ou, S. Segmentation of the Cingulum Bundle in the Human Brain: A New Perspective Based on DSI Tractography and Fiber Dissection Study. Front. Neuroanat. 2016, 10, 84. [Google Scholar] [CrossRef] [Green Version]
- Haber, S.N.; Knutson, B. The Reward Circuit: Linking Primate Anatomy and Human Imaging. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2010, 35, 4–26. [Google Scholar] [CrossRef] [Green Version]
- Coenen, V.A.; Schumacher, L.V.; Kaller, C.; Schlaepfer, T.E.; Reinacher, P.C.; Egger, K.; Urbach, H.; Reisert, M. The anatomy of the human medial forebrain bundle: Ventral tegmental area connections to reward-associated subcortical and frontal lobe regions. Neuroimage Clin. 2018, 18, 770–783. [Google Scholar] [CrossRef]
- Xu, L.; Nan, J.; Lan, Y. The Nucleus Accumbens: A Common Target in the Comorbidity of Depression and Addiction. Front. Neural Circuits 2020, 14, 37. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, W.; Yuan, Y. Aberrant Default Mode Network Underlying the Cognitive Deficits in the Patients With Late-Onset Depression. Front. Aging Neurosci. 2018, 10, 310. [Google Scholar] [CrossRef]
- Long, Z.; Du, L.; Zhao, J.; Wu, S.; Zheng, Q.; Lei, X. Prediction on treatment improvement in depression with resting state connectivity: A coordinate-based meta-analysis. J. Affect. Disord. 2020, 276, 62–68. [Google Scholar] [CrossRef]
- Yang, Y.; Cui, Q.; Lu, F.; Pang, Y.; Chen, Y.; Tang, Q.; Li, D.; Lei, T.; He, Z.; Hu, S.; et al. Default mode network subsystem alterations in bipolar disorder during major depressive episode. J. Affect. Disord. 2021, 281, 856–864. [Google Scholar] [CrossRef]
- Olson, E.A.; Kaiser, R.H.; Pizzagalli, D.A.; Rauch, S.L.; Rosso, I.M. Anhedonia in Trauma-Exposed Individuals: Functional Connectivity and Decision-Making Correlates. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2018, 3, 959–967. [Google Scholar] [CrossRef]
- Kreuzer, P.M.; Downar, J.; de Ridder, D.; Schwarzbach, J.; Schecklmann, M.; Langguth, B. A Comprehensive Review of Dorsomedial Prefrontal Cortex rTMS Utilizing a Double Cone Coil. Neuromodulation J. Int. Neuromodulation Soc. 2019, 22, 851–866. [Google Scholar] [CrossRef]
- Schulze, L.; Feffer, K.; Lozano, C.; Giacobbe, P.; Daskalakis, Z.J.; Blumberger, D.M.; Downar, J. Number of pulses or number of sessions? An open-label study of trajectories of improvement for once-vs. twice-daily dorsomedial prefrontal rTMS in major depression. Brain Stimul. 2018, 11, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Hawton, K.; Casanas, I.C.C.; Haw, C.; Saunders, K. Risk factors for suicide in individuals with depression: A systematic review. J. Affect. Disord. 2013, 147, 17–28. [Google Scholar] [CrossRef]
- Franklin, J.C.; Ribeiro, J.D.; Fox, K.R.; Bentley, K.H.; Kleiman, E.M.; Huang, X.; Musacchio, K.M.; Jaroszewski, A.C.; Chang, B.P.; Nock, M.K. Risk factors for suicidal thoughts and behaviors: A meta-analysis of 50 years of research. Psychol. Bull. 2017, 143, 187. [Google Scholar] [CrossRef]
- Turecki, G.; Brent, D.A. Suicide and suicidal behaviour. Lancet 2016, 387, 1227–1239. [Google Scholar] [CrossRef]
- Malhi, G.S.; Das, P.; Outhred, T.; Bryant, R.A.; Calhoun, V.; Mann, J.J. Default mode dysfunction underpins suicidal activity in mood disorders. Psychol. Med. 2020, 50, 1214–1223. [Google Scholar] [CrossRef]
- Dombrovski, A.Y.; Hallquist, M.N. The decision neuroscience perspective on suicidal behavior: Evidence and hypotheses. Curr. Opin. Psychiatry 2017, 30, 7. [Google Scholar] [CrossRef] [Green Version]
- Marchand, W.R.; Lee, J.N.; Johnson, S.; Thatcher, J.; Gale, P.; Wood, N.; Jeong, E.-K. Striatal and cortical midline circuits in major depression: Implications for suicide and symptom expression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 36, 290–299. [Google Scholar] [CrossRef]
- Reisch, T.; Seifritz, E.; Esposito, F.; Wiest, R.; Valach, L.; Michel, K. An fMRI study on mental pain and suicidal behavior. J. Affect. Disord. 2010, 126, 321–325. [Google Scholar] [CrossRef]
- Clayden, R.C.; Zaruk, A.; Meyre, D.; Thabane, L.; Samaan, Z. The association of attempted suicide with genetic variants in the SLC6A4 and TPH genes depends on the definition of suicidal behavior: A systematic review and meta-analysis. Transl. Psychiatry 2012, 2, e166. [Google Scholar] [CrossRef] [Green Version]
- Thalamuthu, A.; Mills, N.T.; Berger, K.; Minnerup, H.; Grotegerd, D.; Dannlowski, U.; Meinert, S.; Opel, N.; Repple, J.; Gruber, M.; et al. Genome-wide interaction study with major depression identifies novel variants associated with cognitive function. Mol. Psychiatry 2022, 27, 1111–1119. [Google Scholar] [CrossRef]
- Norkeviciene, A.; Gocentiene, R.; Sestokaite, A.; Sabaliauskaite, R.; Dabkeviciene, D.; Jarmalaite, S.; Bulotiene, G. A Systematic Review of Candidate Genes for Major Depression. Medicina 2022, 58, 285. [Google Scholar] [CrossRef]
- Chan, R.F.; Turecki, G.; Shabalin, A.A.; Guintivano, J.; Zhao, M.; Xie, L.Y.; van Grootheest, G.; Kaminsky, Z.A.; Dean, B.; Penninx, B.W.J.H.; et al. Cell Type–Specific Methylome-wide Association Studies Implicate Neurotrophin and Innate Immune Signaling in Major Depressive Disorder. Biol. Psychiatry 2020, 87, 431–442. [Google Scholar] [CrossRef]
- Bai, T.; Wei, Q.; Zu, M.; Xie, W.; Wang, J.; Gong-Jun, J.; Yu, F.; Tian, Y.; Wang, K. Functional plasticity of the dorsomedial prefrontal cortex in depression reorganized by electroconvulsive therapy: Validation in two independent samples. Hum. Brain Mapp. 2019, 40, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Ghosal, S.; Hare, B.; Duman, R.S. Prefrontal Cortex GABAergic Deficits and Circuit Dysfunction in the Pathophysiology and Treatment of Chronic Stress and Depression. Curr. Opin. Behav. Sci. 2017, 14, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.C.; Yao, W.; Hashimoto, K. Brain-derived Neurotrophic Factor (BDNF)-TrkB Signaling in Inflammation-related Depression and Potential Therapeutic Targets. Curr. Neuropharmacol. 2016, 14, 721–731. [Google Scholar] [CrossRef] [Green Version]
- O’Leary, L.A.; Belliveau, C.; Davoli, M.A.; Ma, J.C.; Tanti, A.; Turecki, G.; Mechawar, N. Widespread Decrease of Cerebral Vimentin-Immunoreactive Astrocytes in Depressed Suicides. Front. Psychiatry 2021, 12, 640963. [Google Scholar] [CrossRef]
- Pantazatos, S.P.; Huang, Y.Y.; Rosoklija, G.B.; Dwork, A.J.; Arango, V.; Mann, J.J. Whole-transcriptome brain expression and exon-usage profiling in major depression and suicide: Evidence for altered glial, endothelial and ATPase activity. Mol. Psychiatry 2017, 22, 760–773. [Google Scholar] [CrossRef] [Green Version]
- Labonte, B.; Engmann, O.; Purushothaman, I.; Menard, C.; Wang, J.; Tan, C.; Scarpa, J.R.; Moy, G.; Loh, Y.E.; Cahill, M.; et al. Sex-specific transcriptional signatures in human depression. Nat. Med. 2017, 23, 1102–1111. [Google Scholar] [CrossRef]
- Cabrera, B.; Monroy-Jaramillo, N.; Fries, G.R.; Mendoza-Morales, R.C.; Garcia-Dolores, F.; Mendoza-Larios, A.; Diaz-Otanez, C.; Walss-Bass, C.; Glahn, D.C.; Ostrosky-Wegman, P.; et al. Brain Gene Expression Pattern of Subjects with Completed Suicide and Comorbid Substance Use Disorder. Mol. Neuropsychiatry 2019, 5, 60–73. [Google Scholar] [CrossRef]
- Cabrera-Mendoza, B.; Fresno, C.; Monroy-Jaramillo, N.; Fries, G.R.; Walss-Bass, C.; Glahn, D.C.; Ostrosky-Wegman, P.; Mendoza-Morales, R.C.; Garcia-Dolores, F.; Diaz-Otanez, C.E.; et al. Sex differences in brain gene expression among suicide completers. J. Affect. Disord. 2020, 267, 67–77. [Google Scholar] [CrossRef]
- Li, H.J.; Su, X.; Zhang, L.W.; Zhang, C.Y.; Wang, L.; Li, W.Q.; Yang, Y.F.; Lv, L.X.; Li, M.; Xiao, X. Transcriptomic analyses of humans and mice provide insights into depression. Zool. Res. 2020, 41, 632–643. [Google Scholar] [CrossRef]
- Li, X.; Su, X.; Liu, J.; Li, H.; Li, M.; Li, W.; Luo, X.J. Transcriptome-wide association study identifies new susceptibility genes and pathways for depression. Transl. Psychiatry 2021, 11, 306. [Google Scholar] [CrossRef]
- Romero-Pimentel, A.L.; Almeida, D.; Munoz-Montero, S.; Rangel, C.; Mendoza-Morales, R.; Gonzalez-Saenz, E.E.; Nagy, C.; Chen, G.; Aouabed, Z.; Theroux, J.F.; et al. Integrative DNA Methylation and Gene Expression Analysis in the Prefrontal Cortex of Mexicans who died by Suicide. Int. J. Neuropsychopharmacol. 2021, 24, 935–947. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Chen, M.C.; Gotlib, I.H. Neural systems approaches to understanding major depressive disorder: An intrinsic functional organization perspective. Neurobiol. Dis. 2013, 52, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Drevets, W.C.; Price, J.L.; Furey, M.L. Brain structural and functional abnormalities in mood disorders: Implications for neurocircuitry models of depression. Brain Struct. Funct. 2008, 213, 93–118. [Google Scholar] [CrossRef] [Green Version]
- Biswal, B.; Zerrin Yetkin, F.; Haughton, V.M.; Hyde, J.S. Functional connectivity in the motor cortex of resting human brain using echo-planar MRI. Magn. Reson. Med. 1995, 34, 537–541. [Google Scholar] [CrossRef]
- Greicius, M.D.; Krasnow, B.; Reiss, A.L.; Menon, V. Functional connectivity in the resting brain: A network analysis of the default mode hypothesis. Proc. Natl. Acad. Sci. USA 2003, 100, 253–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greicius, M.D.; Supekar, K.; Menon, V.; Dougherty, R.F. Resting-state functional connectivity reflects structural connectivity in the default mode network. Cereb. Cortex 2009, 19, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Raichle, M.E.; Snyder, A.Z. A default mode of brain function: A brief history of an evolving idea. Neuroimage 2007, 37, 1083–1090; discussion 1097–1099. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L.; Kahn, I.; Snyder, A.Z.; Raichle, M.E.; Buckner, R.L. Evidence for a frontoparietal control system revealed by intrinsic functional connectivity. J. Neurophysiol. 2008, 100, 3328–3342. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Cao, X.; Hou, C.; Li, T.; Cheng, Y.; Jiang, L.; Luo, C.; Li, C.; Yao, D. Effects of Cognitive Training on Resting-State Functional Connectivity of Default Mode, Salience, and Central Executive Networks. Front. Aging Neurosci. 2016, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Geng, X.; Lee, T.M.C. Large-scale functional neural network correlates of response inhibition: An fMRI meta-analysis. Brain Struct. Funct. 2017, 222, 3973–3990. [Google Scholar] [CrossRef] [Green Version]
- Hilger, K.; Ekman, M.; Fiebach, C.J.; Basten, U. Efficient hubs in the intelligent brain: Nodal efficiency of hub regions in the salience network is associated with general intelligence. Intelligence 2017, 60, 10–25. [Google Scholar] [CrossRef]
- Sheline, Y.I.; Price, J.L.; Yan, Z.; Mintun, M.A. Resting-state functional MRI in depression unmasks increased connectivity between networks via the dorsal nexus. Proc. Natl. Acad. Sci. USA 2010, 107, 11020–11025. [Google Scholar] [CrossRef] [Green Version]
- Schurch, N.J.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? Rna 2016, 22, 839–851. [Google Scholar] [CrossRef] [Green Version]
- Ramaker, R.C.; Bowling, K.M.; Lasseigne, B.N.; Hagenauer, M.H.; Hardigan, A.A.; Davis, N.S.; Gertz, J.; Cartagena, P.M.; Walsh, D.M.; Vawter, M.P.; et al. Post-mortem molecular profiling of three psychiatric disorders. Genome Med. 2017, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Jabbi, M.; Arasappan, D.; Eickhoff, S.B.; Strakowski, S.M.; Nemeroff, C.B.; Hofmann, H.A. Neuro-transcriptomic signatures for mood disorder morbidity and suicide mortality. J. Psychiatry Res. 2020, 127, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Underwood, M.D.; Arango, V. Evidence for neurodegeneration and neuroplasticity as part of the neurobiology of suicide. Biol. Psychiatry 2011, 70, 306–307. [Google Scholar] [CrossRef] [PubMed]
- Punzi, G.; Ursini, G.; Chen, Q.; Radulescu, E.; Tao, R.; Huuki, L.A.; Di Carlo, P.; Collado-Torres, L.; Shin, J.H.; Catanesi, R.; et al. Genetics and Brain Transcriptomics of Completed Suicide. Am. J. Psychiatry 2022, 179, 226–241. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, A.; Mamdani, F.; Ernst, C.; Vawter, M.P.; Bunney, W.E.; Lebel, V.; Rehal, S.; Klempan, T.; Gratton, A.; Benkelfat, C.; et al. Global brain gene expression analysis links glutamatergic and GABAergic alterations to suicide and major depression. PLoS ONE 2009, 4, e6585. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Wang, L.; Tang, S.; Yuan, L.; Wu, S.; Du, X.; Xiang, Y.; Qu, X.; Liu, H.; Luo, H.; et al. ITGB4 deficiency in bronchial epithelial cells directs airway inflammation and bipolar disorder-related behavior. J. Neuroinflammation 2018, 15, 246. [Google Scholar] [CrossRef]
- Hlavacova, N.; Wes, P.D.; Ondrejcakova, M.; Flynn, M.E.; Poundstone, P.K.; Babic, S.; Murck, H.; Jezova, D. Subchronic treatment with aldosterone induces depression-like behaviours and gene expression changes relevant to major depressive disorder. Int. J. Neuropsychopharmacol. 2012, 15, 247–265. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.H.; Perlis, R.H.; Jung, J.Y.; Byrne, E.M.; Rueckert, E.; Siburian, R.; Haddad, S.; Mayerfeld, C.E.; Heath, A.C.; Pergadia, M.L.; et al. Multi-locus genome-wide association analysis supports the role of glutamatergic synaptic transmission in the etiology of major depressive disorder. Transl. Psychiatry 2012, 2, e184. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Zeng, B.; Zeng, L.; Du, X.; Li, B.; Huo, R.; Liu, L.; Wang, H.; Dong, M.; Pan, J.; et al. Gut microbiota regulates mouse behaviors through glucocorticoid receptor pathway genes in the hippocampus. Transl. Psychiatry 2018, 8, 187. [Google Scholar] [CrossRef]
- Petrykey, K.; Lippé, S.; Robaey, P.; Sultan, S.; Laniel, J.; Drouin, S.; Bertout, L.; Beaulieu, P.; St-Onge, P.; Boulet-Craig, A.; et al. Influence of genetic factors on long-term treatment related neurocognitive complications, and on anxiety and depression in survivors of childhood acute lymphoblastic leukemia: The Petale study. PLoS ONE 2019, 14, e0217314. [Google Scholar] [CrossRef] [Green Version]
- Klempan, T.A.; Sequeira, A.; Canetti, L.; Lalovic, A.; Ernst, C.; Turecki, G. Altered expression of genes involved in ATP biosynthesis and GABAergic neurotransmission in the ventral prefrontal cortex of suicides with and without major depression. Mol. Psychiatry 2009, 14, 175–189. [Google Scholar] [CrossRef]
- Cotter, D.; Mackay, D.; Chana, G.; Beasley, C.; Landau, S.; Everall, I.P. Reduced neuronal size and glial cell density in area 9 of the dorsolateral prefrontal cortex in subjects with major depressive disorder. Cereb. Cortex 2002, 12, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Öngür, D.; Drevets, W.C.; Price, J.L. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. USA 1998, 95, 13290–13295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajkowska, G.; Miguel-Hidalgo, J.J.; Wei, J.; Dilley, G.; Pittman, S.D.; Meltzer, H.Y.; Overholser, J.C.; Roth, B.L.; Stockmeier, C.A. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiatry 1999, 45, 1085–1098. [Google Scholar] [CrossRef]
- Canela, L.; Fernández-Dueñas, V.; Albergaria, C.; Watanabe, M.; Lluís, C.; Mallol, J.; Canela, E.I.; Franco, R.; Luján, R.; Ciruela, F. The association of metabotropic glutamate receptor type 5 with the neuronal Ca2+-binding protein 2 modulates receptor function. J. Neurochem. 2009, 111, 555–567. [Google Scholar] [CrossRef]
- Zhao, J.; Verwer, R.W.H.; Gao, S.F.; Qi, X.R.; Lucassen, P.J.; Kessels, H.W.; Swaab, D.F. Prefrontal alterations in GABAergic and glutamatergic gene expression in relation to depression and suicide. J. Psychiatr. Res. 2018, 102, 261–274. [Google Scholar] [CrossRef]
- Mazuir, E.; Fricker, D.; Sol-Foulon, N. Neuron-Oligodendrocyte Communication in Myelination of Cortical GABAergic Cells. Life 2021, 11, 216. [Google Scholar] [CrossRef]
- Zhu, Q.; Ke, W.; He, Q.; Wang, X.; Zheng, R.; Li, T.; Luan, G.; Long, Y.S.; Liao, W.P.; Shu, Y. Laminar Distribution of Neurochemically-Identified Interneurons and Cellular Co-expression of Molecular Markers in Epileptic Human Cortex. Neurosci. Bull. 2018, 34, 992–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miczán, V.; Kelemen, K.; Glavinics, J.R.; László, Z.I.; Barti, B.; Kenesei, K.; Kisfali, M.; Katona, I. NECAB1 and NECAB2 are prevalent calcium-binding proteins of CB1/CCK-positive GABAergic interneurons. Cereb. Cortex 2021, 31, 1786–1806. [Google Scholar] [CrossRef]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A meta-analysis of cytokines in major depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Farooq, R.K.; Asghar, K.; Kanwal, S.; Zulqernain, A. Role of inflammatory cytokines in depression: Focus on interleukin-1β. Biomed. Rep. 2017, 6, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Kitagishi, Y.; Kobayashi, M.; Kikuta, K.; Matsuda, S. Roles of PI3K/AKT/GSK3/mTOR pathway in cell signaling of mental illnesses. Depress. Res. Treat. 2012, 2012, 752563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarate, C.A.; Du, J.; Quiroz, J.; Gray, N.A.; Denicoff, K.D.; Singh, J.; Charney, D.S.; Manji, H.K. Regulation of cellular plasticity cascades in the pathophysiology and treatment of mood disorders-Role of the glutamatergic system. Ann. N. Y. Acad. Sci. 2003, 1003, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Nagy, C.; Suderman, M.; Yang, J.; Szyf, M.; Mechawar, N.; Ernst, C.; Turecki, G. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol. Psychiatry 2015, 20, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Mao, X.; Wei, L. Genes and (common) pathways underlying drug addiction. PLoS Comput. Biol. 2008, 4, e2. [Google Scholar] [CrossRef] [Green Version]
- Biernacka, J.M.; Geske, J.; Jenkins, G.D.; Colby, C.; Rider, D.N.; Karpyak, V.M.; Choi, D.-S.; Fridley, B.L. Genome-wide gene-set analysis for identification of pathways associated with alcohol dependence. Int. J. Neuropsychopharmacol. 2013, 16, 271–278. [Google Scholar] [CrossRef] [Green Version]
- Gray, A.L.; Hyde, T.M.; Deep-Soboslay, A.; Kleinman, J.E.; Sodhi, M.S. Sex differences in glutamate receptor gene expression in major depression and suicide. Mol. Psychiatry 2015, 20, 1057–1068. [Google Scholar] [CrossRef]
- Laje, G.; Paddock, S.; Manji, H.; Rush, A.J.; Wilson, A.F.; Charney, D.; McMahon, F.J. Genetic markers of suicidal ideation emerging during citalopram treatment of major depression. Am. J. Psychiatry 2007, 164, 1530–1538. [Google Scholar] [CrossRef] [Green Version]
- Menke, A.; Lucae, S.; Kloiber, S.; Horstmann, S.; Bettecken, T.; Uhr, M.; Ripke, S.; Ising, M.; Müller-Myhsok, B.; Holsboer, F. Genetic markers within glutamate receptors associated with antidepressant treatment-emergent suicidal ideation. Am. J. Psychiatry 2008, 165, 917–918. [Google Scholar] [CrossRef]
- Shaltiel, G.; Maeng, S.; Malkesman, O.; Pearson, B.; Schloesser, R.; Tragon, T.; Rogawski, M.; Gasior, M.; Luckenbaugh, D.; Chen, G. Evidence for the involvement of the kainate receptor subunit GluR6 (GRIK2) in mediating behavioral displays related to behavioral symptoms of mania. Mol. Psychiatry 2008, 13, 858–872. [Google Scholar] [CrossRef]
- Cannon, D.M.; Klaver, J.M.; Peck, S.A.; Rallis-Voak, D.; Erickson, K.; Drevets, W.C. Dopamine type-1 receptor binding in major depressive disorder assessed using positron emission tomography and [11C]NNC-112. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2009, 34, 1277–1287. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.J.; Voleti, B.; Hajszan, T.; Rajkowska, G.; Stockmeier, C.A.; Licznerski, P.; Lepack, A.; Majik, M.S.; Jeong, L.S.; Banasr, M.; et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 2012, 18, 1413–1417. [Google Scholar] [CrossRef] [PubMed]
- Kioka, N.; Ueda, K.; Amachi, T. Vinexin, CAP/ponsin, ArgBP2: A novel adaptor protein family regulating cytoskeletal organization and signal transduction. Cell Struct. Funct. 2002, 27, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Frake, R.A.; Karabiyik, C.; Son, S.M.; Siddiqi, F.H.; Bento, C.F.; Sterk, P.; Vicinanza, M.; Pavel, M.; Rubinsztein, D.C. Vinexin contributes to autophagic decline in brain ageing across species. Cell Death Differ. 2021, 29, 1055–1070. [Google Scholar] [CrossRef] [PubMed]
- Howard, D.M.; Adams, M.J.; Clarke, T.K.; Hafferty, J.D.; Gibson, J.; Shirali, M.; Coleman, J.R.I.; Hagenaars, S.P.; Ward, J.; Wigmore, E.M.; et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat. Neurosci. 2019, 22, 343–352. [Google Scholar] [CrossRef] [Green Version]
- Li, D.L.; London, S.J.; Liu, J.H.; Lee, W.; Jiang, X.J.; Van Den Berg, D.; Bergen, A.W.; Nishita, D.; Waleh, N.; Swan, G.E.; et al. Association of the Calcyon Neuron-Specific Vesicular Protein Gene (CALY) With Adolescent Smoking Initiation in China and California. Am. J. Epidemiol. 2011, 173, 1039–1048. [Google Scholar] [CrossRef] [Green Version]
- Ha, C.M.; Park, D.; Han, J.K.; Jang, J.I.; Park, J.Y.; Hwang, E.M.; Seok, H.; Chang, S. Calcyon forms a novel ternary complex with dopamine D1 receptor through PSD-95 protein and plays a role in dopamine receptor internalization. J. Biol. Chem. 2012, 287, 31813–31822. [Google Scholar] [CrossRef] [Green Version]
- Chander, P.; Kennedy, M.J.; Winckler, B.; Weick, J.P. Neuron-Specific Gene 2 (NSG2) Encodes an AMPA Receptor Interacting Protein That Modulates Excitatory Neurotransmission. Eneuro 2019, 6. [Google Scholar] [CrossRef]
- Davidson, H.T.; Xiao, J.P.; Dai, R.J.; Bergson, C. Calcyon is necessary for activity-dependent AMPA receptor internalization and LTD in CA1 neurons of hippocampus. Eur. J. Neurosci. 2009, 29, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Sakakibara, S.; Nakamura, Y.; Satoh, H.; Okano, H. Rna-binding protein Musashi2: Developmentally regulated expression in neural precursor cells and subpopulations of neurons in mammalian CNS. J. Neurosci. 2001, 21, 8091–8107. [Google Scholar] [CrossRef] [Green Version]
- Imai, T.; Tokunaga, A.; Yoshida, T.; Hashimoto, M.; Mikoshiba, K.; Weinmaster, G.; Nakafuku, M.; Okano, H. The neural RNA-binding protein Musashi1 translationally regulates mammalian numb gene expression by interacting with its mRNA. Mol. Cell. Biol. 2001, 21, 3888–3900. [Google Scholar] [CrossRef] [Green Version]
- Adams, M.E.; Kramarcy, N.; Krall, S.P.; Rossi, S.G.; Rotundo, R.L.; Sealock, R.; Froehner, S.C. Absence of α-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J. Cell Biol. 2000, 150, 1385–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasegawa, M.; Cuenda, A.; Spillantini, M.G.; Thomas, G.M.; Buée-Scherrer, V.; Cohen, P.; Goedert, M. Stress-activated protein kinase-3 interacts with the PDZ domain of α1-syntrophin: A mechanism for specific substrate recognition. J. Biol. Chem. 1999, 274, 12626–12631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de la Torre-Ubieta, L.; Bonni, A. Combinatorial assembly of neurons: From chromatin to dendrites. Trends Cell Biol. 2008, 18, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.; Rousseau, J.; Peng, H.; Aouabed, Z.; Priam, P.; Theroux, J.F.; Jefri, M.; Tanti, A.; Wu, H.; Kolobova, I.; et al. Mutations in ACTL6B Cause Neurodevelopmental Deficits and Epilepsy and Lead to Loss of Dendrites in Human Neurons. Am. J. Hum. Genet. 2019, 104, 815–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morley, J.E. The ascent of cholecystokinin (CCK)—from gut to brain. Life Sci. 1982, 30, 479–493. [Google Scholar] [CrossRef]
- Yin, H.; Pantazatos, S.P.; Galfalvy, H.; Huang, Y.Y.; Rosoklija, G.B.; Dwork, A.J.; Burke, A.; Arango, V.; Oquendo, M.A.; Mann, J.J. A pilot integrative genomics study of GABA and glutamate neurotransmitter systems in suicide, suicidal behavior, and major depressive disorder. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 414–426. [Google Scholar] [CrossRef]
- De la Cruz-Cano, E. Association between FKBP5 and CRHR1 genes with suicidal behavior: A systematic review. Behav. Brain Res. 2017, 317, 46–61. [Google Scholar] [CrossRef]
- Löfberg, C.; Ågren, H.; Harro, J.; Oreland, L. Cholecystokinin in CSF from depressed patients: Possible relations to severity of depression and suicidal behaviour. Eur. Neuropsychopharmacol. 1998, 8, 153–157. [Google Scholar] [CrossRef]
- Jahangard, L.; Solgy, R.; Salehi, I.; Taheri, S.K.; Holsboer-Trachsler, E.; Haghighi, M.; Brand, S. Cholecystokinin (CCK) level is higher among first time suicide attempters than healthy controls, but is not associated with higher depression scores. Psychiatry Res. 2018, 266, 40–46. [Google Scholar] [CrossRef]
- Harro, J.; Marcusson, J.; Oreland, L. Alterations in brain cholecystokinin receptors in suicide victims. Eur. Neuropsychopharmacol. 1992, 2, 57–63. [Google Scholar] [CrossRef]
- Scarr, E.; Beneyto, M.; Meador-Woodruff, J.H.; Dean, B. Cortical glutamatergic markers in schizophrenia. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2005, 30, 1521–1531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokolov, B.P. Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: Evidence on reversible up-regulation by typical neuroleptics. J. Neurochem. 1998, 71, 2454–2464. [Google Scholar] [CrossRef] [PubMed]
- Weickert, C.S.; Fung, S.; Catts, V.; Schofield, P.; Allen, K.; Moore, L.; Newell, K.A.; Pellen, D.; Huang, X.-F.; Catts, S. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol. Psychiatry 2013, 18, 1185–1192. [Google Scholar] [CrossRef] [Green Version]
- Cleva, R.M.; Olive, M.F. Positive Allosteric Modulators of Type 5 Metabotropic Glutamate Receptors (mGluR5) and Their Therapeutic Potential for the Treatment of CNS Disorders. Molecules 2011, 16, 2097–2106. [Google Scholar] [CrossRef] [Green Version]
- Maher, M.P.; Wu, N.; Ravula, S.; Ameriks, M.K.; Savall, B.M.; Liu, C.; Lord, B.; Wyatt, R.M.; Matta, J.A.; Dugovic, C.; et al. Discovery and Characterization of AMPA Receptor Modulators Selective for TARP-γ8. J. Pharmacol. Exp. Ther. 2016, 357, 394–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinero, J.; Bravo, A.; Queralt-Rosinach, N.; Gutierrez-Sacristan, A.; Deu-Pons, J.; Centeno, E.; Garcia-Garcia, J.; Sanz, F.; Furlong, L.I. DisGeNET: A comprehensive platform integrating information on human disease-associated genes and variants. Nucleic. Acids Res. 2017, 45, D833–D839. [Google Scholar] [CrossRef]
- Miraglia del Giudice, E.; Santoro, N.; Fiumani, P.; Dominguez, G.; Kuhar, M.J.; Perrone, L. Adolescents carrying a missense mutation in the CART gene exhibit increased anxiety and depression. Depress. Anxiety 2006, 23, 90–92. [Google Scholar] [CrossRef]
- Ahmadian-Moghadam, H.; Sadat-Shirazi, M.-S.; Zarrindast, M.-R. Cocaine-and amphetamine-regulated transcript (CART): A multifaceted neuropeptide. Peptides 2018, 110, 56–77. [Google Scholar] [CrossRef]
- Wiehager, S.; Beiderbeck, D.I.; Gruber, S.H.; El-Khoury, A.; Wamsteeker, J.; Neumann, I.D.; Petersén, Å.; Mathé, A.A. Increased levels of cocaine and amphetamine regulated transcript in two animal models of depression and anxiety. Neurobiol. Dis. 2009, 34, 375–380. [Google Scholar] [CrossRef]
- Mai, J.K.; Majtanik, M. Human Brain in Standard MNI Space: A Comprehensive Pocket Atlas; Elsevier: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Palkovits, M. Isolated removal of hypothalamic or other brain nuclei of the rat. Brain Res. 1973, 59, 449–450. [Google Scholar] [CrossRef]
- Palkovits, M. Microdissection of Individual Brain Nuclei and Areas. In General Neurochemical Techniques; Boulton, A.A., Baker, G.B., Eds.; Humana Press: Totowa, NJ, USA, 1985; pp. 1–17. [Google Scholar]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic. Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic. Acids Res. 2013, 41, e108. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Vilo, J.; Peterson, H. gprofiler2—An R package for gene list functional enrichment analysis and namespace conversion toolset g:Profiler. F1000Research 2020, 9, ELIXIR-709. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic. Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic. Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic. Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. 4), S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras-López, O.; Moyano, T.C.; Soto, D.C.; Gutiérrez, R.A. Step-by-Step Construction of Gene Co-expression Networks from High-Throughput Arabidopsis RNA Sequencing Data. Methods Mol. Biol. 2018, 1761, 275–301. [Google Scholar] [PubMed]
- Leng, N.; Dawson, J.A.; Thomson, J.A.; Ruotti, V.; Rissman, A.I.; Smits, B.M.; Haag, J.D.; Gould, M.N.; Stewart, R.M.; Kendziorski, C. EBSeq: An empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 2013, 29, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Revelle, W.R. Psych: Procedures for Personality and Psychological Research; Northwestern University: Evanston, IL, USA, 2017. [Google Scholar]
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJournal Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Su, G.; Kuchinsky, A.; Morris, J.H.; States, D.J.; Meng, F. GLay: Community structure analysis of biological networks. Bioinformatics 2010, 26, 3135–3137. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Thul, P.J.; Lindskog, C. The human protein atlas: A spatial map of the human proteome. Protein Sci. 2018, 27, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Dobolyi, A. Central amylin expression and its induction in rat dams. J. Neurochem. 2009, 111, 1490–1500. [Google Scholar] [CrossRef]
- Dobolyi, A.; Ostergaard, E.; Bagó, A.G.; Dóczi, T.; Palkovits, M.; Gál, A.; Molnár, M.J.; Adam-Vizi, V.; Chinopoulos, C. Exclusive neuronal expression of SUCLA2 in the human brain. Brain Struct. Funct. 2015, 220, 135–151. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Gene Symbol | Description | log2 Fold Change | p-Value | Up/Down Regulation | qPCR log2 Fold Change | qPCR p Value | mRNA Expression in Controls (Mean ± SEM) | mRNA Expression in Suicide Victims (Mean ± SEM) | Function |
|---|---|---|---|---|---|---|---|---|---|---|
| ENSG00000164418 | GRIK2 | glutamate ionotropic receptor kainate type subunit 2 | 1.37 | 1.22 × 10−5 | up | 2.35 | 0.007 | 0.002 ± 0.001 | 0.005 ± 0.001 | kainate selective glutamate receptor activity |
| ENSG00000154146 | NRGN | neurogranin | 1.34 | 7.44 × 10−4 | up | 2.16 | 0.005 | 0.083 ± 0.029 | 0.247 ± 0.032 | signal transduction |
| ENSG00000129990 | SYT5 | synaptotagmin 5 | 1.26 | 1.77 × 10−3 | up | 3.76 | 0.038 | 0.001 ± 0.0005 | 0.002 ± 0.0004 | SNARE, syntaxin binding |
| ENSG00000164082 | GRM2 | glutamate metabotropic receptor 2 | 1.19 | 1.70 × 10−3 | up | 2.18 | 0.021 | 0.0003 ± 0.0001 | 0.001 ± 0.0001 | adenylate cyclase inhibiting G protein-coupled glutamate receptor activity |
| ENSG00000171189 | GRIK1 | glutamate ionotropic receptor kainate type subunit 1 | 1.16 | 5.26 × 10−5 | up | 1.59 | 0.021 | 0.007 ± 0.002 | 0.016 ± 0.002 | kainate selective glutamate receptor activity |
| ENSG00000103154 | NECAB2 | N-terminal EF-hand calcium-binding protein 2 | 1.00 | 7.82 × 10−5 | up | 1.05 | 0.037 | 0.022 ± 0.005 | 0.038 ± 0.004 | type 5 metabotropic glutamate receptor binding |
| ENSG00000240583 | AQP1 | aquaporin 1 | −2.59 | 2.29 × 10−7 | down | −1.72 | 0.045 | 0.003 ± 0.001 | 0.001 ± 0.0002 | transmembrane transporter activity |
| ENSG00000143772 | ITPKB | inositol-trisphosphate 3-kinase B | −2.40 | 1.55 × 10−8 | down | −1.57 | 0.036 | 0.019 ± 0.005 | 0.005 ± 0.001 | ATP binding, kinase activity |
| ENSG00000132470 | ITGB4 | integrin subunit beta 4 | −2.26 | 9.70 × 10−7 | down | −2.1 | 0.034 | 0.004 ± 0.001 | 0.001 ± 0.0001 | G protein-coupled receptor binding |
| ENSG00000137491 | SLCO2B1 | solute carrier organic anion transporter family member 2B1 | −1.79 | 9.99 × 10−8 | down | −2.05 | 0.048 | 0.032 ± 0.012 | 0.004 ± 0.0004 | sodium-independent organic anion transmembrane transporter activity |
| ENSG00000152661 | GJA1 | gap junction protein alpha 1 | −1.75 | 2.64 × 10−4 | down | −0.78 | 0.049 | 0.054 ± 0.009 | 0.032 ± 0.004 | glutathione transmembrane transporter activity |
| ENSG00000027075 | PRKCH | protein kinase C eta | −1.71 | 8.74 × 10−7 | down | −2.11 | 0.047 | 0.01 ± 0.003 | 0.001 ± 0.0001 | calcium-dependent protein kinase C activity |
| ENSG00000135821 | GLUL | glutamate-ammonia ligase | −1.51 | 3.07 × 10−4 | down | −1.78 | 0.039 | 8.703 ± 2.713 | 1.807 ± 0.252 | glutamate-ammonia ligase activity |
| ENSG00000160307 | S100B | S100 calcium-binding protein B | −1.39 | 5.78 × 10−6 | down | −1.11 | 0.037 | 0.528 ± 0.128 | 0.198 ± 0.01 | calcium-dependent protein binding |
| Downregulated DEGs | Upregulated DEGs | Function | Depression-Related Pathway |
|---|---|---|---|
| GABRG1, NTSR2, GPR37L1, GABRE, GLRA1, GRIN2C | CARTPT, GABRD, CCK, CRHR1, GRM2, GRIK1, GRIK2 | Signal transduction | Neuroactive ligand-receptor interaction |
| ITPR2, RYR3, ASPH | TRPM2 | Calcium-mediated signaling | Oxytocin signaling pathway |
| SLC6A13, SLC6A11, SLC6A12, GABRG1, GABRE, SLC38A3, SLC38A5 | GABRD | Anion transmembrane transporter activity | GABAergic synapse |
| PDGFRB, FLT4, ERBB2, EGFR, NTRK2, CYSLTR2, FLT1, PTGER1, GRIN2C, NOS3, ADORA2B, VEGFB, ADCY4, FGF8, ADORA2A, FGFR3, GNA14, ASPH, ITPR2, RYR3, ITPKB, PLCD1, TPCN1, PLCD3, FGF1 | CACNA1G, FGF8, P2RX2 | Cell communication | Serotonergic synapse |
| TNFRSF1A, TGFB1, TGFB3, DUSP1, RRAS, HSPB1, PDGFRB, FLT4, ERBB2, GNA12, EGFR, FLT1, VEGFB, ANGPT2, CSF1, MAP4K4, EPHA2, TGFBR2, TGFB2, IL1R1, PGF, FGF1 | CACNA1G, CACNG8, DUSP4, FGF8 | Regulation of cellular process | MAPK signaling pathway |
| Donor | Sex | Age | Post Mortem Interval (PMI) | Cause of Death | Clinical and Pathological Diagnosis |
|---|---|---|---|---|---|
| #1 | female | 48 | 6–7 h | Suicide (drug overdose) | - |
| #2 | male | 71 | 1 h | Suicide (jumping from a height) | Without any clinical care during the past 6 months |
| #3 | male | 48 | 6 h | Suicide (hanging—asphyxia) | Without known drug treatment |
| #4 | female | 65 | 5 h | Suicide (hanging—asphyxia) | Pathological diagnosis: negative status (no pathological sign for any diseases) |
| #5 | male | 31 | 8 h | Suicide (hanging—asphyxia) | Without known drug treatment |
| #6 | female | 49 | 6 h | Suicide (drug overdose) | Without known drug treatment |
| #7 | male | 43 | 4 h | Suicide (hanging) | Without any clinical care |
| #8 | male | 66 | 8–10 h | Suicide (hanging—asphyxia) | Laboratory test: alcohol: negative |
| #9 | male | 42 | 3.5 h | Acute respiratory insufficiency | - |
| #10 | female | 56 | 6 h | Cardiorespiratory insufficiency, edema cerebri | Edema cerebri, coarctatio aortae, hepatitis alcoholica |
| #11 | male | 50 | 5.5 h | Stroke, brain hemorrhage | Large cortical and subcortical hemorrhage in the parietal lobe |
| #12 | male | 68 | 10 h | Acute heart failure | Acute pulmonary edema, serious arteriosclerosis (especially in the heart and kidney), peripheral arterial shunt, cerebral sclerosis. left coronary occlusion |
| #13 | female | 75 | 10 h | Stroke (right side arteria cerebri media) | Diabetes, stroke, hypertonia, mamma carcinoma, emolitio arteriae cerebri mediae lateralis dextri, cortical infarction, general atherosclerosis |
| #14 | male | 64 | 10 h | Stroke (arteria cerebri media on the left side), bronchopneumonia | Cardiomyopathia, coronary sclerosis, hypertonia, infarctus myocardii, bronchopneumonia, cardiorespiratory insufficiency, femoralis amputatio, aphasia, carotis stenosis, pneumonia |
| #15 | male | 90 | 4–5 h | Stroke (cerebri media and posterior) | Stroke, infarctus lacunaris multiplex cerebri, Parkinson’s disease, emolitio, tracheobronchitis, cardiopulmonary insufficiency, carotis stenosis |
| #16 | male | 78 | 10 h | Cardiorespiratory insufficiency | Dementia, diabetes, hypertonia, carotis interna occlusio, polyneuropathia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dóra, F.; Renner, É.; Keller, D.; Palkovits, M.; Dobolyi, Á. Transcriptome Profiling of the Dorsomedial Prefrontal Cortex in Suicide Victims. Int. J. Mol. Sci. 2022, 23, 7067. https://doi.org/10.3390/ijms23137067
Dóra F, Renner É, Keller D, Palkovits M, Dobolyi Á. Transcriptome Profiling of the Dorsomedial Prefrontal Cortex in Suicide Victims. International Journal of Molecular Sciences. 2022; 23(13):7067. https://doi.org/10.3390/ijms23137067
Chicago/Turabian StyleDóra, Fanni, Éva Renner, Dávid Keller, Miklós Palkovits, and Árpád Dobolyi. 2022. "Transcriptome Profiling of the Dorsomedial Prefrontal Cortex in Suicide Victims" International Journal of Molecular Sciences 23, no. 13: 7067. https://doi.org/10.3390/ijms23137067