
Indirect Enantioseparations: Recent Advances in Chiral Metabolomics for Biomedical Research


 ,
, 
Abstract
:1. Introduction
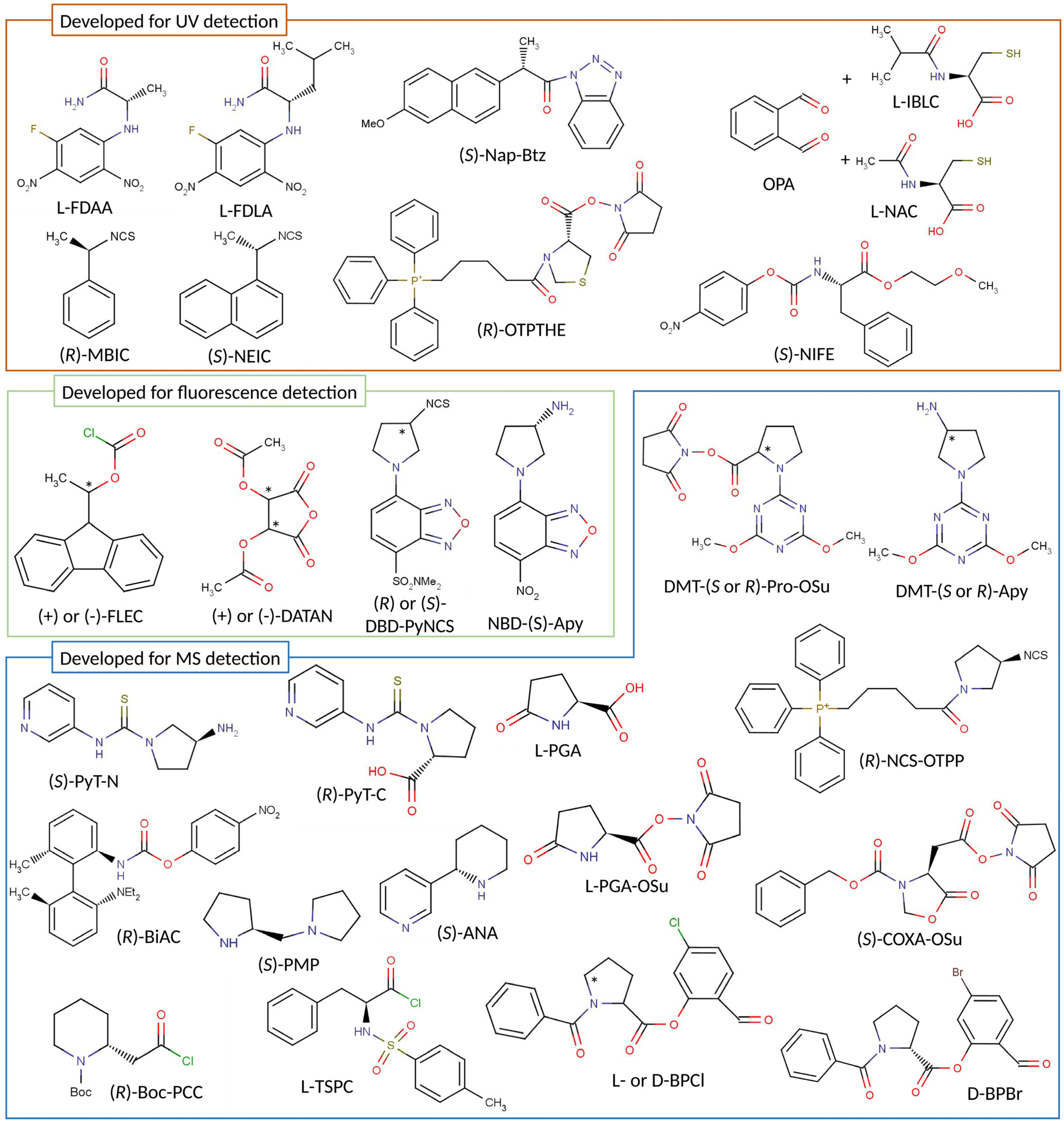
2. Advances in Chiral Derivatization
2.1. Advances and Improvements in Chiral Derivatization

{kind=link}
{kind=link}
{kind=link}
| CDA | Name | Derivatization Moiety | Derivatization Conditions | Commercially Available | Ref. |
|---|---|---|---|---|---|
| L-FDAA | 1-Fluoro-2,4-dinitrophenyl-5-L-alanineamide | Amines | ▪ Alkaline pH (TEA, NaHCO3), 24 h incubation for yield > 95% | Yes | [41,42] |
| α-Hydroxy acids | ▪ In presence of NaH (60% dispersed in oil); Sample in THF | ||||
| OPA | o-Phthalaldehyde/chiral thiols | Primary amines | ▪ Alkaline pH (sodium tetraborate, NaOH) ▪ Chiral thiols: isobuteryl-L-cysteine or N-acetyl-L-cysteine | Yes | [48,49] |
| (+) or (−)-FLEC | (+) or (−)-1-(9-Fluorenyl)ethyl chloroformate | Primary and secondary amines | ▪ Alkaline pH (sodium tetraborate) ▪ FLEC in acetone or ACN ▪ Excess reagent of at least 1:10 will ensure quantitative reaction | Yes | [38] |
| Thiols | |||||
| (S)-NIFE | N-(4-nitrophenoxycarbonyl)-L-phenylalanine 2-methoxyethyl ester | Primary and secondary amines | ▪ Alkaline pH (sodium tetraborate, TEA) ▪ CDA in acetone or ACN ▪ Excess reagent of at least 1:10 will ensure quantitative reaction | Yes | [50] |
| Thiols | |||||
| Phenols | |||||
| (R/S)-DBD-PyNCS | ((R/S)-4-(3- isothiocyanatopyrrolidin-1-yl)-7-(N,N-dimethylaminosulfonyl)-2,1,3-benzoxadiazole | Primary and secondary amines | ▪ In presence of TEA or DMAP, CDA dissolved in ACN | Yes | [32,51] |
| Carboxylic acids | ▪ In aprotic media, using condensation agents | ||||
| NBD-(S)-APy | (S)(+)-4-Nitro-7-(3-aminopyrrolidin-1-yl)-2,1,3-benzoxadiazole | Primary and secondary amines | ▪ In presence of TEA or DMAP, CDA dissolved in ACN | No | [32,51] |
| Carboxylic acids | ▪ In aprotic media, using condensation agents | ||||
| (+)- or (−)-DATAN | (+) or (−)-Diacetyl-L-tartaric anhydride | Amines | ▪ Reacts in aprotic media (CH2Cl2:acetic acid–4:1) | Yes | [52,53] |
| Hydroxyls | |||||
| DMT-3(S or R)-Apy | (S)-1-(4,6-dimethoxy-1,3,5-triazin-2-yl)pyrrolidin-3-amine | Carboxylic acids | ▪ In presence of activation reagents (TPP and DPDS) | No | [54] |
| DMT-1(S or R)-Apy | (S)-1-(4,6-dimethoxy-1,3,5-triazin-2-yl)pyrrolidin-1-amine | Carboxylic acids | ▪ In presence of activation reagents (TPP and DPDS) | No | [54] |
| DMT-(S or R)-Pro-OSu) | ((S)-2,5-dioxopyrrolidin-1-yl-1-(4,6-dimethoxy- 1,3,5-triazin-2-yl) pyrrolidine-2-carboxylate | Amines | ▪ CDA in ACN, in presence of TEA ▪ Room temperature, 40 min | No | [55] |
| (R)-NCS-OTPP | (R)-(5-(3-isothiocyanatopyrrolidin-1-yl)-5-oxopentyl) triphenylphosphonium | Thiols | ▪ In presence of TEA | No | [56,57] |
| (S)-COXA-OSu | (3-[(Benzoyloxy)carbonyl]-5-oxo-1,3-oxazolidin-4-yl)acetate | Amines | ▪ In PBS (100 mM) prepared in ACN | No | [58] |
| L-PGA | L-Pyroglutamic acid | Primary and secondary amines | ▪ In presence of activators (EDC/HOBt) | Yes | [59] |
| L-PGA-OSu | L-Pyroglutamic acid succinidimyl ester | Amines | ▪ CDA in ACN with TEA; sample in ACN | Yes | [60] |
| (R)-BiAC | (R)-4-nitrophenyl N-[2′-(dimethylamino)-6,6′-dimehyl-[1,1′-biphenyl]-2-yl] carbamate | Amines | ▪ In borate buffer (pH 8.8) diluted with ACN | No | [61] |
| (R)-OTPTHE | N-[1-oxo-5-(triphenylphosphonium)pentyl]-(R)-1,3-thiazolidinyl-4-N-hydroxysuccinimide ester bromide salt | Amines | ▪ In ACN containing borate buffer ▪ 60 °C, 30 min | No | [62] |
| D-BPBr | 1-Benzoyl-pyrrolidine-2-carboxylic acid 5-bromo-2-formyl-phenyl ester | Primary amines | ▪ In H2O/ACN solution containing 0.05M PBS | No | [63,64] |
| L- and D-BPCl, | 1-Benzoyl-pyrrolidine-2-carboxylic acid 5-chloro-2-formyl-phenyl ester | Amines | ▪ In H2O/ACN solution containing 0.05 M PBS | No | [63,64] |
| (R)-Boc-PCC | (R)-1-Boc-2-piperidine carbonyl chloride | Amines | ▪ In aqueous solution mixed with acetone. | No | [65] |
| L-TSPC | N-(p-toluenesulfonyl)-L-phenylalanine chloride | Amines | ▪ In presence of TEA or Py | Yes | [66,67] |
| Hydroxyl | ▪ Selective derivatization of hydroxy in anhydrous ACN with Py; 25 °C, 10 min | ||||
| (S)-PMP | (S)(+)-1-(2-pyrrolidinylmethyl)-pyrrolidine | Carboxylic acids | ▪ In presence of activation reagents (TPP and DPDS) | Yes | [23] |
| (S)-Nap-Btz | (S)-naproxen-benzotriazole | Amines | ▪ CDA in ACN; sample in NaHCO3 ▪ In presence of TEA; microwave derivatization (45 s, 600 W) | No | [68] |
| (R)-MBIC | Benzyl-isothiocyanate | Amines | ▪ Sample in NaHCO3 | Yes | [69] |
| (S)-NEIC | Naphtyl-isothiocyanate | Amines | ▪ Sample in NaHCO3; microwave derivatization (60 s, 600 W) | Yes | [69] |
| (S)-ANA | (S)-anabasine | Carboxylic acids | ▪ In presence of condensation agents (DMT-MM) | Yes | [70] |
2.2. New CDAs for UV or FL Detection
2.3. New CDAs for MS Detection
3. Untargeted Methodologies for Chiral Metabolomics
4. Targeted Analysis of Chiral Metabolites
4.1. Approaches Applicable to Chiral Profiling of Proteinogenic Amino Acids
4.2. Targeted Analysis Methods for Amino Acids
4.3. Targeted Analysis Methods for α-hydroxy Acids
5. Diastereomers Discrimination by Ion Mobility
5.1. Enantiopure AAs Complexed with Divalent Metal Ions as Chiral Selectors
5.2. Tert-Butoxycarbonyl Modified Amino Acid as Chiral Selector
5.3. Separation of Diastereomers after CDA Labeling
6. Conclusions and Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hesaka, A.; Sakai, S.; Hamase, K.; Ikeda, T.; Matsui, R.; Mita, M.; Horio, M.; Isaka, Y.; Kimura, T. D-Serine reflects kidney function and diseases. Sci. Rep. 2019, 9, 5104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastings, J.J.A.J.; van Eijk, H.M.; Damink, S.W.O.; Rensen, S.S. D-amino acids in health and disease: A focus on cancer. Nutrients 2019, 11, 2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, A.; Nishikawa, T.; Hayashi, T.; Fujii, N.; Harada, K.; Oka, T.; Takahashi, K. The presence of free D-serine in rat brain. FEBS Lett. 1992, 296, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Errico, F.; Mothet, J.P.; Usiello, A. D-Aspartate: An endogenous NMDA receptor agonist enriched in the developing brain with potential involvement in schizophrenia. J. Pharm. Biomed. Anal. 2014, 116, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K. Emerging role of glutamate in the pathophysiology of major depressive disorder. Brain Res. Rev. 2009, 61, 105–123. [Google Scholar] [CrossRef]
- Hashimoto, K.; Yoshida, T.; Ishikawa, M.; Fujita, Y.; Niitsu, T.; Nakazato, M.; Watanabe, H.; Sasaki, T.; Shiina, A.; Hashimoto, T.; et al. Increased serum levels of serine enantiomers in patients with depression. Acta Neuropsychiatr. 2016, 28, 173–178. [Google Scholar] [CrossRef]
- Hashimoto, K.; Fukushima, T.; Shimizu, E.; Okada, S.I.; Komatsu, N.; Okamura, N.; Koike, K.; Koizumi, H.; Kumakiri, C.; Imai, K.; et al. Possible role of D-serine in the pathophysiology of Alzheimer’s disease. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2004, 28, 385–388. [Google Scholar] [CrossRef]
- Mitchell, J.; Paul, P.; Chen, H.-J.; Morris, A.; Payling, M.; Falchi, M.; Habgood, J.; Panoutsou, S.; Winkler, S.; Tisato, V.; et al. Familial amyotrophic lateral sclerosis is associated with a mutation in D-amino acid oxidase. Proc. Natl. Acad. Sci. USA 2010, 107, 7556–7561. [Google Scholar] [CrossRef] [Green Version]
- Sasabe, J.; Miyoshi, Y.; Suzuki, M.; Mita, M.; Konno, R.; Matsuoka, M.; Hamase, K.; Aiso, S. D-Amino acid oxidase controls motoneuron degeneration through D-serine. Proc. Natl. Acad. Sci. USA 2012, 109, 627–632. [Google Scholar] [CrossRef] [Green Version]
- MacKay, M.A.B.; Kravtsenyuk, M.; Thomas, R.; Mitchell, N.D.; Dursun, S.M.; Baker, G.B. D-serine: Potential therapeutic agent and/or biomarker in schizophrenia and depression? Front. Psychiatry 2019, 10, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Wolosker, H.; Balu, D.T. D-Serine as the gatekeeper of NMDA receptor activity: Implications for the pharmacologic management of anxiety disorders. Transl. Psychiatry 2020, 10, 184. [Google Scholar] [CrossRef] [PubMed]
- Hooi, M.Y.S.; Truscott, R.J.W. Racemisation and human cataract. D-Ser, D-Asp/Asn and D-Thr are higher in the lifelong proteins of cataract lenses than in age-matched normal lenses. Age 2011, 33, 131–141. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Hesaka, A.; Isaka, Y. D-Amino acids and kidney diseases. Clin. Exp. Nephrol. 2020, 24, 404–410. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Hesaka, A.; Isaka, Y. Utility of D-serine monitoring in kidney disease. Biochim. Biophys. Acta-Proteins Proteom. 2020, 1868, 140449. [Google Scholar] [CrossRef]
- Okushima, H.; Iwata, Y.; Hesaka, A.; Sugimori, E.; Ikeda, T.; Nakane, M.; Mita, M.; Hayashi, T.; Isaka, Y.; Kimura, T. Intra-body dynamics of D-serine reflects the origin of kidney diseases. Clin. Exp. Nephrol. 2021, 25, 893–901. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Hamase, K.; Miyoshi, Y.; Yamamoto, R.; Yasuda, K.; Mita, M.; Rakugi, H.; Hayashi, T.; Isaka, Y. Chiral amino acid metabolomics for novel biomarker screening in the prognosis of chronic kidney disease. Sci. Rep. 2016, 6, 26137. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Sato, T.; Enomoto, N.; Ishii, Y.; Sasaki, K.; Yamada, T. High concentrations of D-amino acids in human gastric juice. Amino Acids 2007, 32, 137–140. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Xie, M.; Han, J.; Yuan, D.; Yang, T.; Xie, Y. Development and validation of a rapid, selective, and sensitive LC–MS/MS method for simultaneous determination of D- and L-amino acids in human serum: Application to the study of hepatocellular carcinoma. Anal. Bioanal. Chem. 2018, 410, 2517–2531. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Wang, Y.; Alatrash, N.; Weatherly, C.A.; Roy, D.; MacDonnell, F.M.; Armstrong, D.W. Altered profiles and metabolism of L- and D-amino acids in cultured human breast cancer cells vs. non-tumorigenic human breast epithelial cells. J. Pharm. Biomed. Anal. 2019, 164, 421–429. [Google Scholar] [CrossRef]
- Abdulbagi, M.; Wang, L.; Siddig, O.; Di, B.; Li, B. D-amino acids and D-amino acid-containing peptides: Potential disease biomarkers and therapeutic targets? Biomolecules 2021, 11, 1716. [Google Scholar] [CrossRef]
- Fujii, N.; Takata, T.; Fujii, N.; Aki, K.; Sakaue, H. D-Amino acids in protein: The mirror of life as a molecular index of aging. Biochim. Biophys. Acta-Proteins Proteom. 2018, 1866, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Numako, M.; Takayama, T.; Noge, I.; Kitagawa, Y.; Todoroki, K. Dried Saliva Spot (DSS) as a Convenient and Reliable Sampling for Bioanalysis: An Application for the Diagnosis of Diabetes Mellitus. Anal. Chem. 2016, 88, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Mochizuki, T.; Maeda, T.; Noge, I.; Kitagawa, Y.; Min, J.Z.; Todoroki, K.; Inoue, K.; Toyo, T. Simultaneous determination of DL-lactic acid and DL-3-hydroxybutyric acid enantiomers in saliva of diabetes mellitus patients by high-throughput LC-ESI-MS/MS. Anal. Bioanal. Chem. 2012, 404, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Numako, M.; Toyo’oka, T.; Noge, I.; Kitagawa, Y.; Mizuno, H.; Todoroki, K. Risk assessment of diabetes mellitus using dried saliva spot followed by ultra-performance liquid chromatography with fluorescence and mass spectrometry. Microchem. J. 2018, 142, 202–207. [Google Scholar] [CrossRef]
- Scheijen, J.L.J.M.; Hanssen, N.M.J.; Van De Waarenburg, M.P.H.; Jonkers, D.M.A.E.; Stehouwer, C.D.A.; Schalkwijk, C.G. L(+) and D(−) lactate are increased in plasma and urine samples of type 2 diabetes as measured by a simultaneous quantification of L(+) and D(−) lactate by reversed-phase liquid chromatography tandem mass spectrometry. Exp. Diabetes Res. 2012, 2012, 234812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, H.W.; Nejad, R.; Zhang, W.; Nassiri, F.; Mason, W.; Aldape, K.D.; Zadeh, G.; Chen, E.X. Tissue 2-hydroxyglutarate as a biomarker for isocitrate dehydrogenase mutations in gliomas. Clin. Cancer Res. 2019, 25, 3366–3373. [Google Scholar] [CrossRef] [Green Version]
- Mishra, P.; Tang, W.; Putluri, V.; Dorsey, T.H.; Jin, F.; Wang, F.; Zhu, D.; Amable, L.; Deng, T.; Zhang, S.; et al. ADHFE1 is a breast cancer oncogene and induces metabolic reprogramming. J. Clin. Investig. 2018, 128, 323–340. [Google Scholar] [CrossRef] [Green Version]
- DiNardo, C.D.; Propert, K.J.; Loren, A.W.; Paietta, E.; Sun, Z.; Levine, R.L.; Straley, K.S.; Yen, K.; Patel, J.P.; Agresta, S.; et al. Serum 2-hydroxyglutarate levels predict isocitrate dehydrogenase mutations and clinical outcome in acute myeloid leukemia. Blood 2013, 121, 4917–4924. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Dolowy, M. Application of TLC, HPLC and GC methods to the study of amino acid and peptide enantiomers: A review. Biomed. Chromatogr. 2013, 28, 84–101. [Google Scholar] [CrossRef]
- Ta, H.Y.; Collin, F.; Perquis, L.; Poinsot, V.; Ong-Meang, V.; Couderc, F. Twenty years of amino acid determination using capillary electrophoresis: A review. Anal. Chim. Acta 2021, 1174, 338233. [Google Scholar] [CrossRef]
- Toyo’oka, T. Chiral benzofurazan-derived derivatization reagents for indirect enantioseparations by HPLC. In Chiral Separations: Methods and Protocols; Scriba, G.K.E., Ed.; Humana Press: Totowa, NJ, USA, 2013; Volume 970, pp. 233–248. ISBN 9781627032629. [Google Scholar]
- Furman, C.; Howsam, M.; Lipka, E. Recent developments in separation methods for enantiomeric ratio determination of amino acids specifically involved in cataract and Alzheimer’s disease. TrAC-Trends Anal. Chem. 2021, 141, 116287. [Google Scholar] [CrossRef]
- Ilisz, I.; Berkecz, R.; Péter, A. Application of chiral derivatizing agents in the high-performance liquid chromatographic separation of amino acid enantiomers: A review. J. Pharm. Biomed. Anal. 2008, 47, 1–15. [Google Scholar] [CrossRef]
- Ilisz, I.; Aranyi, A.; Pataj, Z.; Péter, A. Recent advances in the direct and indirect liquid chromatographic enantioseparation of amino acids and related compounds: A review. J. Pharm. Biomed. Anal. 2012, 69, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, Y.; Koga, R.; Oyama, T.; Han, H.; Ueno, K.; Masuyama, K.; Itoh, Y.; Hamase, K. HPLC analysis of naturally occurring free D-amino acids in mammals. J. Pharm. Biomed. Anal. 2012, 69, 42–49. [Google Scholar] [CrossRef]
- Szökő, É.; Vincze, I.; Tábi, T. Chiral separations for D-amino acid analysis in biological samples. J. Pharm. Biomed. Anal. 2016, 130, 100–109. [Google Scholar] [CrossRef]
- Moldovan, R.C.; Bodoki, E.; Servais, A.C.; Crommen, J.; Oprean, R.; Fillet, M. (+) or (−)-1-(9-fluorenyl)ethyl chloroformate as chiral derivatizing agent: A review. J. Chromatogr. A 2017, 1513, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Calderón, C.; Lämmerhofer, M. Enantioselective metabolomics by liquid chromatography-mass spectrometry. J. Pharm. Biomed. Anal. 2022, 207, 114430. [Google Scholar] [CrossRef] [PubMed]
- Marfey, P. Determination of D-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 1984, 49, 591–596. [Google Scholar] [CrossRef] [Green Version]
- Bhushan, R.; Brückner, H. Marfey’s reagent for chiral amino acid analysis: A review. Amino Acids 2004, 27, 231–247. [Google Scholar] [CrossRef]
- Sethi, S.; Martens, J.; Bhushan, R. Assessment and application of Marfey’s reagent and analogs in enantioseparation: A decade’s perspective. Biomed. Chromatogr. 2021, 35, e4990. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.; Lim, C.; Kim, S.; Oh, D.C. Facile determination of the absolute configurations of α-hydroxy acids by chiral derivatization coupled with liquid chromatography-mass spectrometry analysis. J. Chromatogr. A 2013, 1272, 141–144. [Google Scholar] [CrossRef] [PubMed]
- Ayon, N.J.; Sharma, A.D.; Gutheil, W.G. LC-MS/MS-based separation and quantification of Marfey’s reagent derivatized proteinogenic amino acid DL-stereoisomers. J. Am. Soc. Mass Spectrom. 2019, 30, 448–458. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Ikai, Y.; Mayumi, T.; Oka, H.; Suzuki, M.; Harada, K. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: Elucidation of limitations of Marfey’s method and of its separation mechanism. Anal. Chem. 1997, 69, 3346–3352. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yaku, K.; Nakagawa, T. Simultaneous measurement of amino acid enantiomers in aged mouse brain samples by LC/MS/MS combined with derivatization using Nα-(5-fluoro-2,4-dinitrophenyl)-L-leucinamide (L-FDLA). Metabolites 2021, 11, 57. [Google Scholar] [CrossRef]
- Kobayashi, M.; Takano, Y.; Takishima, H.; Sakaitani, S.; Niitsu, M.; Furuchi, T. Simplification of FDLA pre-column derivatization for LC/MS/MS toward separation and detection of D,L-amino acids. Chromatographia 2019, 82, 705–708. [Google Scholar] [CrossRef]
- Molnár-Perl, I. Advancement in the derivatizations of the amino groups with the o-phthaldehyde-thiol and with the 9-fluorenylmethyloxycarbonyl chloride reagents. J. Chromatogr. B 2011, 879, 1241–1269. [Google Scholar] [CrossRef]
- Hanczkó, R.; Jámbor, A.; Perl, A.; Molnár-Perl, I. Advances in the o-phthalaldehyde derivatizations. J. Chromatogr. A 2007, 1163, 25–42. [Google Scholar] [CrossRef]
- Visser, W.F.; Verhoeven-Duif, N.M.; Ophoff, R.; Bakker, S.; Klomp, L.W.; Berger, R.; de Koning, T.J. A sensitive and simple ultra-high-performance-liquid chromatography–tandem mass spectrometry based method for the quantification of D-amino acids in body fluids. J. Chromatogr. A 2011, 1218, 7130–7136. [Google Scholar] [CrossRef]
- Toyo’oka, T. Diagnostic approach to disease using non-invasive samples based on derivatization and LC-ESI-MS/MS. Biol. Pharm. Bull. 2016, 39, 1397–1411. [Google Scholar] [CrossRef] [Green Version]
- Struys, E.A.; Jansen, E.E.W.; Verhoeven, N.M.; Jakobs, C. Measurement of urinary D- and L-2-hydroxyglutarate enantiomers by stable-isotope-dilution liquid chromatography-tandem mass spectrometry after derivatization with diacetly-L-tartaric anhydride. Clin. Chem. 2004, 50, 1391–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poinsignon, V.; Mercier, L.; Nakabayashi, K.; David, M.D.; Lalli, A.; Penard- Lacronique, V.; Quivoron, C.; Saada, V.; De Botton, S.; Broutin, S.; et al. Quantitation of isocitrate dehydrogenase (IDH)-induced D and L enantiomers of 2-hydroxyglutaric acid in biological fluids by a fully validated liquid tandem mass spectrometry method, suitable for clinical applications. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2016, 1022, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Kuwabara, T.; Maeda, T.; Noge, I.; Kitagawa, Y.; Inoue, K.; Todoroki, K.; Min, J.Z.; Toyo’oka, T. Profiling of chiral and achiral carboxylic acid metabolomics: Synthesis and evaluation of triazine-type chiral derivatization reagents for carboxylic acids by LC-ESI-MS/MS and the application to saliva of healthy volunteers and diabetic patients. Anal. Bioanal. Chem. 2015, 407, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, T.; Takayama, T.; Todoroki, K.; Inoue, K.; Min, J.Z.; Toyo’oka, T. Towards the chiral metabolomics: Liquid chromatography-mass spectrometry based DL-amino acid analysis after labeling with a new chiral reagent, (S)-2,5-dioxopyrrolidin-1-yl-1-(4,6-dimethoxy-1,3,5-triazin-2-yl)pyrrolidine-2-carboxylate, and the application. Anal. Chim. Acta 2015, 875, 73–82. [Google Scholar] [CrossRef]
- Ma, Q.; Qi, C.; Li, X.L.; Shi, Q.; Xu, C.Y.; Jin, T.; Min, J.Z. Simultaneous determination of DL-cysteine, DL-homocysteine, and glutathione in saliva and urine by UHPLC-Q-Orbitrap HRMS: Application to studies of oxidative stress. J. Pharm. Biomed. Anal. 2021, 196, 113939. [Google Scholar] [CrossRef]
- Ma, Q.; Man, X.; Xu, C.Y.; Huo, J.; Qi, C.; Shi, Q.; Nan, J.; Min, J.Z. Simultaneous determination of three endogenous chiral thiol compounds in serum from humans at normal and stress states using ultrahigh-performance liquid chromatography coupled to quadrupole-Orbitrap high resolution mass spectrometry. J. Chromatogr. A 2021, 1642, 462028. [Google Scholar] [CrossRef]
- Sakamoto, T.; Furukawa, S.; Nishizawa, T.; Fukuda, M.; Sasaki, M.; Onozato, M.; Uekusa, S.; Ichiba, H.; Fukushima, T. Succinimidyl (3-[(benzyloxy)carbonyl]-5-oxo-1,3-oxazolidin-4-yl)acetate on a triazole-bonded phase for the separation of DL-amino-acid enantiomers and the mass-spectrometric determination of chiral amino acids in rat plasma. J. Chromatogr. A 2019, 1585, 131–137. [Google Scholar] [CrossRef]
- Mochizuki, T.; Taniguchi, S.; Tsutsui, H.; Min, J.Z.; Inoue, K.; Todoroki, K.; Toyo’oka, T. Relative quantification of enantiomers of chiral amines by high-throughput LC-ESI-MS/MS using isotopic variants of light and heavy L-pyroglutamic acids as the derivatization reagents. Anal. Chim. Acta 2013, 773, 76–82. [Google Scholar] [CrossRef]
- Mochizuki, T.; Todoroki, K.; Inoue, K.; Min, J.Z.; Toyo’oka, T. Isotopic variants of light and heavy L-pyroglutamic acid succinimidyl esters as the derivatization reagents for DL-amino acid chiral metabolomics identification by liquid chromatography and electrospray ionization mass spectrometry. Anal. Chim. Acta 2014, 811, 51–59. [Google Scholar] [CrossRef]
- Harada, M.; Karakawa, S.; Yamada, N.; Miyano, H.; Shimbo, K. Biaryl axially chiral derivatizing agent for simultaneous separation and sensitive detection of proteinogenic amino acid enantiomers using liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2019, 1593, 91–101. [Google Scholar] [CrossRef]
- Han, Y.; Jin, M.N.; Xu, C.Y.; Qian, Q.; Nan, J.; Jin, T.; Min, J.Z. Evaluation of chiral separation efficiency of a novel OTPTHE derivatization reagent: Applications to liquid-chromatographic determination of DL-serine in human plasma. Chirality 2019, 31, 1043–1052. [Google Scholar] [CrossRef]
- Shen, K.; Wang, L.; He, Q.; Jin, Z.; Chen, W.; Sun, C.; Pan, Y. Sensitive bromine-labeled probe D-BPBr for simultaneous identification and quantification of chiral amino acids and amino-containing metabolites profiling in human biofluid by HPLC/MS. Anal. Chem. 2020, 92, 1763–1769. [Google Scholar] [CrossRef]
- Huang, R.; Shen, K.; He, Q.; Hu, Y.; Sun, C.; Guo, C.; Pan, Y. Metabolic Profiling of Urinary Chiral Amino-Containing Biomarkers for Gastric Cancer Using a Sensitive Chiral Chlorine-Labeled Probe by HPLC-MS/MS. J. Proteome Res. 2021, 20, 3952–3962. [Google Scholar] [CrossRef]
- Xie, Y.; Alexander, G.M.; Schwartzman, R.J.; Singh, N.; Torjman, M.C.; Goldberg, M.E.; Wainer, I.W.; Moaddel, R. Development and validation of a sensitive LC-MS/MS method for the determination of D-serine in human plasma. J. Pharm. Biomed. Anal. 2014, 89, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.Y.; Xiong, J.; Huang, W.; Ma, Q.; Ci, W.; Feng, Y.Q.; Yuan, B.F. Sensitive Determination of Onco-metabolites of D-and L-2-hydroxyglutarate Enantiomers by Chiral Derivatization Combined with Liquid Chromatography/Mass Spectrometry Analysis. Sci. Rep. 2015, 5, 15217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.Y.; Jiang, X.; Zhou, J.L.; Shi, Z.Q.; Liu, L.F.; Xin, G.Z. A readily 16O-/18O-isotopically-paired chiral derivatization approach for the quantification of 2-HG metabolic panel by liquid chromatography-Tandem mass spectrometry. Anal. Chim. Acta 2019, 1077, 174–182. [Google Scholar] [CrossRef]
- Bhushan, R.; Nagar, H. Indirect enantioseparation of proteinogenic amino acids using naproxen-based chiral derivatizing reagent and HPLC. Biomed. Chromatogr. 2013, 27, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, R.; Dubey, R. Validated high-performance liquid chromatographic enantioseparation of selenomethionine using isothiocyanate based chiral derivatizing reagents. Biomed. Chromatogr. 2012, 26, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Kawasaki, M.; Tadokoro, H.; Ogawa, S.; Tsutsui, H.; Fukushima, T.; Toyo’oka, T. Derivatization of chiral carboxylic acids with (S)-anabasine for increasing detectability and enantiomeric separation in LC/ESI-MS/MS. J. Sep. Sci. 2012, 35, 2840–2846. [Google Scholar] [CrossRef]
- Müller, C.; Fonseca, J.R.; Rock, T.M.; Krauss-Etschmann, S.; Schmitt-Kopplin, P. Enantioseparation and selective detection of D-amino acids by ultra-high-performance liquid chromatography/mass spectrometry in analysis of complex biological samples. J. Chromatogr. A 2014, 1324, 109–114. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Chen, S.; Sun, F.; Luo, P.; Du, Q.; Zhao, S. Indirect chiral separation of tryptophan enantiomers by high performance liquid chromatography with indirect chemiluminiscence detection. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 1006, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Tokuda, M.; Amano, M.; Mikami, K. Simultaneous determination of primary and secondary D- and L-amino acids by reversed-phase high-performance liquid chromatography using pre-column derivatization with two-step labelling method. Biosci. Biotechnol. Biochem. 2017, 81, 1681–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Einarsson, S.; Josefsson, B.; Möller, P.; Sanchez, D. Separation of amino acid enantiomers and chiral amines using precolumn derivatization with (+)-1-(9-fluorenyl)ethyl chloroformate and reversed-phase liquid chromatography. Anal. Chem. 1987, 59, 1191–1195. [Google Scholar] [CrossRef]
- Moldovan, R.-C.; Bodoki, E.; Servais, A.-C.; Crommen, J.; Oprean, R.; Fillet, M. Selectivity evaluation of phenyl based stationary phases for the analysis of amino acid diastereomers by liquid chromatography coupled with mass spectrometry. J. Chromatogr. A 2019, 1590, 80–87. [Google Scholar] [CrossRef]
- Fradi, I.; Farcas, E.; Ben Saïd, A.; Yans, M.-L.; Lamalle, C.; Somsen, G.W.; Prior, A.; de Jong, G.J.; Kallel, M.; Crommen, J.; et al. In-capillary derivatization with (−)-1-(9-fluorenyl)ethyl chloroformate as chiral labeling agent for the electrophoretic separation of amino acids. J. Chromatogr. A 2014, 1363, 338–347. [Google Scholar] [CrossRef]
- Moldovan, R.-C.C.; Bodoki, E.; Kacsó, T.; Servais, A.-C.C.; Crommen, J.; Oprean, R.; Fillet, M. A micellar electrokinetic chromatography–mass spectrometry approach using in-capillary diastereomeric derivatization for fully automatized chiral analysis of amino acids. J. Chromatogr. A 2016, 1467, 400–408. [Google Scholar] [CrossRef]
- Prior, A.; Moldovan, R.-C.C.; Crommen, J.; Servais, A.-C.C.; Fillet, M.; de Jong, G.J.; Somsen, G.W. Enantioselective capillary electrophoresis-mass spectrometry of amino acids in cerebrospinal fluid using a chiral derivatizing agent and volatile surfactant. Anal. Chim. Acta 2016, 940, 150–158. [Google Scholar] [CrossRef]
- Prior, A.; van de Nieuwenhuijzen, E.; de Jong, G.J.; Somsen, G.W. Enantioselective micellar electrokinetic chromatography of DL-amino acids using (+)-1-(9-fluorenyl)-ethyl chloroformate derivatization and UV-induced fluorescence detection. J. Sep. Sci. 2018, 41, 2983–2992. [Google Scholar] [CrossRef] [Green Version]
- Moldovan, R.-C.C.; Bodoki, E.; Servais, A.-C.C.; Chankvetadze, B.; Crommen, J.; Oprean, R.; Fillet, M. Capillary electrophoresis-mass spectrometry of derivatized amino acids for targeted neurometabolomics—pH mediated reversal of diastereomer migration order. J. Chromatogr. A 2018, 1564, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Míguez, R.; Bruyneel, B.; Castro-Puyana, M.; Marina, M.L.; Somsen, G.W.; Domínguez-Vega, E. Chiral Discrimination of DL-Amino Acids by Trapped Ion Mobility Spectrometry after Derivatization with (+)-1-(9-Fluorenyl)ethyl Chloroformate. Anal. Chem. 2019, 91, 3277–3285. [Google Scholar] [CrossRef] [Green Version]
- Hess, S. A Universal HPLC-MS Method to Determine the Stereochemistry of Common and Unusual Amino Acids. In Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2019; Volume 2030, pp. 263–275. ISBN 9781493996391. [Google Scholar]
- Tian, H.; Zheng, N.; Li, S.; Zhang, Y.; Zhao, S.; Wen, F.; Wang, J. Characterization of chiral amino acids from different milk origins using ultra-performance liquid chromatography coupled to ion-mobility mass spectrometry. Sci. Rep. 2017, 7, 46289. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Luo, S.; Tian, R.; Zhang, W.; Sun, B.; Cui, Y. Analysis of endogenous epinephrine and norepinephrine enantiomers in rat plasma and application to a stereoselective pharmacokinetics. J. Pharm. Biomed. Anal. 2020, 177, 112859. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Li, X.; Guo, X.; Cui, Y. Simultaneous determination of 18 D-amino acids in rat plasma by an ultrahigh-performance liquid chromatography-tandem mass spectrometry method: Application to explore the potential relationship between Alzheimer’s disease and D-amino acid level alteration. Anal. Bioanal. Chem. 2016, 408, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Xing, Y.; Guo, X.; Cui, Y. Development of an UPLC–MS/MS method for simultaneous quantitation of 11 D-amino acids in different regions of rat brain: Application to a study on the associations of D-amino acid concentration changes and Alzheimer’s disease. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1058, 40–46. [Google Scholar] [CrossRef]
- Sakamoto, T.; Kuwabara, R.; Takahashi, S.; Onozato, M.; Ichiba, H.; Iizuka, H.; Fukushima, T. Determination of D-serine in human serum by LC-MS/MS using a triazole-bonded column after pre-column derivatization with (S)-4-(3-isothiocyanatopyrrolidin-1-yl)-7-(N, N-dimethylaminosulfonyl)-2,1,3-benzoxadiazole. Anal. Bioanal. Chem. 2016, 408, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Iizuka, H.; Kuwabara, R.; Naito, Y.; Sakamoto, T.; Miyagi, A.; Onozato, M.; Ichiba, H.; Fukushima, T. Determination of L-tryptophan and L-kynurenine derivatized with (R)-4-(3-isothiocyanatopyrrolidin-1-yl)-7-(N,N-dimethylaminosulfonyl)-2,1,3-benzoxadiazole by LC-MS/MS on a triazole-bonded column and their quantification in human serum. Biomed. Chromatogr. 2016, 30, 1481–1486. [Google Scholar] [CrossRef]
- Iizuka, H.; Harashima, T.; Takahashi, S.; Kuwabara, R.; Naito, Y.; Sakamoto, T.; Onozato, M.; Ichiba, H.; Fukushima, T. Chromatographic profiles of tryptophan and kynurenine enantiomers derivatized with (S)-4-(3-isothiocyanatopyrrolidin-1-yl)-7-(N,N-dimethylaminosulfonyl)-2,1,3-benzoxadiazole using LC–MS/MS on a triazole-bonded column. Chirality 2017, 29, 603–609. [Google Scholar] [CrossRef]
- Oldham, W.; Loscalzo, J. Quantification of 2-hydroxyglutarate enantiomers by liquid chromatography-mass spectrometry. Bio.-Protoc. 2016, 6, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Pandey, R.; Collins, M.; Lu, X.; Sweeney, S.R.; Chiou, J.; Lodi, A.; Tiziani, S. Novel strategy for untargeted chiral metabolomics using liquid chromatography-high resolution tandem mass spectrometry. Anal. Chem. 2021, 93, 5805–5814. [Google Scholar] [CrossRef]
- Rakheja, D.; Boriack, R.L.; Mitui, M.; Khokhar, S.; Holt, S.A.; Kapur, P. Papillary thyroid carcinoma shows elevated levels of 2-hydroxyglutarate. Tumor Biol. 2011, 32, 325–333. [Google Scholar] [CrossRef]
- Shim, E.H.; Livi, C.B.; Rakheja, D.; Tan, J.; Benson, D.; Parekh, V.; Kho, E.Y.; Ghosh, A.P.; Kirkman, R.; Velu, S.; et al. L-2-hydroxyglutarate: An epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 2014, 4, 1290–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldham, W.M.; Clish, C.B.; Yang, Y.; Loscalzo, J. Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 2015, 22, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Andrade, J.; Shi, C.; Costa, A.S.H.; Choi, J.; Kim, J.; Doddaballapur, A.; Sugino, T.; Ong, Y.T.; Castro, M.; Zimmermann, B.; et al. Control of endothelial quiescence by FOXO-regulated metabolites. Nat. Cell Biol. 2021, 23, 413–423. [Google Scholar] [CrossRef]
- Mason, S.; Reinecke, C.J.; Kulik, W.; van Cruchten, A.; Solomons, R.; van Furth, A.M.T. Cerebrospinal fluid in tuberculous meningitis exhibits only the L-enantiomer of lactic acid. BMC Infect. Dis. 2016, 16, 251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhushan, R.; Dixit, S. Application of cyanuric chloride-based six new chiral derivatizing reagents having amino acids and amino acid amides as chiral auxiliaries for enantioresolution of proteinogenic amino acids by reversed-phase high-performance liquid chromatography. Amino Acids 2012, 42, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Toyo’oka, T. Chiral metabolomics using triazine-based chiral labeling reagents by UPLC-ESI-MS/MS. In Chiral Separations: Methods and Protocols; Scriba, G.K.E., Ed.; Humana: New York, NY, USA, 2019; Volume 1985, pp. 57–79. ISBN 9781493994380. [Google Scholar]
- Mizuno, H.; Shindo, T.; Ito, K.; Sakane, I.; Miyazaki, Y.; Toyo’oka, T.; Todoroki, K. Development of a selective and sensitive analytical method to detect isomerized aspartic acid residues in crystallin using a combination of derivatization and liquid chromatography mass spectrometry. J. Chromatogr. A 2020, 1623, 461134. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Mochizuki, T.; Todoroki, K.; Min, J.Z.; Mizuno, H.; Inoue, K.; Akatsu, H.; Noge, I.; Toyo’oka, T. A novel approach for LC-MS/MS-based chiral metabolomics fingerprinting and chiral metabolomics extraction using a pair of enantiomers of chiral derivatization reagents. Anal. Chim. Acta 2015, 898, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Takayama, T.; Mizuno, H.; Toyo’oka, T.; Akatsu, H.; Inoue, K.; Todoroki, K. Isotope corrected chiral and achiral nontargeted metabolomics: An approach for high accuracy and precision metabolomics based on derivatization and its application to cerebrospinal fluid of patients with Alzheimer’s disease. Anal. Chem. 2019, 91, 4396–4404. [Google Scholar] [CrossRef]
- Fukui, S.; Sugiyama, E.; Mizuno, H.; Sakane, I.; Asakawa, D.; Saikusa, K.; Nishiya, Y.; Amano, Y.; Takahara, K.; Higo, D.; et al. Rapid chiral discrimination of oncometabolite DL-2-hydroxyglutaric acid using derivatization and field asymmetric waveform ion mobility spectrometry/mass spectrometry. J. Sep. Sci. 2021, 44, 3489–3496. [Google Scholar] [CrossRef]
- Nagao, R.; Tsutsui, H.; Mochizuki, T.; Takayama, T.; Kuwabara, T.; Min, J.Z.; Inoue, K.; Todoroki, K.; Toyo’oka, T. Novel chiral derivatization reagents possessing a pyridylthiourea structure for enantiospecific determination of amines and carboxylic acids in high-throughput liquid chromatography and electrospray-ionization mass spectrometry for chiral metabolomics ide. J. Chromatogr. A 2013, 1296, 111–118. [Google Scholar] [CrossRef]
- Kuwabara, T.; Takayama, T.; Todoroki, K.; Inoue, K.; Min, J.Z.; Toyo’oka, T. Evaluation of a series of prolylamidepyridines as the chiral derivatization reagents for enantioseparation of carboxylic acids by LC-ESI-MS/MS and the application to human saliva. Anal. Bioanal. Chem. 2014, 406, 2641–2649. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Karakawa, S.; Miyano, H.; Shimbo, K. Simultaneous analysis of D,L-amino acids in human urine using a chirality-switchable biaryl axial tag and liquid chromatography electrospray ionization tandem mass spectrometry. Symmetry 2020, 12, 913. [Google Scholar] [CrossRef]
- Cheng, Q.Y.; Xiong, J.; Wang, F.; Yuan, B.F.; Feng, Y.Q. Chiral derivatization coupled with liquid chromatography/mass spectrometry for determining ketone metabolites of hydroxybutyrate enantiomers. Chinese Chem. Lett. 2018, 29, 115–118. [Google Scholar] [CrossRef]
- Zhang, J.D.; Mohibul Kabir, K.M.; Lee, H.E.; Donald, W.A. Chiral recognition of amino acid enantiomers using high-definition differential ion mobility mass spectrometry. Int. J. Mass Spectrom. 2018, 428, 1–7. [Google Scholar] [CrossRef]
- Zhang, J.D.; Kabir, K.M.M.; Donald, W.A. Metal-ion free chiral analysis of amino acids as small as proline using high-definition differential ion mobility mass spectrometry. Anal. Chim. Acta 2018, 1036, 172–178. [Google Scholar] [CrossRef]
- Zhang, S.; Shi, J.; Shan, C.; Huang, C.; Wu, Y.; Ding, R.; Xue, Y.; Liu, W.; Zhou, Q.; Zhao, Y.; et al. Stable isotope N-phosphoryl amino acids labeling for quantitative profiling of amine-containing metabolites using liquid chromatography mass spectrometry. Anal. Chim. Acta 2017, 978, 24–34. [Google Scholar] [CrossRef]
- Min, J.Z.; Hatanaka, S.; Yu, H.F.; Higashi, T.; Inagaki, S.; Toyo’oka, T. Determination of DL-amino acids, derivatized with R(−)-4-(3-isothiocyanatopyrrolidin-1-yl)-7-(N,N-dimethylaminosulfonyl)-2,1,3-benzoxadiazole, in nail of diabetic patients by UPLC-ESI-TOF-MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2011, 879, 3220–3228. [Google Scholar] [CrossRef]
- McGarry, A.; Gaughan, J.; Hackmyer, C.; Lovett, J.; Khadeer, M.; Shaikh, H.; Pradhan, B.; Ferraro, T.N.; Wainer, I.W.; Moaddel, R. Cross-sectional analysis of plasma and CSF metabolomic markers in Huntington’s disease for participants of varying functional disability: A pilot study. Sci. Rep. 2020, 10, 20490. [Google Scholar] [CrossRef]
- Jones, P.M.; Boriack, R.; Struys, E.A.; Rakheja, D. Measurement of oncometabolites D-2-hydroxyglutaric acid and L-2-hydroxyglutaric acid. Methods Mol. Biol. 2017, 1633, 219–234. [Google Scholar]
- Uetrecht, C.; Rose, R.J.; van Duijn, E.; Lorenzen, K.; Heck, A.J.R. Ion mobility mass spectrometry of proteins and protein assemblies. Chem. Soc. Rev. 2010, 39, 1633–1655. [Google Scholar] [CrossRef]
- Lapthorn, C.; Pullen, F.; Chowdhry, B.Z. Ion mobility spectrometry-mass spectrometry (IMS-MS) of small molecules: Separating and assigning structures to ions. Mass Spectrom. Rev. 2013, 32, 43–71. [Google Scholar] [CrossRef] [Green Version]
- Paglia, G.; Smith, A.J.; Astarita, G. Ion mobility mass spectrometry in the omics era: Challenges and opportunities for metabolomics and lipidomics. Mass Spectrom. Rev. 2021; in press. [Google Scholar] [CrossRef]
- Richardson, K.; Langridge, D.; Giles, K. Fundamentals of travelling wave ion mobility revisited: I. Smoothly moving waves. Int. J. Mass Spectrom. 2018, 428, 71–80. [Google Scholar] [CrossRef]
- Zhang, J.D.; Mohibul Kabir, K.M.; Donald, W.A. Ion-Mobility Mass Spectrometry for Chiral Analysis of Small Molecules. In Comprehensive Analytical Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; Volume 83, pp. 51–81. [Google Scholar]
- Michelmann, K.; Silveira, J.A.; Ridgeway, M.E.; Park, M.A. Fundamentals of trapped ion mobility spectrometry. J. Am. Soc. Mass Spectrom. 2014, 26, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Swearingen, K.E.; Moritz, R.L. High-field asymmetric waveform ion mobility spectrometry for mass spectrometry-based proteomics. Expert Rev. Proteom. 2012, 9, 505–517. [Google Scholar] [CrossRef] [PubMed]
- Aksenov, A.A.; Kapron, J.; Davis, C.E. Predicting compensation voltage for singly-charged ions in high-field asymmetric waveform ion mobility spectrometry (FAIMS). J. Am. Soc. Mass Spectrom. 2012, 23, 1794–1798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodds, J.N.; May, J.C.; McLean, J.A. Correlating Resolving Power, Resolution, and Collision Cross Section: Unifying Cross-Platform Assessment of Separation Efficiency in Ion Mobility Spectrometry. Anal. Chem. 2017, 89, 12176–12184. [Google Scholar] [CrossRef] [Green Version]
- Domalain, V.; Hubert-Roux, M.; Tognetti, V.; Joubert, L.; Lange, C.M.; Rouden, J.; Afonso, C. Enantiomeric differentiation of aromatic amino acids using traveling wave ion mobility-mass spectrometry. Chem. Sci. 2014, 5, 3234–3239. [Google Scholar] [CrossRef]
- Mie, A.; Jörntén-Karlsson, M.; Axelsson, B.; Ray, A.; Reimann, C.T. Enantiomer Separation of Amino Acids by Complexation with Chiral Reference Compounds and High-Field Asymmetric Waveform Ion Mobility Spectrometry: Preliminary Results and Possible Limitations. Anal. Chem. 2007, 79, 2850–2858. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Yao, Z.P. Chiral differentiation of amino acids through binuclear copper bound tetramers by ion mobility mass spectrometry. Anal. Chim. Acta 2017, 981, 62–70. [Google Scholar] [CrossRef]
- Will, J.M.; Behrens, A.; Macke, M.; Quarles, C.D.; Karst, U. Automated Chiral Analysis of Amino Acids Based on Chiral Derivatization and Trapped Ion Mobility-Mass Spectrometry. Anal. Chem. 2021, 93, 878–885. [Google Scholar] [CrossRef]
- May, J.C.; Knochenmuss, R.; Fjeldsted, J.C.; McLean, J.A. Resolution of Isomeric Mixtures in Ion Mobility Using a Combined Demultiplexing and Peak Deconvolution Technique. Anal. Chem. 2020, 92, 9482–9492. [Google Scholar] [CrossRef] [PubMed]
- Belov, M.E.; Clowers, B.H.; Prior, D.C.; Danielson, W.F.; Liyu, A.V.; Petritis, B.O.; Smith, R.D. Dynamically multiplexed ion mobility time-of-flight mass spectrometry. Anal. Chem. 2008, 80, 5873–5883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demelenne, A.; Nys, G.; Nix, C.; Fjeldsted, J.C.; Crommen, J.; Fillet, M. Separation of phosphorothioated oligonucleotide diastereomers using multiplexed drift tube ion mobility mass spectrometry. Anal. Chim. Acta 2021, 1191, 339297. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogos, L.-G.; Pralea, I.-E.; Moldovan, R.-C.; Iuga, C.-A. Indirect Enantioseparations: Recent Advances in Chiral Metabolomics for Biomedical Research. Int. J. Mol. Sci. 2022, 23, 7428. https://doi.org/10.3390/ijms23137428
Bogos L-G, Pralea I-E, Moldovan R-C, Iuga C-A. Indirect Enantioseparations: Recent Advances in Chiral Metabolomics for Biomedical Research. International Journal of Molecular Sciences. 2022; 23(13):7428. https://doi.org/10.3390/ijms23137428
Chicago/Turabian StyleBogos, Luisa-Gabriela, Ioana-Ecaterina Pralea, Radu-Cristian Moldovan, and Cristina-Adela Iuga. 2022. "Indirect Enantioseparations: Recent Advances in Chiral Metabolomics for Biomedical Research" International Journal of Molecular Sciences 23, no. 13: 7428. https://doi.org/10.3390/ijms23137428