
PD1 Deficiency Modifies Cardiac Immunity during Baseline Conditions and in Reperfused Acute Myocardial Infarction


 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
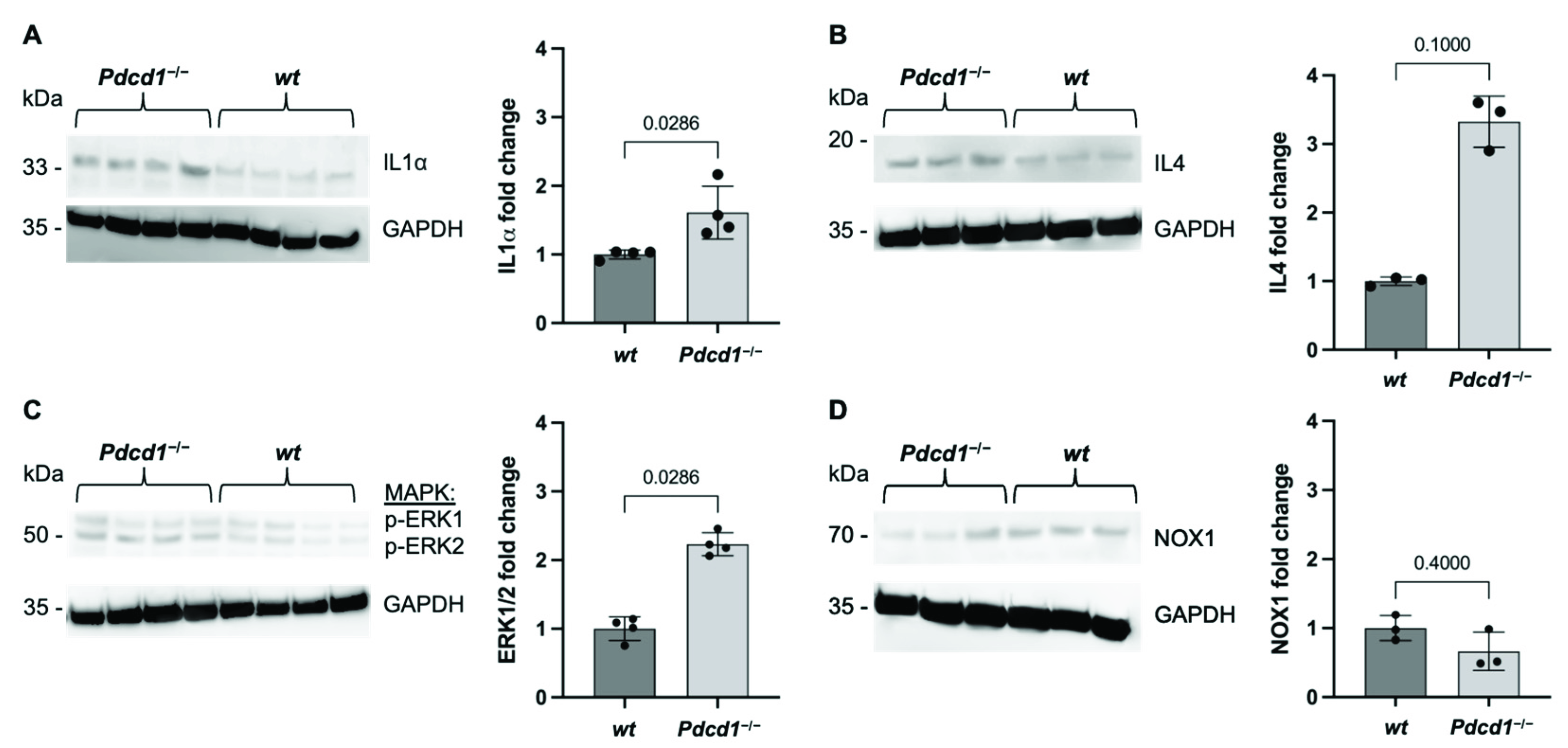
2.1. Cardiac PD1 Deficiency during Baseline Conditions
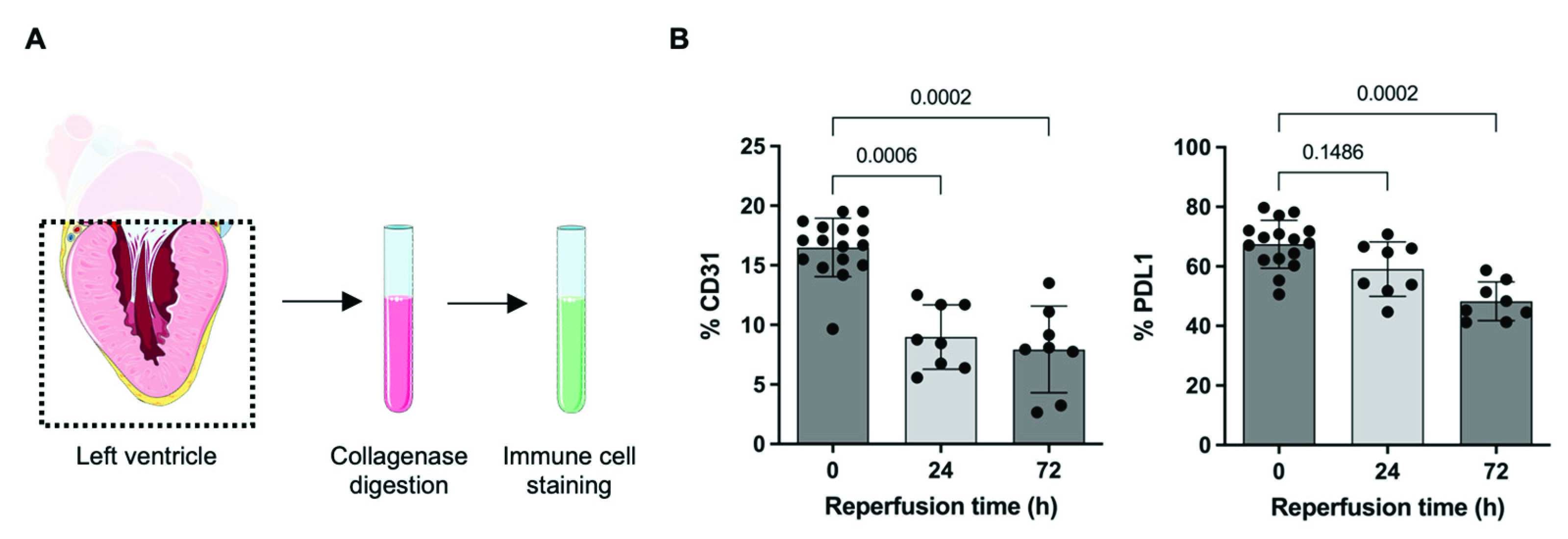
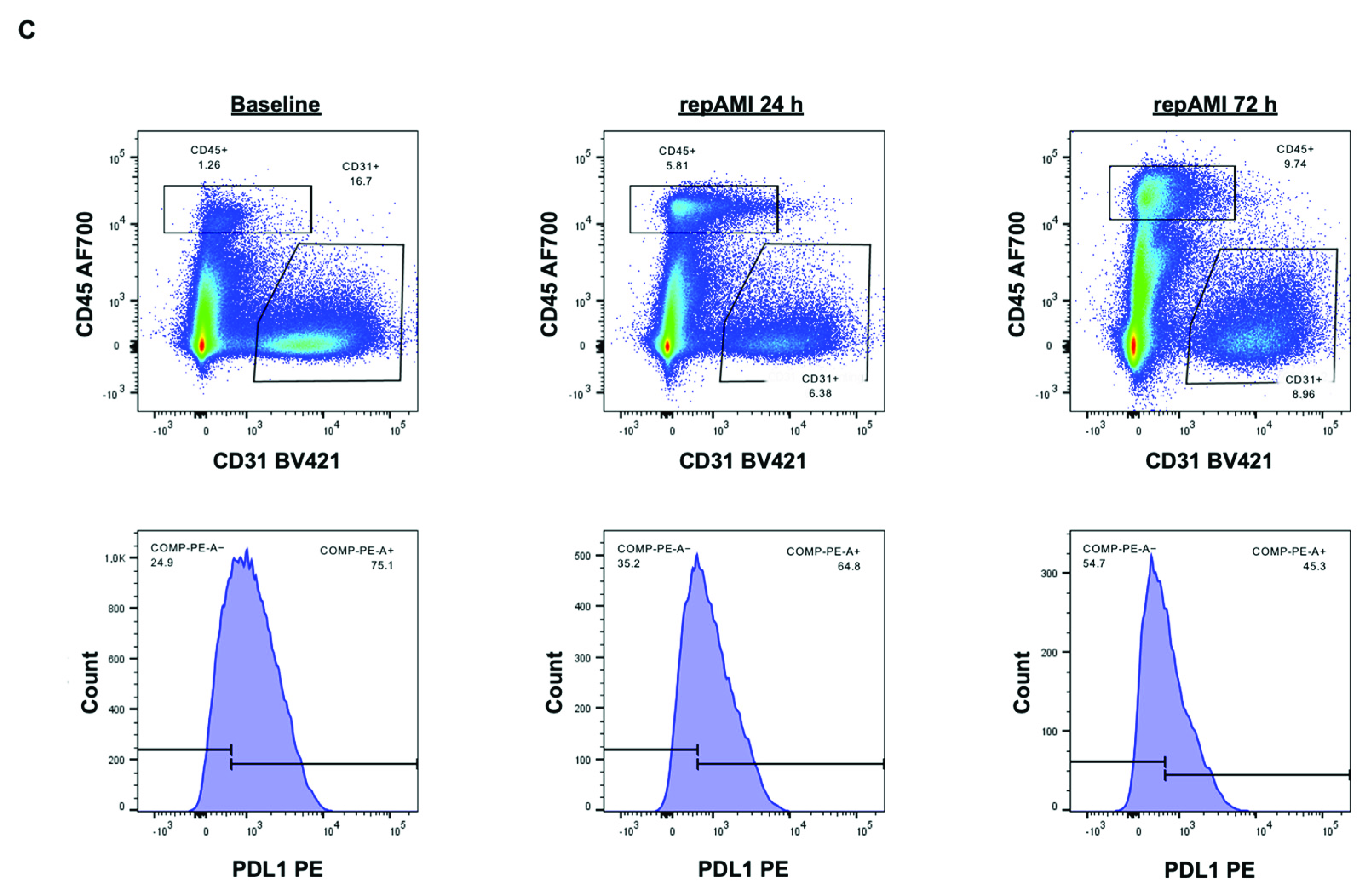
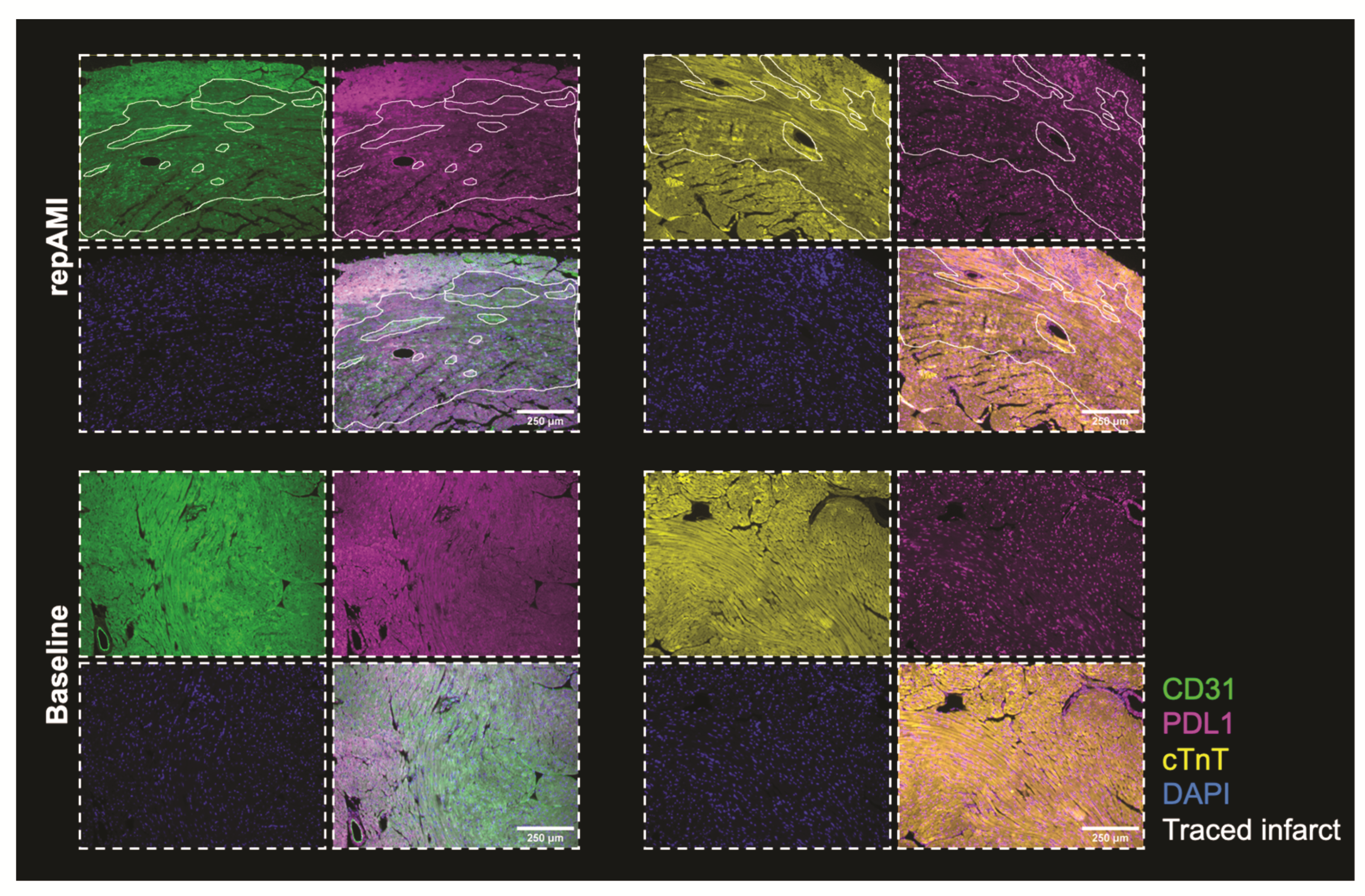
2.2. PDL1 Expression during repAMI
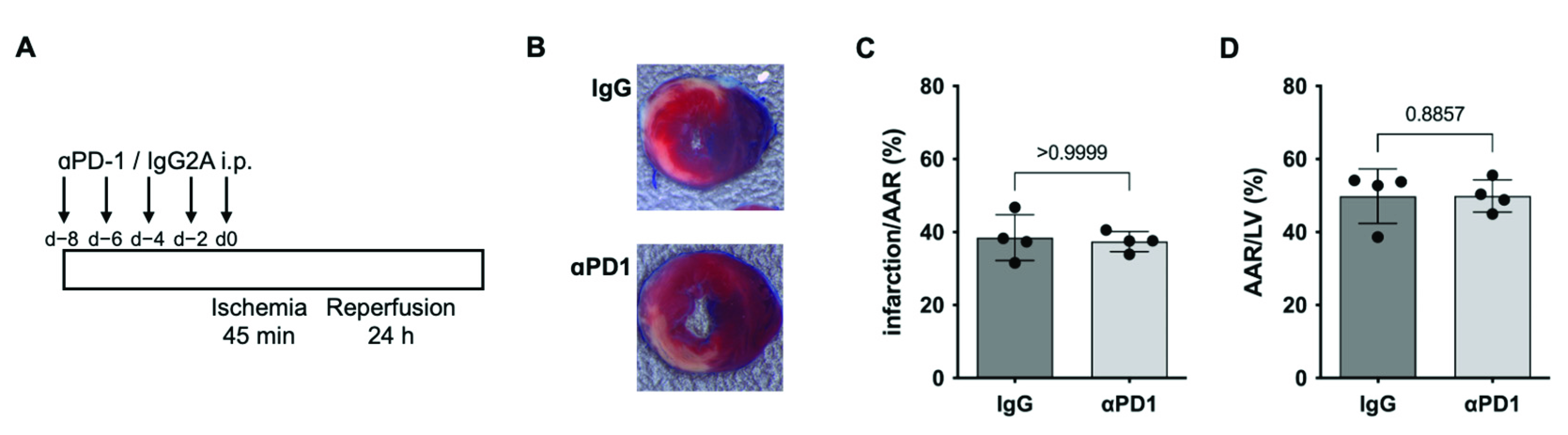
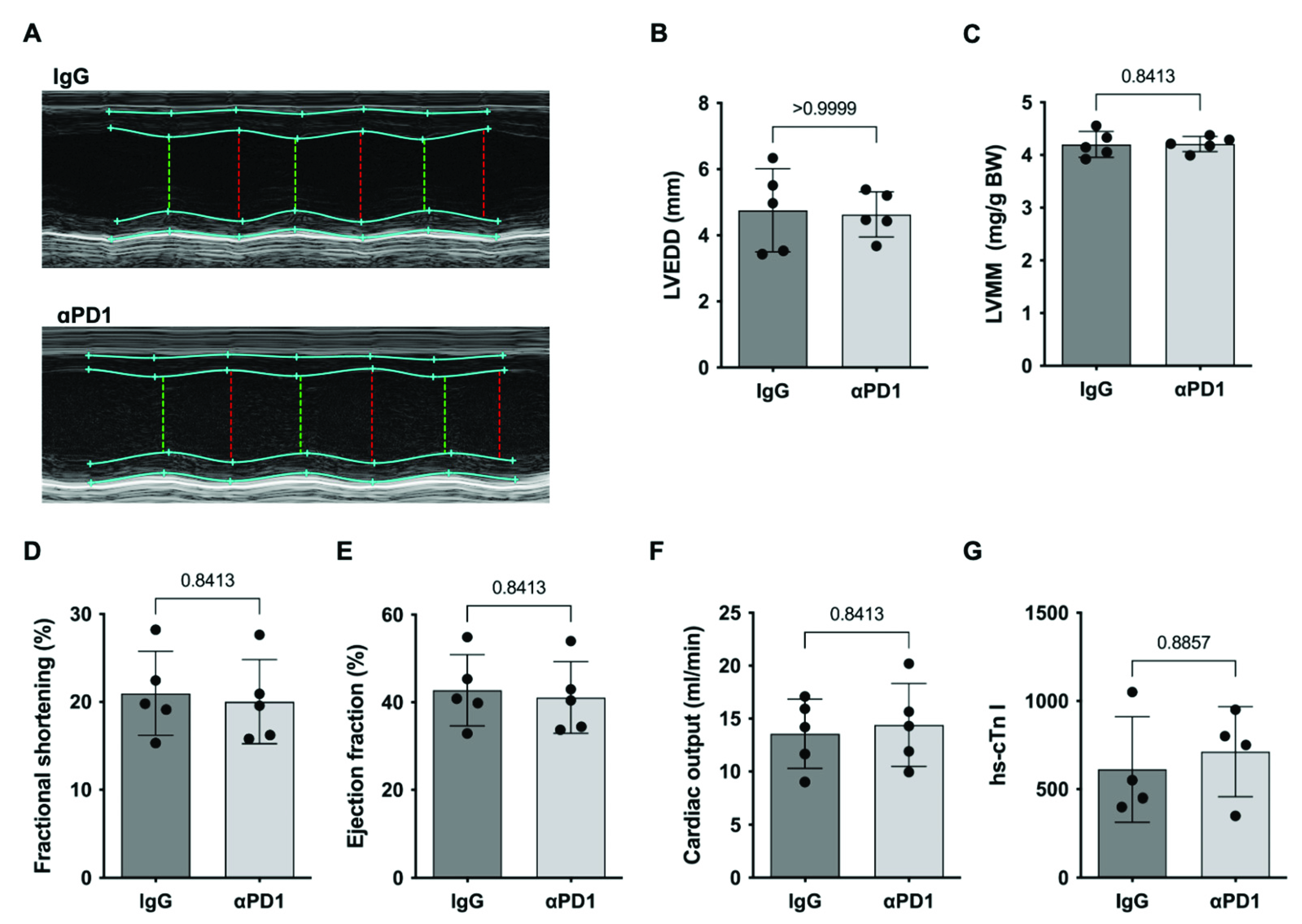
2.3. PD1-Depletion during repAMI
3. Discussion
Limitations
4. Materials and Methods
4.1. Ischemia/Reperfusion (I/R) Heart Injury
4.2. 2,3,5-Triphenyltetrazolium Chloride (TTC) Evan’s Blue Staining
4.3. Echocardiography
4.4. Flow Cytometry
4.5. Western Blot and Plasma Analysis
4.6. Immunohistochemistry
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 18 January 2022).
- Heusch, G. Myocardial ischaemia–reperfusion injury and cardioprotection in perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Grabie, N.; Lichtman, A.H.; Padera, R. T cell checkpoint regulators in the heart. Cardiovasc. Res. 2019, 115, 869–877. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 2017, 18, 153–167. [Google Scholar] [CrossRef]
- Lyon, A.R.; Yousaf, N.; Battisti, N.M.L.; Moslehi, J.; Larkin, J. Immune checkpoint inhibitors and cardiovascular toxicity. Lancet Oncol. 2018, 19, e447–e458. [Google Scholar] [CrossRef]
- Michel, L.; Rassaf, T.; Totzeck, M. Cardiotoxicity from immune checkpoint inhibitors. IJC Hear. Vasc. 2019, 25, 100420. [Google Scholar] [CrossRef]
- Rassaf, T.; Committee for Clinical Cardiovascular Medicine of the German Cardiac Society; Totzeck, M.; Backs, J.; Bokemeyer, C.; Hallek, M.; Hilfiker-Kleiner, D.; Hochhaus, A.; Lüftner, D.; Müller, O.J.; et al. Onco-Cardiology: Consensus Paper of the German Cardiac Society, the German Society for Pediatric Cardiology and Congenital Heart Defects and the German Society for Hematology and Medical Oncology. Clin. Res. Cardiol. 2020, 109, 1197–1222. [Google Scholar] [CrossRef]
- Johnson, D.B.; Balko, J.M.; Compton, M.L.; Chalkias, S.; Gorham, J.; Xu, Y.; Hicks, M.; Puzanov, I.; Alexander, M.R.; Bloomer, T.L.; et al. Fulminant Myocarditis with Combination Immune Checkpoint Blockade. N. Engl. J. Med. 2016, 375, 1749–1755. [Google Scholar] [CrossRef]
- Michel, L.; Helfrich, I.; Hendgen-Cotta, U.B.; Mincu, R.-I.; Korste, S.; Mrotzek, S.M.; Spomer, A.; Odersky, A.; Rischpler, C.; Herrmann, K.; et al. Targeting early stages of cardiotoxicity from anti-PD1 immune checkpoint inhibitor therapy. Eur. Hear. J. 2021, 43, 316–329. [Google Scholar] [CrossRef]
- Grabie, N.; Gotsman, I.; Dacosta, R.; Pang, H.; Stavrakis, G.; Butte, M.J.; Keir, M.E.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Endothelial Programmed Death-1 Ligand 1 (PD-L1) Regulates CD8 + T-Cell–Mediated Injury in the Heart. Circulation 2007, 116, 2062–2071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baban, B.; Liu, J.Y.; Qin, X.; Weintraub, N.; Mozaffari, M.S. Upregulation of Programmed Death-1 and Its Ligand in Cardiac Injury Models: Interaction with GADD153. PLoS ONE 2015, 10, e0124059. [Google Scholar] [CrossRef] [PubMed]
- Michel, L.; Totzeck, M.; Rassaf, T. Cardiac dysfunction from cancer and cancer therapy: New pathways for the prevention of late cardiotoxicity. Basic Res. Cardiol. 2021, 116, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Foks, A.; Kuiper, J. Immune checkpoint proteins: Exploring their therapeutic potential to regulate atherosclerosis. J. Cereb. Blood Flow Metab. 2017, 174, 3940–3955. [Google Scholar] [CrossRef] [Green Version]
- Drobni, Z.D.; Alvi, R.M.; Taron, J.; Zafar, A.; Murphy, S.P.; Rambarat, P.K.; Mosarla, R.C.; Lee, C.; Zlotoff, D.A.; Raghu, V.K. Association Between Immune Checkpoint Inhibitors with Cardiovascular Events and Atherosclerotic Plaque. Circulation 2020, 142, 2299–2311. [Google Scholar] [CrossRef]
- Bu, D.-X.; Tarrio, M.; Maganto-Garcia, E.; Stavrakis, G.; Tajima, G.; Lederer, J.A.; Jarolim, P.; Freeman, G.J.; Sharpe, A.H.; Lichtman, A.H. Impairment of the Programmed Cell Death-1 Pathway Increases Atherosclerotic Lesion Development and Inflammation. Arter. Thromb. Vasc. Biol. 2011, 31, 1100–1107. [Google Scholar] [CrossRef] [Green Version]
- Totzeck, M.; Lutgens, E.; Neilan, T.G. Are we underestimating the potential for cardiotoxicity related to immune checkpoint inhibitors? Eur. Heart J. 2021, 42, 1632–1635. [Google Scholar] [CrossRef]
- D’Souza, M.; Nielsen, D.; Svane, I.M.; Iversen, K.; Rasmussen, P.V.; Madelaire, C.; Fosbøl, E.; Køber, L.; Gustafsson, F.; Andersson, C.; et al. The risk of cardiac events in patients receiving immune checkpoint inhibitors: A nationwide Danish study. Eur. Hear. J. 2020, 42, 1621–1631. [Google Scholar] [CrossRef]
- Xia, W.; Zou, C.; Chen, H.; Xie, C.; Hou, M. Immune checkpoint inhibitor induces cardiac injury through polarizing macrophages via modulating microRNA-34a/Kruppel-like factor 4 signaling. Cell Death Dis. 2020, 11, 575. [Google Scholar] [CrossRef]
- Varricchi, G.; Galdiero, M.R.; Tocchetti, C.G. Novel actors on the stage of cardiac dysfunction induced by anti-PD1 oncological treatments. Eur. Hear. J. 2021, 43, 330–332. [Google Scholar] [CrossRef]
- Du, S.; Zhou, L.; Alexander, G.S.; Park, K.; Yang, L.; Wang, N.; Zaorsky, N.G.; Ma, X.; Wang, Y.; Dicker, A.P.; et al. PD-1 Modulates Radiation-Induced Cardiac Toxicity through Cytotoxic T Lymphocytes. J. Thorac. Oncol. 2017, 13, 510–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppenheim, J.J.; Matsushima, K.; Yoshimura, T.; Leonard, E.J.; Neta, R. Relationship between interleukin 1 (IL1), tumor necrosis factor (TNF) and a neutrophil attracting peptide (NAP-1). Agents Actions 1989, 26, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Walsh, L.J.; Trinchieri, G.; Waldorf, H.A.; Whitaker, D.; Murphy, G.F. Human dermal mast cells contain and release tumor necrosis factor alpha, which induces endothelial leukocyte adhesion molecule 1. Proc. Natl. Acad. Sci. USA 1991, 88, 4220–4224. [Google Scholar] [CrossRef] [Green Version]
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Park, P.H.; McMullen, M.R.; Huang, H.; Thakur, V.; Nagy, L.E. Short-term treatment of RAW264.7 macrophages with adiponectin increases tumor necrosis factor-alpha (TNF-alpha) expression via ERK1/2 activation and Egr-1 expression: Role of TNF-alpha in adiponectin-stimulated interleukin-10 production. J. Biol. Chem. 2007, 282, 21695–21703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusters, P.J.H.; Lutgens, E.; Seijkens, T.T.P. Exploring immune checkpoints as potential therapeutic targets in atherosclerosis. Cardiovasc. Res. 2018, 114, 368–377. [Google Scholar] [CrossRef]
- Poels, K.; van Leent, M.M.; Boutros, C.; Tissot, H.; Roy, S.; Meerwaldt, A.E.; Toner, Y.C.; Reiche, M.E.; Kusters, P.J.; Malinova, T.; et al. Immune Checkpoint Inhibitor Therapy Aggravates T Cell–Driven Plaque Inflammation in Atherosclerosis. JACC CardioOncol. 2020, 2, 599–610. [Google Scholar] [CrossRef]
- Keir, M.E.; Freeman, G.J.; Sharpe, A.H. PD-1 Regulates Self-Reactive CD8+ T Cell Responses to Antigen in Lymph Nodes and Tissues. J. Immunol. 2007, 179, 5064–5070. [Google Scholar] [CrossRef] [Green Version]
- Totzeck, M.; Hendgen-Cotta, U.B.; French, B.A.; Rassaf, T. A practical approach to remote ischemic preconditioning and ischemic preconditioning against myocardial ischemia/reperfusion injury. J. Biol. Methods 2016, 3, e57. [Google Scholar] [CrossRef] [Green Version]
- Merz, S.F.; Korste, S.; Bornemann, L.; Michel, L.; Stock, P.; Squire, A.; Soun, C.; Engel, D.R.; Detzer, J.; Lorchner, H.; et al. Contemporaneous 3D characterization of acute and chronic myocardial I/R injury and response. Nat. Commun. 2019, 10, 2312. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Michel, L.; Korste, S.; Spomer, A.; Hendgen-Cotta, U.B.; Rassaf, T.; Totzeck, M. PD1 Deficiency Modifies Cardiac Immunity during Baseline Conditions and in Reperfused Acute Myocardial Infarction. Int. J. Mol. Sci. 2022, 23, 7533. https://doi.org/10.3390/ijms23147533
Michel L, Korste S, Spomer A, Hendgen-Cotta UB, Rassaf T, Totzeck M. PD1 Deficiency Modifies Cardiac Immunity during Baseline Conditions and in Reperfused Acute Myocardial Infarction. International Journal of Molecular Sciences. 2022; 23(14):7533. https://doi.org/10.3390/ijms23147533
Chicago/Turabian StyleMichel, Lars, Sebastian Korste, Armin Spomer, Ulrike Barbara Hendgen-Cotta, Tienush Rassaf, and Matthias Totzeck. 2022. "PD1 Deficiency Modifies Cardiac Immunity during Baseline Conditions and in Reperfused Acute Myocardial Infarction" International Journal of Molecular Sciences 23, no. 14: 7533. https://doi.org/10.3390/ijms23147533
APA StyleMichel, L., Korste, S., Spomer, A., Hendgen-Cotta, U. B., Rassaf, T., & Totzeck, M. (2022). PD1 Deficiency Modifies Cardiac Immunity during Baseline Conditions and in Reperfused Acute Myocardial Infarction. International Journal of Molecular Sciences, 23(14), 7533. https://doi.org/10.3390/ijms23147533