
Synthesis of Antibacterial Hybrid Hydroxyapatite/Collagen/Polysaccharide Bioactive Membranes and Their Effect on Osteoblast Culture

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Hydrogel Preparation for Synthesis of Biopolymeric Membranes
2.3. Inorganic Phase Incorporation and Preparation of Hybrid Membranes
2.4. Compositional and Morphological Characterization
2.5. In Vitro Bioactivity Tests
2.6. Mechanical Properties
2.7. Swelling and Degradability
2.8. Wettability and Surface Free Energy
2.9. Incorporation of Silver Nanoparticles (AgNPs) into the Biopolymeric Membranes
2.10. In Vitro Toxicity to Osteoblast Culture
2.11. Antimicrobial Activity Assay
2.12. AgNP Release from the Membranes In Vitro
2.13. Statistical Analyses
3. Results and Discussion
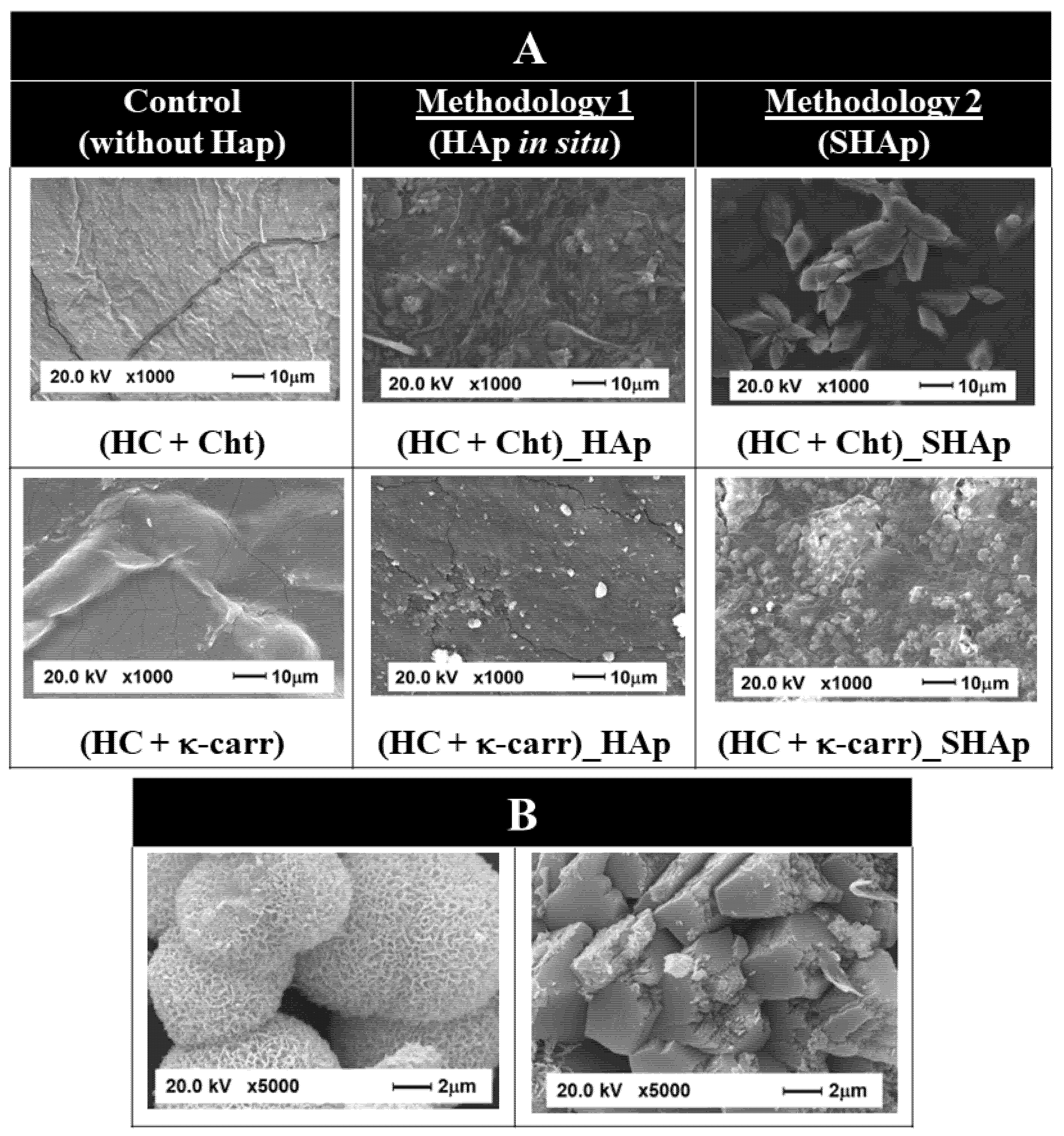
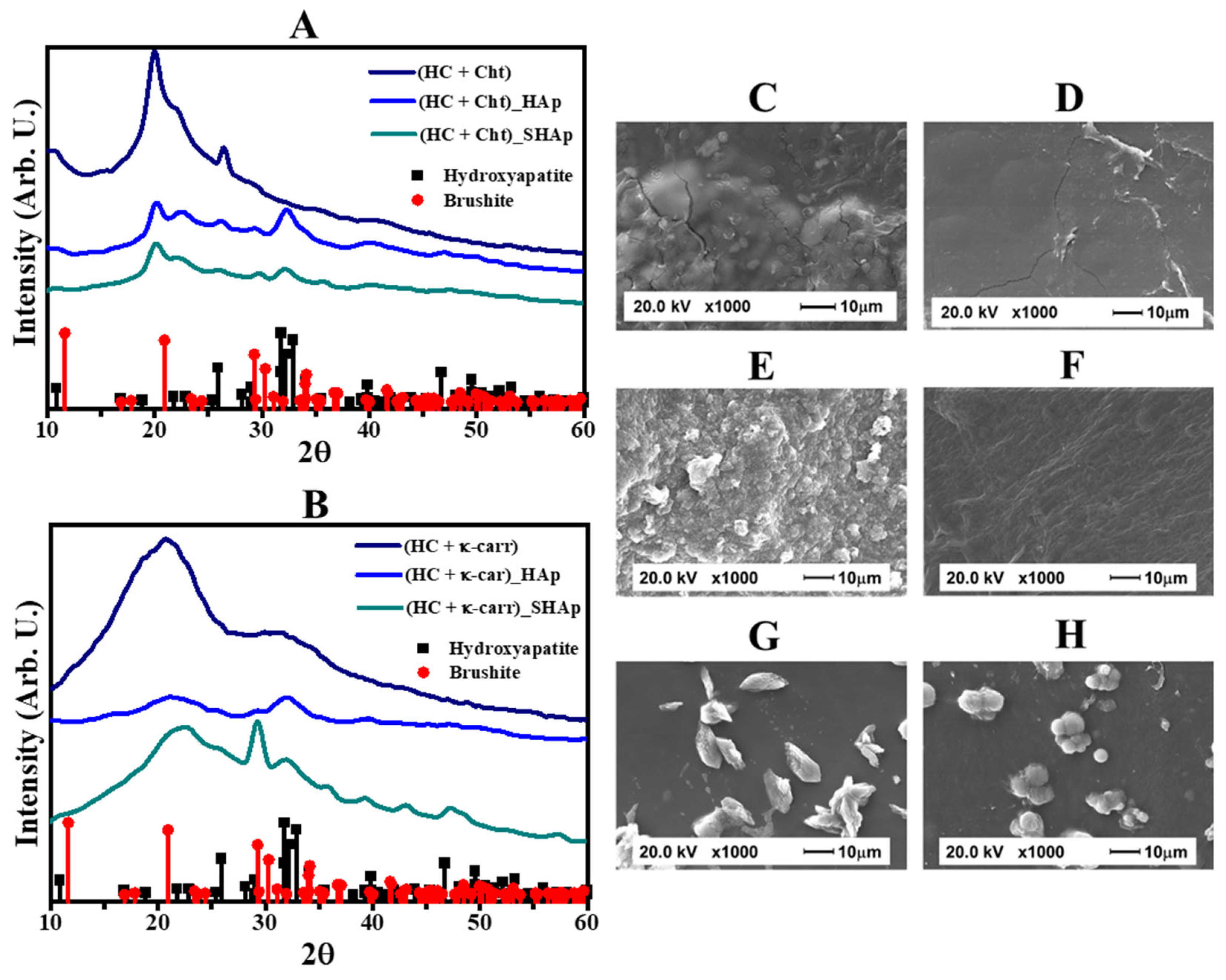
3.1. Composition and Morphology of the Hybrid Membranes
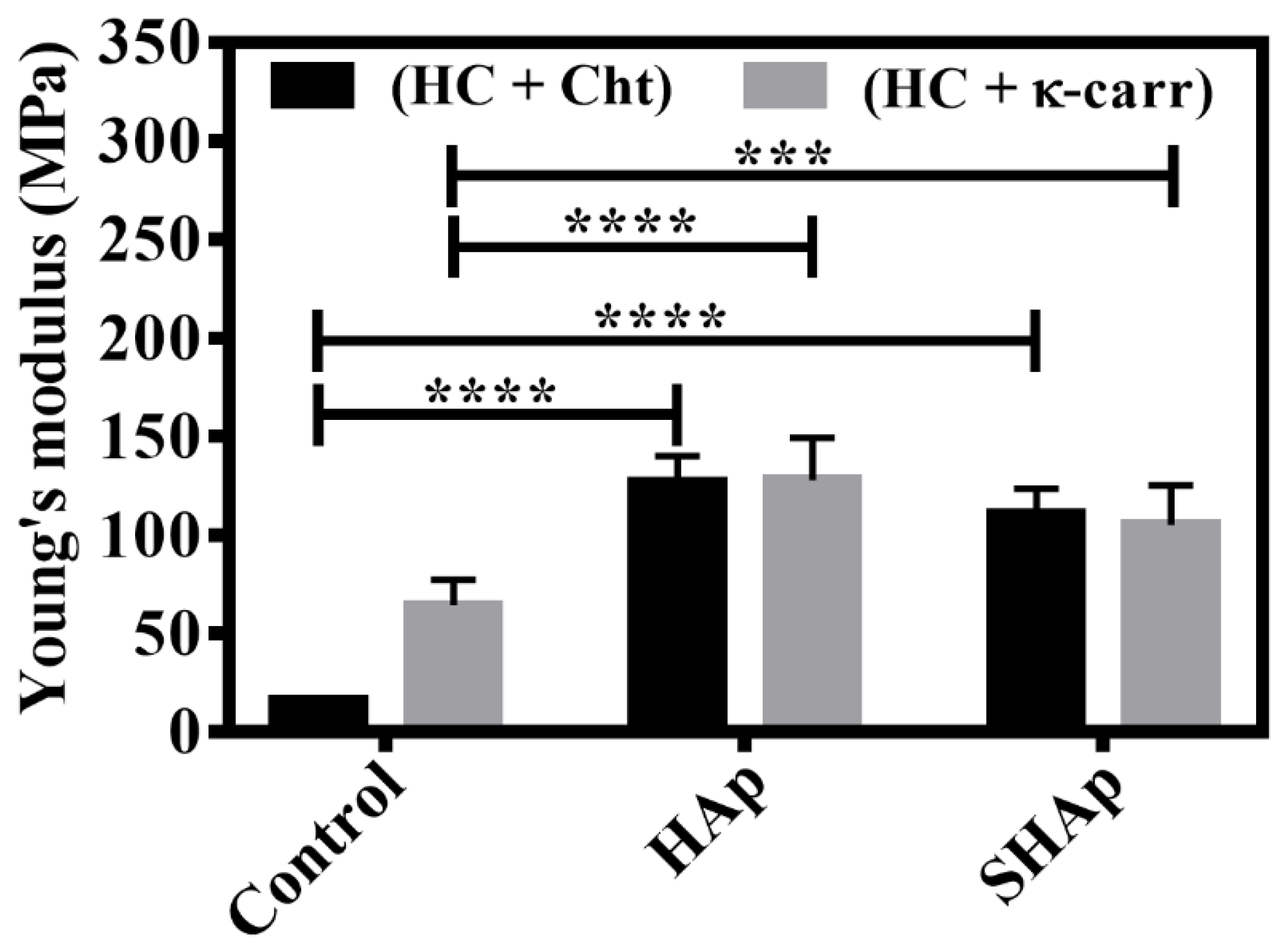
3.2. Mechanical Properties
3.3. Physical-Chemical Properties: Surface Wettability, Surface Free Energy, and Water Absorption Ability
3.4. In Vitro Bioactivity Evaluation of the Hybrid Membranes
3.5. Biological Properties of the Membranes
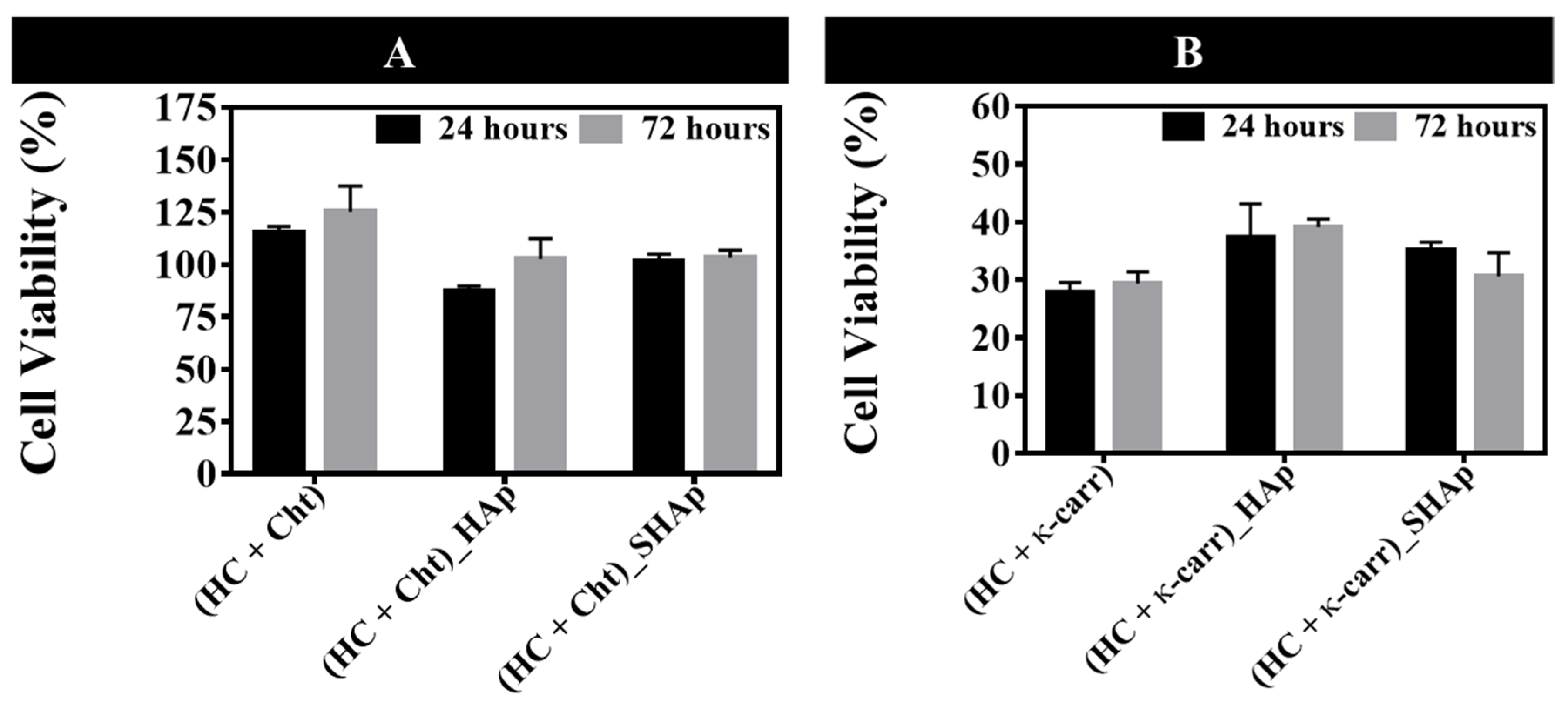
3.5.1. Osteoblasts Culture
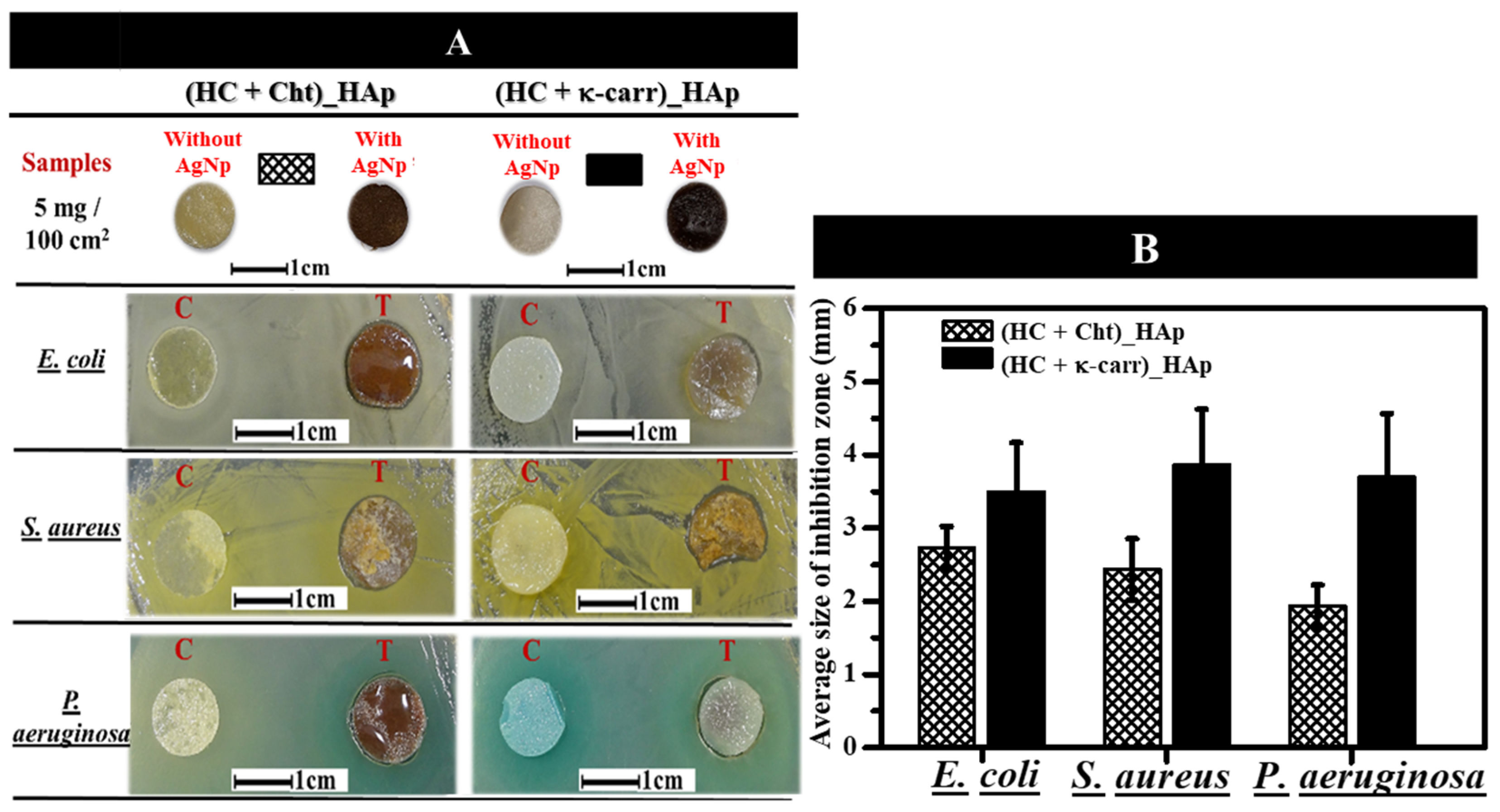
3.5.2. Antimicrobial Activity of the Membranes in the Presence of Silver Nanoparticles (AgNPs)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agarwal, R.; García, A.J. Biomaterial strategies for engineering implants for enhanced osseointegration and bone repair. Adv. Drug Deliv. Rev. 2015, 94, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesan, J.; Kim, S.-K. Marine Algae Derived Polysaccharides for Bone Tissue Regeneration. In Marine Algae Extracts; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2015; Volume 2, pp. 509–522. ISBN 9783527679577. [Google Scholar]
- Venkatesan, J.; Bhatnagar, I.; Manivasagan, P.; Kang, K.-H.; Kim, S.-K. Alginate composites for bone tissue engineering: A review. Int. J. Biol. Macromol. 2015, 72, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; He, J.; Peng, S.; Gao, C.; Zhao, Z.; Xiong, S.; Shuai, C. Characterizations and interfacial reinforcement mechanisms of multicomponent biopolymer based scaffold. Mater. Sci. Eng. C 2019, 100, 809–825. [Google Scholar] [CrossRef] [PubMed]
- Goonoo, N.; Khanbabaee, B.; Steuber, M.; Bhaw-Luximon, A.; Jonas, U.; Pietsch, U.; Jhurry, D.; Schönherr, H. κ-Carrageenan Enhances the Biomineralization and Osteogenic Differentiation of Electrospun Polyhydroxybutyrate and Polyhydroxybutyrate Valerate Fibers. Biomacromolecules 2017, 18, 1563–1573. [Google Scholar] [CrossRef] [PubMed]
- Nourmohammadi, J.; Roshanfar, F.; Farokhi, M.; Haghbin Nazarpak, M. Silk fibroin/kappa-carrageenan composite scaffolds with enhanced biomimetic mineralization for bone regeneration applications. Mater. Sci. Eng. C 2017, 76, 951–958. [Google Scholar] [CrossRef]
- Mota, J.; Yu, N.; Caridade, S.G.; Luz, G.M.; Gomes, M.E.; Reis, R.L.; Jansen, J.A.; Walboomers, X.F.; Mano, J.F. Chitosan/bioactive glass nanoparticle composite membranes for periodontal regeneration. Acta Biomater. 2012, 8, 4173–4180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.-J.; Shin, D.-S.; Kim, H.-E.; Kim, H.-W.; Koh, Y.-H.; Jang, J.-H. Membrane of hybrid chitosan–silica xerogel for guided bone regeneration. Biomaterials 2009, 30, 743–750. [Google Scholar] [CrossRef]
- Veríssimo, D.M.; Leitão, R.F.C.; Ribeiro, R.A.; Figueiró, S.D.; Sombra, A.S.B.; Góes, J.C.; Brito, G.A.C. Polyanionic collagen membranes for guided tissue regeneration: Effect of progressive glutaraldehyde cross-linking on biocompatibility and degradation. Acta Biomater. 2010, 6, 4011–4018. [Google Scholar] [CrossRef]
- Kwon, K.-J.; Seok, H. Silk Protein-Based Membrane for Guided Bone Regeneration. Appl. Sci. 2018, 8, 1214. [Google Scholar] [CrossRef] [Green Version]
- Naumenko, E.; Zakirova, E.; Guryanov, I.; Akhatova, F.; Sergeev, M.; Valeeva, A.; Fakhrullin, R. Composite biodegradable polymeric matrix doped with halloysite nanotubes for the repair of bone defects in dogs. Clays Clay Miner. 2021, 69, 522–532. [Google Scholar] [CrossRef]
- Gorgieva, S.; Vuherer, T.; Kokol, V. Autofluorescence-aided assessment of integration and μ-structuring in chitosan/gelatin bilayer membranes with rapidly mineralized interface in relevance to guided tissue regeneration. Mater. Sci. Eng. C 2018, 93, 226–241. [Google Scholar] [CrossRef]
- Vaquette, C.; Pilipchuk, S.P.; Bartold, P.M.; Hutmacher, D.W.; Giannobile, W.V.; Ivanovski, S. Tissue Engineered Constructs for Periodontal Regeneration: Current Status and Future Perspectives. Adv. Healthc. Mater. 2018, 7, 1800457. [Google Scholar] [CrossRef]
- Pandele, A.M.; Neacsu, P.; Cimpean, A.; Staras, A.I.; Miculescu, F.; Iordache, A.; Voicu, S.I.; Thakur, V.K.; Toader, O.D. Cellulose acetate membranes functionalized with resveratrol by covalent immobilization for improved osseointegration. Appl. Surf. Sci. 2018, 438, 2–13. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Guo, J.L.; Wang, J.; Mikos, A.G.; Zhang, S. Hierarchically designed bone scaffolds: From internal cues to external stimuli. Biomaterials 2019, 218, 119334. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Luo, D.; Wang, T. Hierarchical Structures of Bone and Bioinspired Bone Tissue Engineering. Small 2016, 12, 4611–4632. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, Z.; Qureshi, J.; Alshahrani, A.M.; Nassar, H.; Ikeda, Y.; Glogauer, M.; Ganss, B. Collagen based barrier membranes for periodontal guided bone regeneration applications. Odontology 2017, 105, 1–12. [Google Scholar] [CrossRef]
- Caridade, S.G.; Merino, E.G.; Alves, N.M.; Bermudez, V.d.Z.; Boccaccini, A.R.; Mano, J.F. Chitosan membranes containing micro or nano-size bioactive glass particles: Evolution of biomineralization followed by in situ dynamic mechanical analysis. J. Mech. Behav. Biomed. Mater. 2013, 20, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansari, Z.; Kalantar, M.; Soriente, A.; Fasolino, I.; Kharaziha, M.; Ambrosio, L.; Raucci, M.G. In-Situ Synthesis and Characterization of Chitosan/Hydroxyapatite Nanocomposite Coatings to Improve the Bioactive Properties of Ti6Al4V Substrates. Materials 2020, 13, 3772. [Google Scholar] [CrossRef]
- Yan, Y.; Zhang, X.; Li, C.; Huang, Y.; Ding, Q.; Pang, X. Preparation and characterization of chitosan-silver/hydroxyapatite composite coatings onTiO2 nanotube for biomedical applications. Appl. Surf. Sci. 2015, 332, 62–69. [Google Scholar] [CrossRef]
- Jegal, S.-H.; Park, J.-H.; Kim, J.-H.; Kim, T.-H.; Shin, U.S.; Kim, T.-I.; Kim, H.-W. Functional composite nanofibers of poly(lactide–co-caprolactone) containing gelatin–apatite bone mimetic precipitate for bone regeneration. Acta Biomater. 2011, 7, 1609–1617. [Google Scholar] [CrossRef]
- Swetha, M.; Sahithi, K.; Moorthi, A.; Srinivasan, N.; Ramasamy, K.; Selvamurugan, N. Biocomposites containing natural polymers and hydroxyapatite for bone tissue engineering. Int. J. Biol. Macromol. 2010, 47, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Nwe, N.; Furuike, T.; Tamura, H. Selection of a biopolymer based on attachment, morphology and proliferation of fibroblast NIH/3T3 cells for the development of a biodegradable tissue regeneration template: Alginate, bacterial cellulose and gelatin. Process Biochem. 2010, 45, 457–466. [Google Scholar] [CrossRef]
- Schweizer, S.; Bick, A.; Subramanian, L.; Krokidis, X. Influences on the stability of collagen triple-helix. Fluid Phase Equilib. 2014, 362, 113–117. [Google Scholar] [CrossRef]
- Ferreira, A.M.; Gentile, P.; Chiono, V.; Ciardelli, G. Collagen for bone tissue regeneration. Acta Biomater. 2012, 8, 3191–3200. [Google Scholar] [CrossRef]
- Ficai, A.; Albu, M.G.; Birsan, M.; Sonmez, M.; Ficai, D.; Trandafir, V.; Andronescu, E. Collagen hydrolysate based collagen/hydroxyapatite composite materials. J. Mol. Struct. 2013, 1037, 154–159. [Google Scholar] [CrossRef]
- Ramadass, S.K.; Perumal, S.; Gopinath, A.; Nisal, A.; Subramanian, S.; Madhan, B. Sol–Gel Assisted Fabrication of Collagen Hydrolysate Composite Scaffold: A Novel Therapeutic Alternative to the Traditional Collagen Scaffold. ACS Appl. Mater. Interfaces 2014, 6, 15015–15025. [Google Scholar] [CrossRef]
- Pei, Y.; Yang, J.; Liu, P.; Xu, M.; Zhang, X.; Zhang, L. Fabrication, properties and bioapplications of cellulose/collagen hydrolysate composite films. Carbohydr. Polym. 2013, 92, 1752–1760. [Google Scholar] [CrossRef]
- Li, J.; Yang, B.; Qian, Y.; Wang, Q.; Han, R.; Hao, T.; Shu, Y.; Zhang, Y.; Yao, F.; Wang, C. Iota-carrageenan/chitosan/gelatin scaffold for the osteogenic differentiation of adipose-derived MSCs in vitro. J. Biomed. Mater. Res.-Part B Appl. Biomater. 2015, 103, 1498–1510. [Google Scholar] [CrossRef]
- Mozumder, M.S.; Mairpady, A.; Mourad, A.H.I. Polymeric nanobiocomposites for biomedical applications. J. Biomed. Mater. Res.-Part B Appl. Biomater. 2017, 105, 1241–1259. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and chitosan: Properties and applications. Prog. Polym. Sci. 2006, 31, 603–632. [Google Scholar] [CrossRef]
- Singh, R.; Shitiz, K.; Singh, A. Chitin and chitosan: Biopolymers for wound management. Int. Wound J. 2017, 14, 1276–1289. [Google Scholar] [CrossRef] [PubMed]
- Deepthi, S.; Venkatesan, J.; Kim, S.K.; Bumgardner, J.D.; Jayakumar, R. An overview of chitin or chitosan/nano ceramic composite scaffolds for bone tissue engineering. Int. J. Biol. Macromol. 2016, 93, 1338–1353. [Google Scholar] [CrossRef] [PubMed]
- Kassab, Z.; Aziz, F.; Hannache, H.; Ben Youcef, H.; El Achaby, M. Improved mechanical properties of k-carrageenan-based nanocomposite films reinforced with cellulose nanocrystals. Int. J. Biol. Macromol. 2019, 123, 1248–1256. [Google Scholar] [CrossRef] [PubMed]
- González Ocampo, J.I.; Machado de Paula, M.M.; Bassous, N.J.; Lobo, A.O.; Ossa Orozco, C.P.; Webster, T.J. Osteoblast responses to injectable bone substitutes of kappa-carrageenan and nano hydroxyapatite. Acta Biomater. 2019, 83, 425–434. [Google Scholar] [CrossRef]
- Feng, W.; Feng, S.; Tang, K.; He, X.; Jing, A.; Liang, G. A novel composite of collagen-hydroxyapatite/kappa-carrageenan. J. Alloys Compd. 2017, 693, 482–489. [Google Scholar] [CrossRef]
- Nogueira, L.F.B.; Maniglia, B.C.; Pereira, L.S.; Tapia-Blácido, D.R.; Ramos, A.P. Formation of carrageenan-CaCO3 bioactive membranes. Mater. Sci. Eng. C 2016, 58, 1–6. [Google Scholar] [CrossRef]
- Mihaila, S.M.; Gaharwar, A.K.; Reis, R.L.; Marques, A.P.; Gomes, M.E.; Khademhosseini, A. Photocrosslinkable Kappa -Carrageenan Hydrogels for Tissue Engineering Applications. Adv. Healthc. Mater. 2013, 2, 895–907. [Google Scholar] [CrossRef]
- Song, J.M.; Shin, S.H.; Kim, Y.D.; Lee, J.Y.; Baek, Y.J.; Yoon, S.Y.; Kim, H.S. Comparative study of chitosan/fibroin-hydroxyapatite and collagen membranes for guided bone regeneration in rat calvarial defects: Micro-computed tomography analysis. Int. J. Oral Sci. 2014, 6, 87–93. [Google Scholar] [CrossRef] [Green Version]
- Bakan, F.; Laçin, O.; Sarac, H. A novel low temperature sol–gel synthesis process for thermally stable nano crystalline hydroxyapatite. Powder Technol. 2013, 233, 295–302. [Google Scholar] [CrossRef]
- Doostmohammadi, A.; Monshi, A.; Salehi, R.; Fathi, M.H.; Karbasi, S.; Pieles, U.; Daniels, A.U. Preparation, chemistry and physical properties of bone-derived hydroxyapatite particles having a negative zeta potential. Mater. Chem. Phys. 2012, 132, 446–452. [Google Scholar] [CrossRef]
- Ma, M.-G. Hierarchically nanostructured hydroxyapatite: Hydrothermal synthesis, morphology control, growth mechanism, and biological activity. Int. J. Nanomed. 2012, 7, 1781–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity ? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Kokubo, T. Design of bioactive bone substitutes based on biomineralization process. Mater. Sci. Eng. C 2005, 25, 97–104. [Google Scholar] [CrossRef]
- Zadpoor, A.A. Relationship between in vitro apatite-forming ability measured using simulated body fluid and in vivo bioactivity of biomaterials. Mater. Sci. Eng. C. Mater. Biol. Appl. 2014, 35, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Chen, W.; Yu, D.; Lei, Y.; Ke, Q.; Guo, Y.; Zhu, Z. Fabrication of silver nanoparticle-doped hydroxyapatite coatings with oriented block arrays for enhancing bactericidal effect and osteoinductivity. J. Mech. Behav. Biomed. Mater. 2016, 61, 345–359. [Google Scholar] [CrossRef]
- Tian, B.; Chen, W.; Dong, Y.; Marymont, J.V.; Lei, Y.; Ke, Q.; Guo, Y.; Zhu, Z. Silver nanoparticle-loaded hydroxyapatite coating: Structure, antibacterial properties, and capacity for osteogenic induction in vitro. RSC Adv. 2016, 6, 8549–8562. [Google Scholar] [CrossRef]
- Pauksch, L.; Hartmann, S.; Rohnke, M.; Szalay, G.; Alt, V.; Schnettler, R.; Lips, K.S. Biocompatibility of silver nanoparticles and silver ions in primary human mesenchymal stem cells and osteoblasts. Acta Biomater. 2014, 10, 439–449. [Google Scholar] [CrossRef]
- Zepon, K.M.; Marques, M.S.; da Silva Paula, M.M.; Morisso, F.D.P.; Kanis, L.A. Facile, green and scalable method to produce carrageenan-based hydrogel containing in situ synthesized AgNPs for application as wound dressing. Int. J. Biol. Macromol. 2018, 113, 51–58. [Google Scholar] [CrossRef]
- Patrascu, J.M.; Nedelcu, I.A.; Sonmez, M.; Ficai, D.; Ficai, A.; Vasile, B.S.; Ungureanu, C.; Albu, M.G.; Andor, B.; Andronescu, E.; et al. Composite Scaffolds Based on Silver Nanoparticles for Biomedical Applications. J. Nanomater. 2015, 2015, 587989. [Google Scholar] [CrossRef] [Green Version]
- Saravanan, S.; Nethala, S.; Pattnaik, S.; Tripathi, A.; Moorthi, A.; Selvamurugan, N. Preparation, characterization and antimicrobial activity of a bio-composite scaffold containing chitosan/nano-hydroxyapatite/nano-silver for bone tissue engineering. Int. J. Biol. Macromol. 2011, 49, 188–193. [Google Scholar] [CrossRef]
- Andrade, F.A.C.; Vercik, L.C.d.O.; Monteiro, F.J.; da Silva Rigo, E.C. Preparation, characterization and antibacterial properties of silver nanoparticles–hydroxyapatite composites by a simple and eco-friendly method. Ceram. Int. 2016, 42, 2271–2280. [Google Scholar] [CrossRef]
- Albers, C.E.; Hofstetter, W.; Siebenrock, K.A.; Landmann, R.; Klenke, F.M. In vitro cytotoxicity of silver nanoparticles on osteoblasts and osteoclasts at antibacterial concentrations. Nanotoxicology 2013, 7, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Cüneyt Tas, A. Synthesis of biomimetic Ca-hydroxyapatite powders at 37 °C in synthetic body fluids. Biomaterials 2000, 21, 1429–1438. [Google Scholar] [CrossRef]
- Nogueira, L.F.B.; Maniglia, B.C.; Blácido, D.R.T.; Ramos, A.P. Organic–inorganic collagen/iota-carrageenan/hydroxyapatite hybrid membranes are bioactive materials for bone regeneration. J. Appl. Polym. Sci. 2019, 136, 48004. [Google Scholar] [CrossRef]
- Ramos, A.P.; Espimpolo, D.M.; Zaniquelli, M.E.D. Influence of the type of phospholipid head and of the conformation of the polyelectrolyte on the growth of calcium carbonate thin films on LB/LbL matrices. Colloids Surfaces B Biointerfaces 2012, 95, 178–185. [Google Scholar] [CrossRef]
- Fang, W.; Zhang, H.; Yin, J.; Yang, B.; Zhang, Y.; Li, J.; Yao, F. Hydroxyapatite Crystal Formation in the Presence of Polysaccharide. Cryst. Growth Des. 2016, 16, 1247–1255. [Google Scholar] [CrossRef]
- Kokubo, T.; Kushitani, H.; Sakka, S.; Kitsugi, T.; Yamamuro, T. Solutions able to reproduce in vivo surface-structure changes in bioactive glass-ceramic A-W3. J. Biomed. Mater. Res. 1990, 24, 721–734. [Google Scholar] [CrossRef]
- Oyane, A.; Kim, H.-M.; Furuya, T.; Kokubo, T.; Miyazaki, T.; Nakamura, T. Preparation and assessment of revised simulated body fluids. J. Biomed. Mater. Res. 2003, 65A, 188–195. [Google Scholar] [CrossRef]
- ASTM D882-12; Standard Test Method for Tensile Properties of Thin Plastic Sheeting; ASTM International: West Conshohocken, PA, USA, 2018.
- ASTM D882-18; Stand Test Method for Tensile Properties of Thin Plast Sheeting. ASTM International: West Conshohocken, PA, USA, 2018. [CrossRef]
- Rudawska, A.; Jacniacka, E. Analysis for determining surface free energy uncertainty by the Owen–Wendt method. Int. J. Adhes. Adhes. 2009, 29, 451–457. [Google Scholar] [CrossRef]
- Feng, B.; Weng, J.; Yang, B.C.; Qu, S.X.; Zhang, X.D. Characterization of surface oxide films on titanium and adhesion of osteoblast. Biomaterials 2003, 24, 4663–4670. [Google Scholar] [CrossRef]
- Mulfinger, L.; Solomon, S.D.; Bahadory, M.; Jeyarajasingam, A.V.; Rutkowsky, S.A.; Boritz, C. Synthesis and Study of Silver Nanoparticles. J. Chem. Educ. 2007, 84, 322. [Google Scholar] [CrossRef]
- Kim, J.S.; Kuk, E.; Yu, K.N.; Kim, J.-H.; Park, S.J.; Lee, H.J.; Kim, S.H.; Park, Y.K.; Park, Y.H.; Hwang, C.-Y.; et al. Antimicrobial effects of silver nanoparticles. Nanomed. Nanotechnol. Biol. Med. 2007, 3, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- de Faria, A.N.; Zancanela, D.C.; Ramos, A.P.; Torqueti, M.R.; Ciancaglini, P. Estrogen and phenol red free medium for osteoblast culture: Study of the mineralization ability. Cytotechnology 2016, 68, 1623–1632. [Google Scholar] [CrossRef] [Green Version]
- Yamini, D.; Devanand Venkatasubbu, G.; Kumar, J.; Ramakrishnan, V. Raman scattering studies on PEG functionalized hydroxyapatite nanoparticles. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2014, 117, 299–303. [Google Scholar] [CrossRef]
- Zając, A.; Hanuza, J.; Wandas, M.; Dymińska, L. Determination of N-acetylation degree in chitosan using Raman spectroscopy. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2015, 134, 114–120. [Google Scholar] [CrossRef]
- Pereira, L.; Amado, A.M.; Critchley, A.T.; Van De Velde, F.; Ribeiro-claro, P.J.A. Identification of selected seaweed polysaccharides (phycocolloids) by vibrational spectroscopy (FTIR-ATR and FT-Raman). Food Hydrocoll. 2009, 23, 1903–1909. [Google Scholar] [CrossRef] [Green Version]
- Bannerman, A.; Williams, R.L.; Cox, S.C.; Grover, L.M. Visualising phase change in a brushite-based calcium phosphate ceramic. Sci. Rep. 2016, 6, 32671. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ouyang, Z.; Ren, Z.; Li, J.; Zhang, P.; Wei, G.; Su, Z. Self-assembled peptide nanofibers on graphene oxide as a novel nanohybrid for biomimetic mineralization of hydroxyapatite. Carbon 2015, 89, 20–30. [Google Scholar] [CrossRef]
- Li, Z.; Wen, T.; Su, Y.; Wei, X.; He, C.; Wang, D. Hollow hydroxyapatite spheres fabrication with three-dimensional hydrogel template. CrystEngComm 2014, 16, 4202–4209. [Google Scholar] [CrossRef]
- Falini, G.; Fermani, S.; Goisis, M.; Manganelli, G. Calcite Morphology and Aggregation in the Presence of Comb-like Polymers Adsorbed on Cement Particles. Cryst. Growth Des. 2009, 9, 2240–2247. [Google Scholar] [CrossRef]
- Kasha, A.; Al-Hashim, H.; Abdallah, W.; Taherian, R.; Sauerer, B. Effect of Ca2+, Mg2+ and SO42− ions on the zeta potential of calcite and dolomite particles aged with stearic acid. Colloids Surfaces A Physicochem. Eng. Asp. 2015, 482, 290–299. [Google Scholar] [CrossRef]
- Tovani, C.B.; Gloter, A.; Azaïs, T.; Selmane, M.; Ramos, A.P.; Nassif, N. Formation of stable strontium-rich amorphous calcium phosphate: Possible effects on bone mineral. Acta Biomater 2019, 92, 315–324. [Google Scholar] [CrossRef]
- Toshima, T.; Hamai, R.; Tafu, M.; Takemura, Y.; Fujita, S.; Chohji, T.; Tanda, S.; Li, S.; Qin, G.W. Morphology control of brushite prepared by aqueous solution synthesis. J. Asian Ceram. Soc. 2014, 2, 52–56. [Google Scholar] [CrossRef]
- Teng, S.-H.; Lee, E.-J.; Yoon, B.-H.; Shin, D.-S.; Kim, H.-E.; Oh, J.-S. Chitosan/nanohydroxyapatite composite membranes via dynamic filtration for guided bone regeneration. J. Biomed. Mater. Res. Part A 2009, 88A, 569–580. [Google Scholar] [CrossRef]
- Isikli, C.; Hasirci, V.; Hasirci, N. Development of porous chitosan-gelatin/hydroxyapatite composite scaffolds for hard tissue-engineering applications. J. Tissue Eng. Regen. Med. 2012, 6, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Gentleman, M.M.; Gentleman, E. The role of surface free energy in osteoblast–biomaterial interactions. Int. Mater. Rev. 2014, 59, 417–429. [Google Scholar] [CrossRef]
- Rhim, J.-W. Physical-Mechanical Properties of Agar/κ-Carrageenan Blend Film and Derived Clay Nanocomposite Film. J. Food Sci. 2012, 77, N66–N73. [Google Scholar] [CrossRef]
- Yegappan, R.; Selvaprithiviraj, V.; Amirthalingam, S.; Jayakumar, R. Carrageenan based hydrogels for drug delivery, tissue engineering and wound healing. Carbohydr. Polym. 2018, 198, 385–400. [Google Scholar] [CrossRef]
- Salgueiro, A.M.; Daniel-da-Silva, A.L.; Fateixa, S.; Trindade, T. κ-Carrageenan hydrogel nanocomposites with release behavior mediated by morphological distinct Au nanofillers. Carbohydr. Polym. 2013, 91, 100–109. [Google Scholar] [CrossRef]
- Ishizaki, T.; Saito, N.; Takai, O. Correlation of cell adhesive behaviors on superhydrophobic, superhydrophilic, and micropatterned superhydrophobic/superhydrophilic surfaces to their surface chemistry. Langmuir 2010, 26, 8147–8154. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.J.; Clegg, R.E.; Leavesley, D.I.; Pearcy, M.J. Mediation of biomaterial-cell interactions by adsorbed proteins: A review. Tissue Eng. 2005, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Rupp, F.; Scheideler, L.; Olshanska, N.; de Wild, M.; Wieland, M.; Geis-Gerstorfer, J. Enhancing surface free energy and hydrophilicity through chemical modification of microstructured titanium implant surfaces. J. Biomed. Mater. Res. Part A 2006, 76A, 323–334. [Google Scholar] [CrossRef] [PubMed]
- López-Pérez, P.M.; Marques, A.P.; da Silva, R.M.P.; Pashkuleva, I.; Reis, R.L. Effect of chitosan membrane surface modification via plasma induced polymerization on the adhesion of osteoblast-like cells. J. Mater. Chem. 2007, 17, 4064. [Google Scholar] [CrossRef] [Green Version]
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12. [Google Scholar] [CrossRef]
- Lim Soo, P.; Cho, J.; Grant, J.; Ho, E.; Piquette-Miller, M.; Allen, C. Drug release mechanism of paclitaxel from a chitosan–lipid implant system: Effect of swelling, degradation and morphology. Eur. J. Pharm. Biopharm. 2008, 69, 149–157. [Google Scholar] [CrossRef]
- Khoshakhlagh, P.; Rabiee, S.M.; Kiaee, G.; Heidari, P.; Miri, A.K.; Moradi, R.; Moztarzadeh, F.; Ravarian, R. Development and characterization of a bioglass/chitosan composite as an injectable bone substitute. Carbohydr. Polym. 2017, 157, 1261–1271. [Google Scholar] [CrossRef]
- González Ocampo, J.I.; Bassous, N.; Ossa Orozco, C.P.; Webster, T.J. Evaluation of cytotoxicity and antimicrobial activity of an injectable bone substitute of carrageenan and nano hydroxyapatite. J. Biomed. Mater. Res. Part A 2018, 106, 2984–2993. [Google Scholar] [CrossRef]
- Franci, G.; Falanga, A.; Galdiero, S.; Palomba, L.; Rai, M.; Morelli, G.; Galdiero, M. Silver Nanoparticles as Potential Antibacterial Agents. Molecules 2015, 20, 8856–8874. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zheng, Y.; Song, W.; Luan, J.; Wen, X.; Wu, Z.; Chen, X.; Wang, Q.; Guo, S. In situ synthesis of silver-nanoparticles/bacterial cellulose composites for slow-released antimicrobial wound dressing. Carbohydr. Polym. 2014, 102, 762–771. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Membrane/Components | HC | Cht | κ-carr | HAp In Situ | SHAp | |
|---|---|---|---|---|---|---|
| Control | (HC + Cht) | ✓ | ✓ | 0 | 0 | 0 |
| (HC + κ-carr) | ✓ | 0 | ✓ | 0 | 0 | |
| Methodology 1 | (HC + Cht)_HAp | ✓ | ✓ | 0 | ✓ | 0 |
| (HC + κ-carr)_HAp | ✓ | 0 | ✓ | ✓ | 0 | |
| Methodology 2 | (HC + Cht)_SHAp | ✓ | ✓ | 0 | 0 | ✓ |
| (HC + κ-carr)_SHAp | ✓ | 0 | ✓ | 0 | ✓ | |
| Membrane | θwater | γS (mJ.m−2) | γSP (mJ.m−2) | γSD (mJ.m−2) | Water Absorption (%Wa) |
|---|---|---|---|---|---|
| (HC + Cht) | 72.38 ± 8.16 | 36.12 ± 8.45 | 9.57 ± 5.24 | 26.56 ± 6.63 | 143.5 ± 0.6 |
| (HC + Cht)_HAp | 17.85 ± 1.79 | 70.47 ± 1.71 | 61.04 ± 1.6 | 9.43 ± 0.58 | 1342.2 ± 0.8 |
| (HC + Cht)_SHAp | 53.62 ± 3.19 | 53.9 ± 5.94 | 46.47 ± 5.04 | 7.02 ± 2.42 | 160.1 ± 0.3 |
| (HC + κ-carr) | 58.99 ± 7.66 | 36.73 ± 8.22 | 15.96 ± 6.16 | 20.77 ± 5.44 | 611.4 ± 0.5 |
| (HC + κ-carr)_HAp | 17.58 ± 5.35 | 68.87 ± 3.8 | 56.33 ± 3.6 | 12.54 ± 1.23 | 616.5 ± 0.7 |
| (HC + κ-carr)_SHAp | 23.58 ± 5.89 | 65.21 ± 5.24 | 49.41 ± 4.83 | 15.8 ± 2.04 | 733.9 ± 0.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nogueira, L.F.B.; Eufrásio Cruz, M.A.; Aguilar, G.J.; Tapia-Blácido, D.R.; da Silva Ferreira, M.E.; Maniglia, B.C.; Bottini, M.; Ciancaglini, P.; Ramos, A.P. Synthesis of Antibacterial Hybrid Hydroxyapatite/Collagen/Polysaccharide Bioactive Membranes and Their Effect on Osteoblast Culture. Int. J. Mol. Sci. 2022, 23, 7277. https://doi.org/10.3390/ijms23137277
Nogueira LFB, Eufrásio Cruz MA, Aguilar GJ, Tapia-Blácido DR, da Silva Ferreira ME, Maniglia BC, Bottini M, Ciancaglini P, Ramos AP. Synthesis of Antibacterial Hybrid Hydroxyapatite/Collagen/Polysaccharide Bioactive Membranes and Their Effect on Osteoblast Culture. International Journal of Molecular Sciences. 2022; 23(13):7277. https://doi.org/10.3390/ijms23137277
Chicago/Turabian StyleNogueira, Lucas Fabrício Bahia, Marcos Antônio Eufrásio Cruz, Guilherme José Aguilar, Delia Rita Tapia-Blácido, Márcia Eliana da Silva Ferreira, Bianca Chieregato Maniglia, Massimo Bottini, Pietro Ciancaglini, and Ana Paula Ramos. 2022. "Synthesis of Antibacterial Hybrid Hydroxyapatite/Collagen/Polysaccharide Bioactive Membranes and Their Effect on Osteoblast Culture" International Journal of Molecular Sciences 23, no. 13: 7277. https://doi.org/10.3390/ijms23137277
APA StyleNogueira, L. F. B., Eufrásio Cruz, M. A., Aguilar, G. J., Tapia-Blácido, D. R., da Silva Ferreira, M. E., Maniglia, B. C., Bottini, M., Ciancaglini, P., & Ramos, A. P. (2022). Synthesis of Antibacterial Hybrid Hydroxyapatite/Collagen/Polysaccharide Bioactive Membranes and Their Effect on Osteoblast Culture. International Journal of Molecular Sciences, 23(13), 7277. https://doi.org/10.3390/ijms23137277