
Targeting the Microenvironment for Treating Multiple Myeloma

 , ,
, ,
Abstract
:1. Introduction
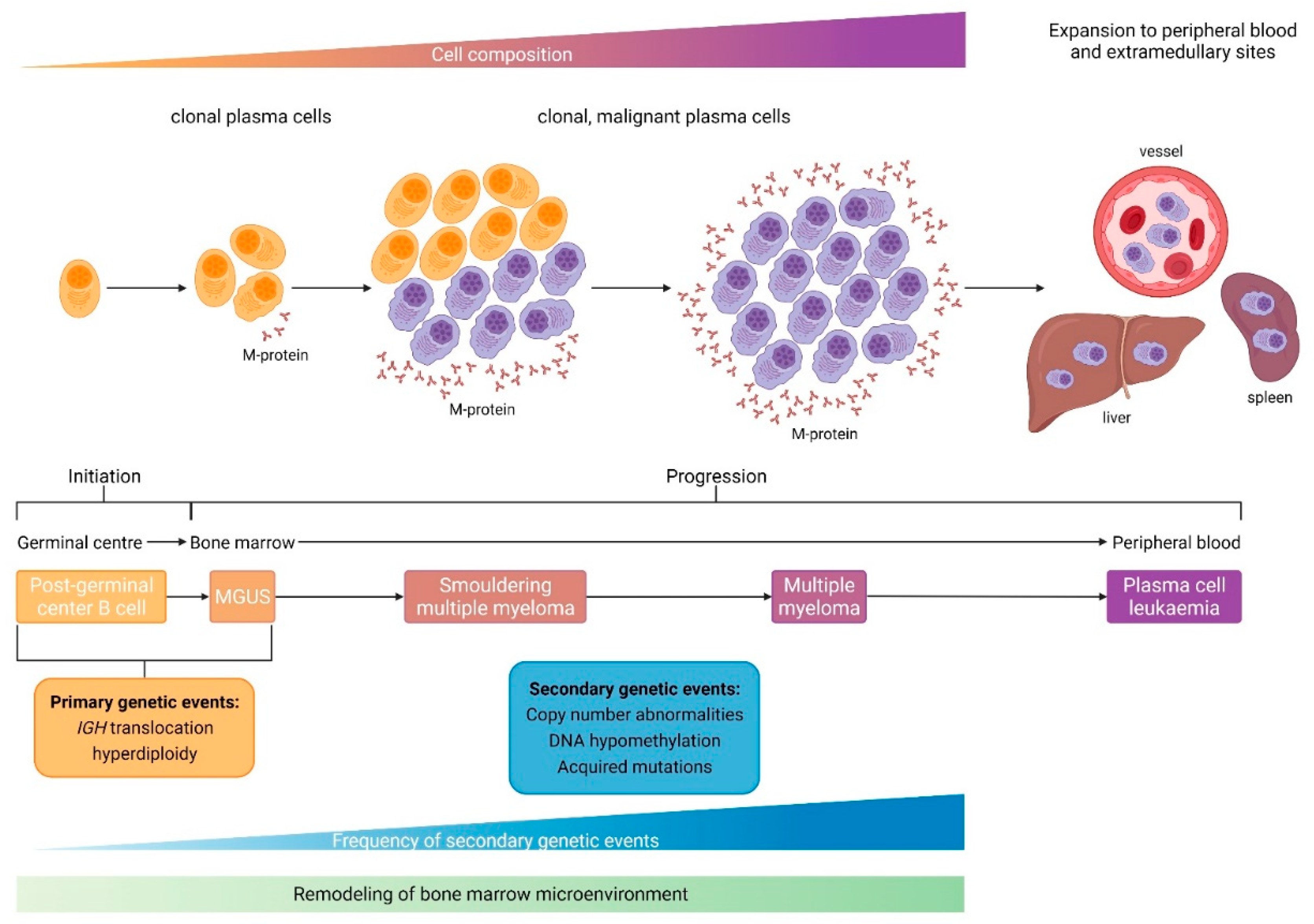
2. The Role of Microenvironment and Immunology
3. Immunosuppressive BMME and Immune Exhaustion
4. From Bench to Bedside: Therapeutical Targeting of the BMME in MM
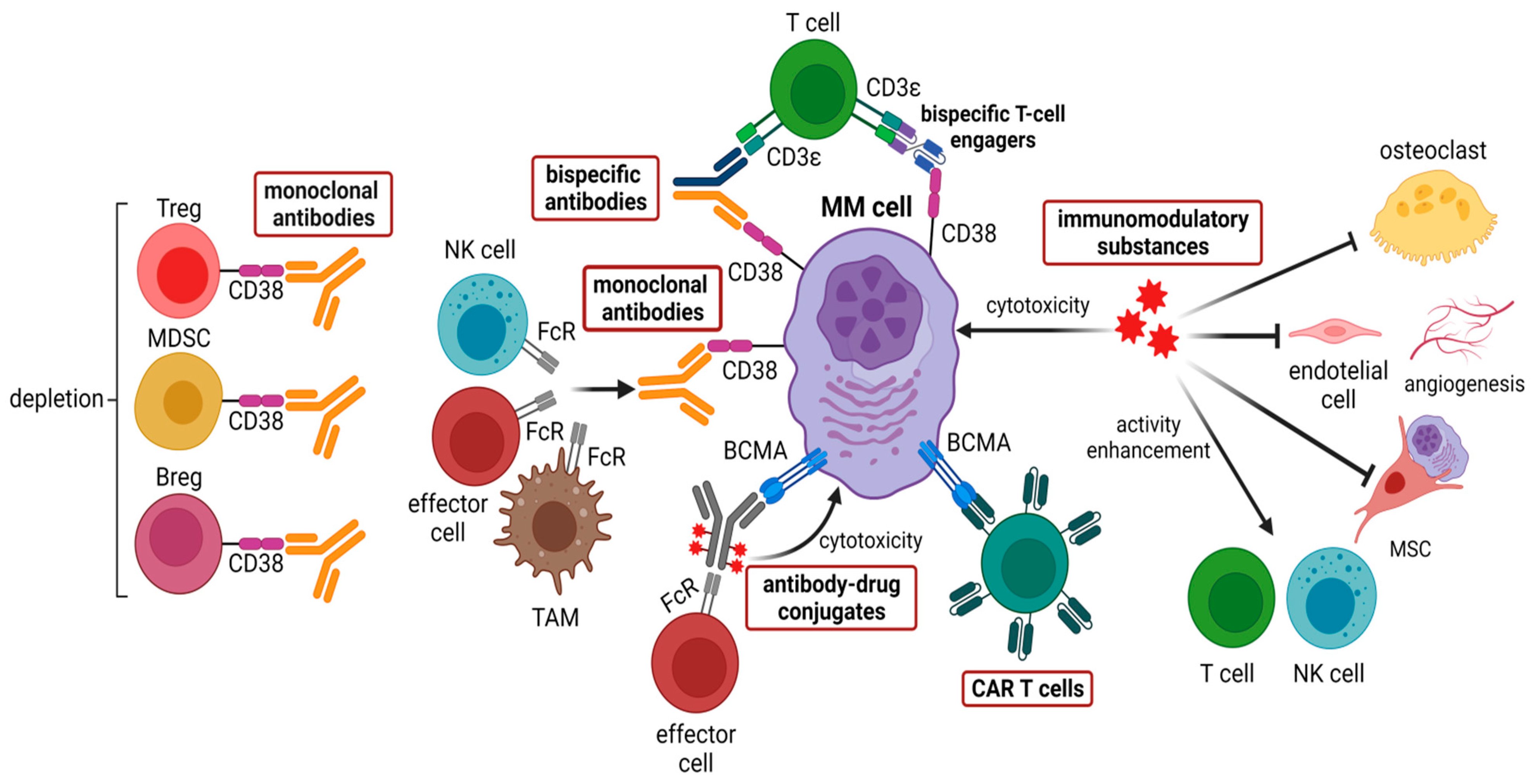
4.1. Anti-CD38 Therapies
4.2. Immunomodulatory Substances (IMiDs)
4.3. CAR T Cells
4.4. Bispecific Antibodies
4.5. Antibody-Drug Conjugates Targeting BCMA
4.6. BMME Mediated Therapy Resistance
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADC | antibody-drug conjugate |
| ADCC | antibody-dependent cytotoxicity |
| APRIL | A proliferation-inducing ligand |
| BCMA | B cell maturation antigen |
| BM | bone marrow |
| BMME | bone marrow microenvironment |
| Breg | regulatory B cell |
| BsAb | bispecific antibody |
| CAR | chimeric antigen receptor |
| CR | complete response |
| CRAB | hypercalcemia (C), renal failure (R), anemia (A), bone lesions (B) |
| CRS | cytokine release syndrome |
| CTL | cytotoxic T lymphocytes |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| DC | dendritic cell |
| EMD | extra-medullary disease |
| FDA | food and drug administration |
| FOXP3 | forkhead box P3 |
| GC | germinal center |
| GvHD | graft-versus-host-disease |
| HLA | human leukocyte antigen |
| ICAM-1 | intercellular Adhesion Molecule 1 |
| IDO | indoleamine 2,3-dioxygenase |
| IFN | Interferon |
| Ig | immunoglobulin |
| IL | interleukin |
| IMiDs | immunomulatory drugs |
| LAG-3 | lymphocyte-activation gene 3 |
| mAb | monoclonal antibody |
| MDSC | myeloid-derived suppressor cells |
| MGUS | monoclonal gammopathy of undetermined significance |
| MHC | major histocompatibility complex |
| MM | multiple myeloma |
| MMAF | monomethyl auristatin F |
| mOS | median overall survival |
| MRD | minimal residual disease |
| MSC | mesenchymal stromal cell |
| NDMM | newly diagnosed multiple myeloma |
| NGS | next-generation sequencing |
| NO | nitric oxide |
| NR | not reported |
| NTE | not transplant eligible |
| ORR | overall response rate |
| PD-1 | programmed cell death protein 1 |
| PD-L1/2 | programmed cell death protein 1 ligand 1/2 |
| PGE2 | prostaglandin E2 |
| PI | proteasome inhibitor |
| ROS | reactive oxygen species |
| RRMM | relapsed/refractory multiple myeloma |
| sFLC | serum free light chain |
| SMM | smouldering multiple myeloma |
| STAT3 | signal transducer and activator of transcription 3 |
| TACI | transmembrane activator, and calcium modulator and cyclophilin ligand interactor |
| TAM | tumor-associated macrophage |
| TAM | tumor-associated macrophage |
| TE | transplant eligible |
| TGF-β | transforming growth factor beta |
| Th | T helper cell |
| TIGIT | T cell immunoglobulin and ITIM domain |
| TIM-3 | T cell immunoglobulin mucin-3 |
| Treg | regulatory T cell |
| TRL4 | toll-like receptor 4 |
| VCAM-1 | vascular cell adhesion molecule 1 |
| VEGF | vascular endothelial growth factor |
| VGPR | very good partial response |
References
- Kumar, S.K.; Rajkumar, V.; Kyle, R.A.; Van Duin, M.; Sonneveld, P.; Mateos, M.V.; Gay, F.; Anderson, K.C. Multiple Myeloma. Nat. Rev. Dis. Primers 2017, 3, 17046. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Anderson, K.C. Understanding Biology to Tackle the Disease: Multiple Myeloma from Bench to Bedside, and Back. CA Cancer J. Clin. 2014, 64, 422–444. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental Regulation of Tumor Progression and Metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- van Nieuwenhuijzen, N.; Spaan, I.; Raymakers, R.; Peperzak, V. From MGUS to Multiple Myeloma, a Paradigm for Clonal Evolution of Premalignant Cells. Cancer Res. 2018, 78, 2449–2456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlyn, C.; Morgan, G.J. Evolutionary Biology of High-Risk Multiple Myeloma. Nat. Rev. Cancer 2017, 17, 543–556. [Google Scholar] [CrossRef] [PubMed]
- Ghobrial, I.M.; Detappe, A.; Anderson, K.C.; Steensma, D.P. The Bone-Marrow Niche in MDS and MGUS: Implications for AML and MM. Nat. Rev. Clin. Oncol. 2018, 15, 219–233. [Google Scholar] [CrossRef] [PubMed]
- Mikulasova, A.; Wardell, C.P.; Murison, A.; Boyle, E.M.; Jackson, G.H.; Smetana, J.; Kufova, Z.; Pour, L.; Sandecka, V.; Almasi, M.; et al. The Spectrum of Somatic Mutations in Monoclonal Gammopathy of Undetermined Significance Indicates a Less Complex Genomic Landscape than That in Multiple Myeloma. Haematologica 2017, 102, 1617–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledergor, G.; Weiner, A.; Zada, M.; Wang, S.Y.; Cohen, Y.C.; Gatt, M.E.; Snir, N.; Magen, H.; Koren-Michowitz, M.; Herzog-Tzarfati, K.; et al. Single Cell Dissection of Plasma Cell Heterogeneity in Symptomatic and Asymptomatic Myeloma. Nat. Med. 2018, 24, 1867–1876. [Google Scholar] [CrossRef]
- Hoang, P.H.; Cornish, A.J.; Dobbins, S.E.; Kaiser, M.; Houlston, R.S. Mutational Processes Contributing to the Development of Multiple Myeloma. Blood Cancer J. 2019, 9, 60. [Google Scholar] [CrossRef]
- Avigan, D.; Rosenblatt, J. Current Treatment for Multiple Myeloma. N. Engl. J. Med. 2014, 371, 961–962. [Google Scholar] [CrossRef] [Green Version]
- Mikhael, J.; Ismaila, N.; Cheung, M.C.; Costello, C.; Dhodapkar, M.V.; Kumar, S.; Lacy, M.; Lipe, B.; Little, R.F.; Nikonova, A.; et al. Treatment of Multiple Myeloma: ASCO and CCO Joint Clinical Practice Guideline. J. Clin. Oncol. 2019, 37, 1228–1263. [Google Scholar] [CrossRef]
- Lomas, O.C.; Tahri, S.; Ghobrial, I.M. The Microenvironment in Myeloma. Curr. Opin. Oncol. 2020, 32, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Schürch, C.M.; Rasche, L.; Frauenfeld, L.; Weinhold, N.; Fend, F. A Review on Tumor Heterogeneity and Evolution in Multiple Myeloma: Pathological, Radiological, Molecular Genetics, and Clinical Integration. Virchows Archiv. 2020, 476, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Holthof, L.C.; Mutis, T. Challenges for Immunotherapy in Multiple Myeloma: Bone Marrow Microenvironment-Mediated Immune Suppression and Immune Resistance. Cancers 2020, 12, 988. [Google Scholar] [CrossRef] [PubMed]
- Franssen, L.E.; Mutis, T.; Lokhorst, H.M.; van de Donk, N.W.C.J. Immunotherapy in Myeloma: How Far Have We Come? Ther. Adv. Hematol. 2019, 10, 2040620718822660. [Google Scholar] [CrossRef] [PubMed]
- Görgün, G.T.; Whitehill, G.; Anderson, J.L.; Hideshima, T.; Maguire, C.; Laubach, J.; Raje, N.; Munshi, N.C.; Richardson, P.G.; Anderson, K.C. Tumor-Promoting Immune-Suppressive Myeloid-Derived Suppressor Cells in the Multiple Myeloma Microenvironment in Humans. Blood 2013, 121, 2975–2987. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, I.R.; Martner, A.; Pisklakova, A.; Condamine, T.; Chase, T.; Vogl, T.; Roth, J.; Gabrilovich, D.; Nefedova, Y. Myeloid-Derived Suppressor Cells Regulate Growth of Multiple Myeloma by Inhibiting T Cells in Bone Marrow. J. Immunol. 2013, 190, 3815–3823. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Kawano, Y.; Zavidij, O.; Park, J.; Moschetta, M.; Kokubun, K.; Mouhieddine, T.H.; Manier, S.; Mishima, Y.; Murakami, N.; Bustoros, M.; et al. Blocking IFNAR1 Inhibits Multiple Myeloma-Driven Treg Expansion and Immunosuppression. J. Clin. Investig. 2018, 128, 2487–2499. [Google Scholar] [CrossRef]
- Zorn, E.; Nelson, E.A.; Mohseni, M.; Porcheray, F.; Kim, H.; Litsa, D.; Bellucci, R.; Raderschall, E.; Canning, C.; Soiffer, R.J.; et al. IL-2 Regulates FOXP3 Expression in Human CD4+CD25+ Regulatory T Cells through a STAT-Dependent Mechanism and Induces the Expansion of These Cells in Vivo. Blood 2006, 108, 1571–1579. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Zhang, L.; Acharya, C.; An, G.; Wen, K.; Qiu, L.; Munshi, N.C.; Tai, Y.T.; Anderson, K.C. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin. Cancer Res. 2017, 23, 4290–4300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejcik, J.; Casneuf, T.; Nijhof, I.S.; Verbist, B.; Bald, J.; Plesner, T.; Syed, K.; Liu, K.; van de Donk, N.W.C.J.; Weiss, B.M.; et al. Daratumumab Depletes CD38+ Immune Regulatory Cells, Promotes T-Cell Expansion, and Skews T-Cell Repertoire in Multiple Myeloma. Blood 2016, 128, 384–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giallongo, C.; Tibullo, D.; Camiolo, G.; Parrinello, N.L.; Romano, A.; Puglisi, F.; Barbato, A.; Conticello, C.; Lupo, G.; Anfuso, C.D.; et al. TLR4 Signaling Drives Mesenchymal Stromal Cells Commitment to Promote Tumor Microenvironment Transformation in Multiple Myeloma. Cell Death Dis. 2019, 10, 704. [Google Scholar] [CrossRef] [Green Version]
- André, T.; Najar, M.; Stamatopoulos, B.; Pieters, K.; Pradier, O.; Bron, D.; Meuleman, N.; Lagneaux, L. Immune Impairments in Multiple Myeloma Bone Marrow Mesenchymal Stromal Cells. Cancer Immunol. Immunother. 2015, 64, 213–224. [Google Scholar] [CrossRef]
- Tai, Y.T.; Lin, L.; Xing, L.; Cho, S.F.; Yu, T.; Acharya, C.; Wen, K.; Hsieh, P.A.; Dulos, J.; van Elsas, A.; et al. APRIL Signaling via TACI Mediates Immunosuppression by T Regulatory Cells in Multiple Myeloma: Therapeutic Implications. Leukemia 2019, 33, 426–438. [Google Scholar] [CrossRef]
- Minnie, S.A.; Hill, G.R. Immunotherapy of Multiple Myeloma. J. Clin. Investig. 2020, 130, 1565–1575. [Google Scholar] [CrossRef]
- Yadav, M.; Green, C.; Ma, C.; Robert, A.; Glibicky, A.; Nakamura, R.; Sumiyoshi, T.; Meng, R.; Chu, Y.-W.; Wu, J.; et al. Tigit, CD226 and PD-L1/PD-1 Are Highly Expressed By Marrow-Infiltrating T Cells in Patients with Multiple Myeloma. Blood 2016, 128, 2102. [Google Scholar] [CrossRef]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma Cells from Multiple Myeloma Patients Express B7-H1 (PD-L1) and Increase Expression after Stimulation with IFN-γ and TLR Ligands via a MyD88-, TRAF6-, and MEK-Dependent Pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillerey, C.; Harjunpää, H.; Carrié, N.; Kassem, S.; Teo, T.; Miles, K.; Krumeich, S.; Weulersse, M.; Cuisinier, M.; Stannard, K.; et al. TIGIT Immune Checkpoint Blockade Restores CD81 T-Cell Immunity against Multiple Myeloma. Blood 2018, 132, 1689–1694. [Google Scholar] [CrossRef] [Green Version]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Blacklock, H.; Schjesvold, F.; Oriol, A.; Simpson, D.; George, A.; Goldschmidt, H.; Larocca, A.; Chanan-Khan, A.; Sherbenou, D.; et al. Pembrolizumab plus Pomalidomide and Dexamethasone for Patients with Relapsed or Refractory Multiple Myeloma (KEYNOTE-183): A Randomised, Open-Label, Phase 3 Trial. Lancet Haematol. 2019, 6, e459–e469. [Google Scholar] [CrossRef]
- Costello, C. The Future of Checkpoint Inhibition in Multiple Myeloma? Lancet Haematol. 2019, 6, e439–e440. [Google Scholar] [CrossRef]
- Strassl, I.; Schreder, M.; Steiner, N.; Rudzki, J.; Agis, H.; Künz, T.; Müser, N.; Willenbacher, W.; Petzer, A.; Neumeister, P.; et al. The Agony of Choice—Where to Place the Wave of BCMA-Targeted Therapies in the Multiple Myeloma Treatment Puzzle in 2022 and Beyond. Cancers 2021, 13, 4701. [Google Scholar] [CrossRef] [PubMed]
- Radocha, J.; van de Donk, N.W.C.J.; Weisel, K. Monoclonal Antibodies and Antibody Drug Conjugates in Multiple Myeloma. Cancers 2021, 13, 1571. [Google Scholar] [CrossRef] [PubMed]
- Mateos, M.V.; Cavo, M.; Blade, J.; Dimopoulos, M.A.; Suzuki, K.; Jakubowiak, A.; Knop, S.; Doyen, C.; Lucio, P.; Nagy, Z.; et al. Overall Survival with Daratumumab, Bortezomib, Melphalan, and Prednisone in Newly Diagnosed Multiple Myeloma (ALCYONE): A Randomised, Open-Label, Phase 3 Trial. The Lancet 2020, 395, 132–141. [Google Scholar] [CrossRef]
- Bonello, F.; Grasso, M.; D’agostino, M.; Celeghini, I.; Castellino, A.; Boccadoro, M.; Bringhen, S. The Role of Monoclonal Antibodies in the First-Line Treatment of Transplant-Ineligible Patients with Newly Diagnosed Multiple Myeloma. Pharmaceuticals 2021, 14, 20. [Google Scholar] [CrossRef]
- Facon, T.; Kumar, S.; Plesner, T.; Orlowski, R.Z.; Moreau, P.; Bahlis, N.; Basu, S.; Nahi, H.; Hulin, C.; Quach, H.; et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. N. Engl. J. Med. 2019, 380, 2104–2115. [Google Scholar] [CrossRef]
- Kumar, S.K.; Facon, T.; Usmani, S.Z.; Plesner, T.; Orlowski, R.Z.; Touzeau, C.; Basu, S.; Bahlis, N.J.; Goldschmidt, H.; O’Dwyer, M.E.; et al. Updated Analysis of Daratumumab Plus Lenalidomide and Dexamethasone (D-Rd) Versus Lenalidomide and Dexamethasone (Rd) in Patients with Transplant-Ineligible Newly Diagnosed Multiple Myeloma (NDMM): The Phase 3 Maia Study. Blood 2020, 136, 24–26. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Hulin, C.; Arnulf, B.; Belhadj, K.; Benboubker, L.; Béné, M.C.; Broijl, A.; Caillon, H.; Caillot, D.; et al. Bortezomib, Thalidomide, and Dexamethasone with or without Daratumumab before and after Autologous Stem-Cell Transplantation for Newly Diagnosed Multiple Myeloma (CASSIOPEIA): A Randomised, Open-Label, Phase 3 Study. The Lancet 2019, 394, 29–38. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Dimopoulos, M.A.; White, D.J.; Benboubker, L.; Cook, G.; Leiba, M.; Ho, P.J.; Kim, K.; Takezako, N.; Moreau, P.; et al. Daratumumab plus Lenalidomide and Dexamethasone in Relapsed/Refractory Multiple Myeloma: Extended Follow-up of POLLUX, a Randomized, Open-Label, Phase 3 Study. Leukemia 2020, 34, 1875–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, A.; Lentzsch, S.; Weisel, K.; Avet-Loiseau, H.; Mark, T.M.; Spicka, I.; Masszi, T.; Lauri, B.; Levin, M.D.; Bosi, A.; et al. Daratumumab plus Bortezomib and Dexamethasone versus Bortezomib and Dexamethasone in Relapsed or Refractory Multiple Myeloma: Updated Analysis of CASTOR. Haematologica 2018, 103, 2079–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimopoulos, M.; Quach, H.; Mateos, M.V.; Landgren, O.; Leleu, X.; Siegel, D.; Weisel, K.; Yang, H.; Klippel, Z.; Zahlten-Kumeli, A.; et al. Carfilzomib, Dexamethasone, and Daratumumab versus Carfilzomib and Dexamethasone for Patients with Relapsed or Refractory Multiple Myeloma (CANDOR): Results from a Randomised, Multicentre, Open-Label, Phase 3 Study. Lancet 2020, 396, 186–197. [Google Scholar] [CrossRef]
- Bonello, F.; Mina, R.; Boccadoro, M.; Gay, F. Therapeutic Monoclonal Antibodies and Antibody Products: Current Practices and Development in Multiple Myeloma. Cancers 2020, 12, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreau, P.; Dimopoulos, M.A.; Mikhael, J.; Yong, K.; Capra, M.; Facon, T.; Hajek, R.; Špička, I.; Baker, R.; Kim, K.; et al. Isatuximab, Carfilzomib, and Dexamethasone in Relapsed Multiple Myeloma (IKEMA): A Multicentre, Open-Label, Randomised Phase 3 Trial. The Lancet 2021, 397, 2361–2371. [Google Scholar] [CrossRef]
- Attal, M.; Richardson, P.G.; Rajkumar, S.V.; San-Miguel, J.; Beksac, M.; Spicka, I.; Leleu, X.; Schjesvold, F.; Moreau, P.; Dimopoulos, M.A.; et al. Isatuximab plus Pomalidomide and Low-Dose Dexamethasone versus Pomalidomide and Low-Dose Dexamethasone in Patients with Relapsed and Refractory Multiple Myeloma (ICARIA-MM): A Randomised, Multicentre, Open-Label, Phase 3 Study. The Lancet 2019, 394, 2096–2107. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Moreau, P.; Terpos, E.; Mateos, M.V.; Zweegman, S.; Cook, G.; Delforge, M.; Hájek, R.; Schjesvold, F.; Cavo, M.; et al. Multiple Myeloma: EHA-ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2021, 32, 309–322. [Google Scholar] [CrossRef]
- Holstein, S.A.; McCarthy, P.L. Immunomodulatory Drugs in Multiple Myeloma: Mechanisms of Action and Clinical Experience. Drugs 2017, 77, 505–520. [Google Scholar] [CrossRef]
- Durie, B.G.M.; Hoering, A.; Sexton, R.; Abidi, M.H.; Epstein, J.; Rajkumar, S.V.; Dispenzieri, A.; Kahanic, S.P.; Thakuri, M.C.; Reu, F.J.; et al. Longer Term Follow-up of the Randomized Phase III Trial SWOG S0777: Bortezomib, Lenalidomide and Dexamethasone vs. Lenalidomide and Dexamethasone in Patients (Pts) with Previously Untreated Multiple Myeloma without an Intent for Immediate Autologous Stem. Blood Cancer J. 2020, 10, 53. [Google Scholar] [CrossRef]
- McCarthy, P.L.; Holstein, S.A.; Petrucci, M.T.; Richardson, P.G.; Hulin, C.; Tosi, P.; Bringhen, S.; Musto, P.; Anderson, K.C.; Caillot, D.; et al. Lenalidomide Maintenance after Autologous Stem-Cell Transplantation in Newly Diagnosed Multiple Myeloma: A Meta-Analysis. J. Clin. Oncol. 2017, 35, 3279–3289. [Google Scholar] [CrossRef]
- Attal, M.; Lauwers-Cances, V.; Marit, G.; Caillot, D.; Moreau, P.; Facon, T.; Stoppa, A.M.; Hulin, C.; Benboubker, L.; Garderet, L.; et al. Lenalidomide Maintenance after Stem-Cell Transplantation for Multiple Myeloma. N. Engl. J. Med. 2012, 366, 1782–1791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCarthy, P.L.; Owzar, K.; Hofmeister, C.C.; Hurd, D.D.; Hassoun, H.; Richardson, P.G.; Giralt, S.; Stadtmauer, E.A.; Weisdorf, D.J.; Vij, R.; et al. Lenalidomide after Stem-Cell Transplantation for Multiple Myeloma. N. Engl. J. Med. 2012, 366, 1770–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, D.S.; Dimopoulos, M.A.; Ludwig, H.; Facon, T.; Goldschmidt, H.; Jakubowiak, A.; San-Miguel, J.; Obreja, M.; Blaedel, J.; Stewart, A.K. Improvement in Overall Survival with Carfilzomib, Lenalidomide, and Dexamethasone in Patients with Relapsed or Refractory Multiple Myeloma. J. Clin. Oncol. 2018, 36, 728–734. [Google Scholar] [CrossRef] [PubMed]
- Richardson, P.G.; Oriol, A.; Beksac, M.; Liberati, A.M.; Galli, M.; Schjesvold, F.; Lindsay, J.; Weisel, K.; White, D.; Facon, T.; et al. Pomalidomide, Bortezomib, and Dexamethasone for Patients with Relapsed or Refractory Multiple Myeloma Previously Treated with Lenalidomide (OPTIMISMM): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2019, 20, 781–794. [Google Scholar] [CrossRef]
- Duong, C.P.M.; Yong, C.S.M.; Kershaw, M.H.; Slaney, C.Y.; Darcy, P.K. Cancer Immunotherapy Utilizing Gene-Modified T Cells: From the Bench to the Clinic. Mol. Immunol. 2015, 67, 46–57. [Google Scholar] [CrossRef]
- Davies, D.M.; Maher, J. Adoptive T-Cell Immunotherapy of Cancer Using Chimeric Antigen Receptor-Grafted T Cells. Arch. Immunol. Ther. Exp. 2010, 58, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Curran, K.J.; Pegram, H.J.; Brentjens, R.J. Chimeric Antigen Receptors for T Cell Immunotherapy: Current Understanding and Future Directions. J. Gene Med. 2012, 14, 405–415. [Google Scholar] [CrossRef] [Green Version]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177ra38. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Kassim, S.H.; Somerville, R.P.T.; Carpenter, R.O.; Maryalice, S.S.; Yang, J.C.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated with Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J. Clin. Oncol. 2015, 33, 540–549. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Munoz, J.; Goy, A.; Locke, F.L.; Jacobson, C.A.; Hill, B.T.; Timmerman, J.M.; Holmes, H.; Jaglowski, S.; Flinn, I.W.; et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N. Engl. J. Med. 2020, 382, 1331–1342. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Yakoub-Agha, I.; Chabannon, C.; Bader, P.; Basak, G.W.; Bonig, H.; Ciceri, F.; Corbacioglu, S.; Duarte, R.F.; Einsele, H.; Hudecek, M.; et al. Management of Adults and Children Undergoing Chimeric Antigen Receptor T-Cell Therapy: Best Practice Recommendations of the European Society for Blood and Marrow Transplantation (EBMT) and the Joint Accreditation Committee of ISCT and EBMT (JACIE). Haematologica 2020, 105, 297–316. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schubert, M.L.; Schmitt, M.; Wang, L.; Ramos, C.A.; Jordan, K.; Müller-Tidow, C.; Dreger, P. Side-Effect Management of Chimeric Antigen Receptor (CAR) T-Cell Therapy. Ann. Oncol. 2021, 32, 34–48. [Google Scholar] [CrossRef]
- Carpenter, R.O.; Evbuomwan, M.O.; Pittaluga, S.; Rose, J.J.; Raffeld, M.; Yang, S.; Gress, R.E.; Hakim, F.T.; Kochenderfer, J.N. B-Cell Maturation Antigen Is a Promising Target for Adoptive T-Cell Therapy of Multiple Myeloma. Clin. Cancer Res. 2013, 19, 2048–2060. [Google Scholar] [CrossRef] [Green Version]
- Ghermezi, M.; Li, M.; Vardanyan, S.; Harutyunyan, N.M.; Gottlieb, J.; Berenson, A.; Spektor, T.M.; Andreu-Vieyra, C.; Petraki, S.; Sanchez, E.; et al. Serum B-Cell Maturation Antigen: A Novel Biomarker to Predict Outcomes for Multiple Myeloma Patients. Haematologica 2017, 102, 785–795. [Google Scholar] [CrossRef] [Green Version]
- Seckinger, A.; Delgado, J.A.; Moser, S.; Moreno, L.; Neuber, B.; Grab, A.; Lipp, S.; Merino, J.; Prosper, F.; Emde, M.; et al. Target Expression, Generation, Preclinical Activity, and Pharmacokinetics of the BCMA-T Cell Bispecific Antibody EM801 for Multiple Myeloma Treatment. Cancer Cell 2017, 31, 396–410. [Google Scholar] [CrossRef] [Green Version]
- Palma, B.D.; Marchica, V.; Catarozzo, M.T.; Giuliani, N.; Accardi, F. Monoclonal and Bispecific Anti-BCMA Antibodies in Multiple Myeloma. J. Clin. Med. 2020, 9, 3022. [Google Scholar] [CrossRef]
- Mohyuddin, G.R.; Banerjee, R.; Alam, Z.; Berger, K.E.; Chakraborty, R. Rethinking Mechanisms of Neurotoxicity with BCMA Directed Therapy. Crit. Rev. Oncol. Hematol. 2021, 166, 103453. [Google Scholar] [CrossRef]
- Lin, Y.; Raje, N.S.; Berdeja, J.G.; Siegel, D.S.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Massaro, M.; et al. Idecabtagene Vicleucel (Ide-Cel, Bb2121), a BCMA-Directed CAR T Cell Therapy, in Patients with Relapsed and Refractory Multiple Myeloma: Updated Results from Phase 1 CRB-401 Study. Blood 2020, 136, 26–27. [Google Scholar] [CrossRef]
- Raje, N.; Berdeja, J.; Lin, Y.; Siegel, D.; Jagannath, S.; Madduri, D.; Liedtke, M.; Rosenblatt, J.; Maus, M.V.; Turka, A.; et al. Anti-BCMA CAR T-Cell Therapy Bb2121 in Relapsed or Refractory Multiple Myeloma. N. Engl. J. Med. 2019, 380, 1726–1737. [Google Scholar] [CrossRef]
- Mailankody, S.; Matous, J.V.; Liedtke, M.; Sidana, S.; Malik, S.; Nath, R.; Oluwole, O.O.; Karski, E.E.; Lovelace, W.; Zhou, X.; et al. Universal: An Allogeneic First-in-Human Study of the Anti-Bcma ALLO-715 and the Anti-CD52 ALLO-647 in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 24–25. [Google Scholar] [CrossRef]
- Larry, D.; Anderson, J.; Munshi, N.C.; Shah, N.; Jagannath, S.; Berdeja, J.G.; Lonial, S.; Raje, N.S.; Siegel, D.S.D.; Lin, Y.; et al. Idecabtagene Vicleucel (Ide-Cel, Bb2121), a BCMA-Directed CAR T Cell Therapy, in Relapsed and Refractory Multiple Myeloma: Updated KarMMa Results. J. Clin. Oncol. 2021, 39, 8016. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A.; et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-Y.; Zhao, W.-H.; Liu, J.; Chen, Y.-X.; Cao, X.-M.; Yang, Y.; Zhang, Y.-L.; Wang, F.-X.; Zhang, P.-Y.; Lei, B.; et al. Long-Term Follow-up of a Phase 1, First-in-Human Open-Label Study of LCAR-B38M, a Structurally Differentiated Chimeric Antigen Receptor T (CAR-T) Cell Therapy Targeting B-Cell Maturation Antigen (BCMA), in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2019, 134, 579. [Google Scholar] [CrossRef]
- Lin, Y.; Martin, T.; Cohen, A.D.; Jakubowiak, A.; Jasielec, J.; Usmani, S.Z.; Madduri, D.; Agha, M.; Stewart, A.K.; Singh, I.; et al. Cytokine Release Syndrome in Patients with Relapsed/Refractory Multiple Myeloma Treated with Ciltacabtagene Autoleucel in the Phase 1b/2 CARTITUDE-1 Study. Blood 2020, 136, 45–46. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene Autoleucel, a B-Cell Maturation Antigen-Directed Chimeric Antigen Receptor T-Cell Therapy in Patients with Relapsed or Refractory Multiple Myeloma (CARTITUDE-1): A Phase 1b/2 Open-Label Study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Mailankody, S.; Jakubowiak, A.J.; Htut, M.; Costa, L.J.; Lee, K.; Ganguly, S.; Kaufman, J.L.; Siegel, D.S.D.; Bensinger, W.; Cota, M.; et al. Orvacabtagene Autoleucel (Orva-Cel), a B-Cell Maturation Antigen (BCMA)-Directed CAR T Cell Therapy for Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Update of the Phase 1/2 EVOLVE Study (NCT03430011). J. Clin. Oncol. 2020, 38, 8504. [Google Scholar] [CrossRef]
- Alsina, M.; Shah, N.; Raje, N.S.; Jagannath, S.; Madduri, D.; Kaufman, J.L.; Siegel, D.S.; Munshi, N.C.; Rosenblatt, J.; Lin, Y.; et al. Updated Results from the Phase I CRB-402 Study of Anti-Bcma CAR-T Cell Therapy Bb21217 in Patients with Relapsed and Refractory Multiple Myeloma: Correlation of Expansion and Duration of Response with T Cell Phenotypes. Blood 2020, 136, 25–26. [Google Scholar] [CrossRef]
- Brudno, J.N.; Maric, I.; Hartman, S.D.; Rose, J.J.; Wang, M.; Lam, N.; Stetler-Stevenson, M.; Salem, D.; Yuan, C.; Pavletic, S.; et al. T Cells Genetically Modified to Express an Anti–B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Poor-Prognosis Relapsed Multiple Myeloma. J. Clin. Oncol. 2018, 36, 2267–2280. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.A.; Shi, V.; Maric, I.; Wang, M.; Stroncek, D.F.; Rose, J.J.; Brudno, J.N.; Stetler-Stevenson, M.; Feldman, S.A.; Hansen, B.G.; et al. T Cells Expressing an Anti–B-Cell Maturation Antigen Chimeric Antigen Receptor Cause Remissions of Multiple Myeloma. Blood 2016, 128, 1688–1700. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B Cell Maturation Antigen–Specific CAR T Cells Are Clinically Active in Multiple Myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.K.; Baz, R.C.; Orlowski, R.Z.; Anderson, L.D.; Ma, H.; Shrewsbury, A.; Croghan, K.A.; Bilgi, M.; Kansagra, A.; Kapoor, P.; et al. Results from Lummicar-2: A Phase 1b/2 Study of Fully Human B-Cell Maturation Antigen-Specific CAR T Cells (CT053) in Patients with Relapsed and/or Refractory Multiple Myeloma. Blood 2020, 136, 28–29. [Google Scholar] [CrossRef]
- An, G.; Sui, W.; Wang, T.; Qu, X.; Zhang, X.; Yang, J.; Zhang, Y.; Zhang, L.; Zhu, J.; Zheng, C.; et al. An Anti-Bcma CAR T-Cell Therapy (C-CAR088) Shows Promising Safety and Efficacy Profile in Relapsed or Refractory Multiple Myeloma. Blood 2020, 136, 29–30. [Google Scholar] [CrossRef]
- Caraccio, C.; Krishna, S.; Phillips, D.J.; Schürch, C.M. Bispecific Antibodies for Multiple Myeloma: A Review of Targets, Drugs, Clinical Trials, and Future Directions. Front. Immunol. 2020, 11, 501. [Google Scholar] [CrossRef]
- Lejeune, M.; Köse, M.C.; Duray, E.; Einsele, H.; Beguin, Y.; Caers, J. Bispecific, T-Cell-Recruiting Antibodies in B-Cell Malignancies. Front. Immunol. 2020, 11, 762. [Google Scholar] [CrossRef]
- Verkleij, C.P.; Frerichs, K.A.; Broekmans, M.; Absalah, S.; Maas-Bosman, P.W.; Kruyswijk, S.; Nijhof, I.S.; Mutis, T.; Zweegman, S.; van de Donk, N.W. T-Cell Redirecting Bispecific Antibodies Targeting BCMA for the Treatment of Multiple Myeloma. Oncotarget 2020, 11, 4076–4081. [Google Scholar] [CrossRef]
- Kumar, S.; Rajkumar, S.V. BiTEing the Tumor. J. Clin. Oncol. 2020, 38, 2077–2079. [Google Scholar] [CrossRef]
- Tian, Z.; Liu, M.; Zhang, Y.; Wang, X. Bispecific T Cell Engagers: An Emerging Therapy for Management of Hematologic Malignancies. J. Hematol. Oncol. 2021, 14, 75. [Google Scholar] [CrossRef]
- Sanchez, L.; Dardac, A.; Madduri, D.; Richard, S.; Richter, J. B-Cell Maturation Antigen (BCMA) in Multiple Myeloma: The New Frontier of Targeted Therapies. Ther. Adv. Hematol. 2021, 12, 2040620721989585. [Google Scholar] [CrossRef] [PubMed]
- Geis, M.; Nowotny, B.; Bohn, M.D.; Kouhestani, D.; Einsele, H.; Bumm, T.; Stuhler, G. Combinatorial Targeting of Multiple Myeloma by Complementing T Cell Engaging Antibody Fragments. Commun. Biol. 2021, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Einsele, H.; Danhof, S. Bispecific Antibodies: A New Era of Treatment for Multiple Myeloma. J. Clin. Med. 2020, 9, 2166. [Google Scholar] [CrossRef] [PubMed]
- Topp, M.S.; Duell, J.; Zugmaier, G.; Attal, M.; Moreau, P.; Langer, C.; Krönke, J.; Facon, T.; Salnikov, A.V.; Lesley, R.; et al. Anti–B-Cell Maturation Antigen Bite Molecule AMG 420 Induces Responses in Multiple Myeloma. J. Clin. Oncol. 2020, 38, 775–783. [Google Scholar] [CrossRef]
- Harrison, S.J.; Minnema, M.C.; Lee, H.C.; Spencer, A.; Kapoor, P.; Madduri, D.; Larsen, J.; Ailawadhi, S.; Kaufman, J.L.; Raab, M.S.; et al. A Phase 1 First in Human (FIH) Study of AMG 701, an Anti-B-Cell Maturation Antigen (BCMA) Half-Life Extended (HLE) BiTE® (Bispecific T-Cell Engager) Molecule, in Relapsed/Refractory (RR) Multiple Myeloma (MM). Blood 2020, 136, 28–29. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Garfall, A.L.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Rosinol, L.; Chari, A.; Bhutani, M.; Karlin, L.; et al. Teclistamab, a B-Cell Maturation Antigen × CD3 Bispecific Antibody, in Patients with Relapsed or Refractory Multiple Myeloma (MajesTEC-1): A Multicentre, Open-Label, Single-Arm, Phase 1 Study. Lancet 2021, 398, 665–674. [Google Scholar] [CrossRef]
- Krishnan, A.Y.; Garfall, A.L.; Mateos, M.-V.; van de Donk, N.W.C.J.; Nahi, H.; San-Miguel, J.F.; Oriol, A.; Rosiñol, L.; Chari, A.; Bhutani, M.; et al. Updated Phase 1 Results of Teclistamab, a B-Cell Maturation Antigen (BCMA) × CD3 Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma (MM). J. Clin. Oncol. 2021, 39, 8007. [Google Scholar] [CrossRef]
- Usmani, S.Z.; Mateos, M.-V.; Nahi, H.; Krishnan, A.Y.; van de Donk, N.W.C.J.; Miguel, J.S.; Oriol, A.; Rosiñol, L.; Chari, A.; Adams, H.; et al. Phase I Study of Teclistamab, a Humanized B-Cell Maturation Antigen (BCMA) x CD3 Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma (R/R MM). J. Clin. Oncol. 2020, 38, 100. [Google Scholar] [CrossRef]
- Madduri, D.; Rosko, A.; Brayer, J.; Zonder, J.; Bensinger, W.I.; Li, J.; Xu, L.; Adriaens, L.; Chokshi, D.; Zhang, W.; et al. REGN5458, a BCMA x CD3 Bispecific Monoclonal Antibody, Induces Deep and Durable Responses in Patients with Relapsed/Refractory Multiple Myeloma (RRMM). Blood 2020, 136, 41–42. [Google Scholar] [CrossRef]
- Rodriguez, C.; D’Souza, A.; Shah, N.; Voorhees, P.M.; Buelow, B.; Vij, R.; Kumar, S.K. Initial Results of a Phase I Study of TNB-383B, a BCMA x CD3 Bispecific T-Cell Redirecting Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 43–44. [Google Scholar] [CrossRef]
- Bahlis, N.J.; Raje, N.S.; Costello, C.; Dholaria, B.R.; Solh, M.M.; Levy, M.Y.; Tomasson, M.H.; Dube, H.; Liu, F.; Liao, K.H.; et al. Efficacy and Safety of Elranatamab (PF-06863135), a B-Cell Maturation Antigen (BCMA)-CD3 Bispecific Antibody, in Patients with Relapsed or Refractory Multiple Myeloma (MM). J. Clin. Oncol. 2021, 39, 8006. [Google Scholar] [CrossRef]
- Costa, L.J.; Wong, S.W.; Bermúdez, A.; de la Rubia, J.; Mateos, M.-V.; Ocio, E.M.; Rodríguez-Otero, P.; San-Miguel, J.; Li, S.; Sarmiento, R.; et al. First Clinical Study of the B-Cell Maturation Antigen (BCMA) 2+1 T Cell Engager (TCE) CC-93269 in Patients (Pts) with Relapsed/Refractory Multiple Myeloma (RRMM): Interim Results of a Phase 1 Multicenter Trial. Blood 2019, 134, 143. [Google Scholar] [CrossRef]
- Richter, J.R.; Landgren, C.O.; Kauh, J.S.; Back, J.; Salhi, Y.; Reddy, V.; Bayever, E.; Berdeja, J.G. Phase 1, Multicenter, Open-Label Study of Single-Agent Bispecific Antibody t-Cell Engager GBR 1342 in Relapsed/Refractory Multiple Myeloma. J. Clin. Oncol. 2018, 36, TPS3132. [Google Scholar] [CrossRef]
- de Zafra, C.L.Z.; Fajardo, F.; Zhong, W.; Bernett, M.J.; Muchhal, U.S.; Moore, G.L.; Stevens, J.; Case, R.; Pearson, J.T.; Liu, S.; et al. Targeting Multiple Myeloma with AMG 424, a Novel Anti-CD38/CD3 Bispecific T-Cell-Recruiting Antibody Optimized for Cytotoxicity and Cytokine Release. Clin. Cancer Res. 2019, 25, 3921–3933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillarisetti, K.; Edavettal, S.; Mendonça, M.; Li, Y.; Tornetta, M.; Babich, A.; Majewski, N.; Husovsky, M.; Reeves, D.; Walsh, E.; et al. A T-Cell–Redirecting Bispecific G-Protein–Coupled Receptor Class 5 Member D x CD3 Antibody to Treat Multiple Myeloma. Blood 2020, 135, 1232–1243. [Google Scholar] [CrossRef] [PubMed]
- Mancia, S.S.; Farrell, A.; Louw, K.; Florendo, E.; Aronson, E.; Purcell, K.; Catamero, D.D.; Escalon, J.; Thomas, J.; Aponte, A.; et al. Characterization and Management of Oral and Dermatological Toxicities in Patients Receiving the CD3 X GPRC5D Bispecific Antibody Talquetamab (JNJ-64407564) for the Treatment of Relapsed and/or Refractory Multiple Myeloma. Blood 2021, 138, 1658. [Google Scholar] [CrossRef]
- Cohen, A.D.; Harrison, S.J.; Krishnan, A.; Fonseca, R.; Forsberg, P.A.; Spencer, A.; Berdeja, J.G.; Laubach, J.P.; Li, M.; Choeurng, V.; et al. Initial Clinical Activity and Safety of BFCR4350A, a FcRH5/CD3 T-Cell-Engaging Bispecific Antibody, in Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 42–43. [Google Scholar] [CrossRef]
- Lambert, J.M.; Morris, C.Q. Antibody-Drug Conjugates (ADCs) for Personalized Treatment of Solid Tumors: A Review. Adv. Ther. 2017, 34, 1015–1035. [Google Scholar] [CrossRef] [Green Version]
- Ryan, M.C.; Hering, M.; Peckham, D.; McDonagh, C.F.; Brown, L.; Kim, K.M.; Meyer, D.L.; Zabinski, R.F.; Grewal, I.S.; Carter, P.J. Antibody Targeting of B-Cell Maturation Antigen on Malignant Plasma Cells. Mol. Cancer Ther. 2007, 6, 3009–3018. [Google Scholar] [CrossRef] [Green Version]
- Tai, Y.T.; Mayes, P.A.; Acharya, C.; Zhong, M.Y.; Cea, M.; Cagnetta, A.; Craigen, J.; Yates, J.; Gliddon, L.; Fieles, W.; et al. Novel Anti–B-Cell Maturation Antigen Antibody-Drug Conjugate (GSK2857916) Selectively Induces Killing of Multiple Myeloma. Blood 2014, 123, 3128–3138. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab Mafodotin for Relapsed or Refractory Multiple Myeloma (DREAMM-2): A Two-Arm, Randomised, Open-Label, Phase 2 Study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- Kumar, S.K.; Migkou, M.; Bhutani, M.; Spencer, A.; Ailawadhi, S.; Kalff, A.; Walcott, F.; Pore, N.; Gibson, D.; Wang, F.; et al. Phase 1, First-in-Human Study of MEDI2228, a BCMA-Targeted ADC in Patients with Relapsed/Refractory Multiple Myeloma. Blood 2020, 136, 26–27. [Google Scholar] [CrossRef]
- Figueroa-Vazquez, V.; Ko, J.; Breunig, C.; Baumann, A.; Giesen, N.; Palfi, A.; Muller, C.; Lutz, C.; Hechler, T.; Kulke, M.; et al. HDP-101, an Anti-Bcma Antibody–Drug Conjugate, Safely Delivers Amanitin to Induce Cell Death in Proliferating and Resting Multiple Myeloma Cells. Mol. Cancer Ther. 2021, 20, 367–378. [Google Scholar] [CrossRef]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human Bone Marrow Stromal Cells Suppress T-Lymphocyte Proliferation Induced by Cellular or Nonspecific Mitogenic Stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.; Jung, K.; Jeon, Y.; Yun, S.; Kim, T.W.; Choi, I. Knockdown of the Interleukin-6 Receptor Alpha Chain of Dendritic Cell Vaccines Enhances the Therapeutic Potential against IL-6 Producing Tumors. Vaccine 2010, 29, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Ohno, Y.; Kitamura, H.; Takahashi, N.; Ohtake, J.; Kaneumi, S.; Sumida, K.; Homma, S.; Kawamura, H.; Minagawa, N.; Shibasaki, S.; et al. IL-6 down-Regulates HLA Class II Expression and IL-12 Production of Human Dendritic Cells to Impair Activation of Antigen-Specific CD4+ T Cells. Cancer Immunol. Immunother. 2016, 65, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumida, K.; Wakita, D.; Narita, Y.; Masuko, K.; Terada, S.; Watanabe, K.; Satoh, T.; Kitamura, H.; Nishimura, T. Anti-IL-6 Receptor MAb Eliminates Myeloid-Derived Suppressor Cells and Inhibits Tumor Growth by Enhancing T-Cell Responses. Eur. J. Immunol. 2012, 42, 2060–2072. [Google Scholar] [CrossRef]
- Chen, L.; Willis, S.N.; Wei, A.; Smith, B.J.; Fletcher, J.I.; Hinds, M.G.; Colman, P.M.; Day, C.L.; Adams, J.M.; Huang, D.C.S. Differential Targeting of Prosurvival Bcl-2 Proteins by Their BH3-Only Ligands Allows Complementary Apoptotic Function. Mol. Cell 2005, 17, 393–403. [Google Scholar] [CrossRef]
- Roy, P.; Sarkar, U.A.; Basak, S. The NF-ΚB Activating Pathways in Multiple Myeloma. Biomedicines 2018, 6, 59. [Google Scholar] [CrossRef] [Green Version]
- Shehata, M.; Schnabl, S.; Demirtas, D.; Hilgarth, M.; Hubmann, R.; Ponath, E.; Badrnya, S.; Lehner, C.; Hoelbl, A.; Duechler, M.; et al. Reconstitution of PTEN Activity by CK2 Inhibitors and Interference with the PI3-K/Akt Cascade Counteract the Antiapoptotic Effect of Human Stromal Cells in Chronic Lymphocytic Leukemia. Blood 2010, 116, 2513–2521. [Google Scholar] [CrossRef]
- Di Marzo, L.; Desantis, V.; Solimando, A.G.; Ruggieri, S.; Annese, T.; Nico, B.; Fumarulo, R.; Vacca, A.; Frassanito, M.A.; di Marzo, L.; et al. Microenvironment Drug Resistance in Multiple Myeloma: Emerging New Players. Oncotarget 2016, 7, 60698–60711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutton, V.R.; Davis, J.E.; Cancilla, M.; Johnstone, R.W.; Ruefli, A.A.; Sedelies, K.; Browne, K.A.; Trapani, J.A. Initiation of Apoptosis by Granzyme B Requires Direct Cleavage of Bid, but Not Direct Granzyme B–Mediated Caspase Activation. J. Exp. Med. 2000, 192, 1403–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalán, E.; Jaime-Sánchez, P.; Aguilo, N.; Simon, M.M.; Froelich, C.J.; Pardo, J. Mouse Cytotoxic T Cell-Derived Granzyme B Activates the Mitochondrial Cell Death Pathway in a Bim-Dependent Fashion. J. Biol. Chem. 2015, 290, 6868–6877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, S.P.; Brunet, M.; Martin, S.J. Granzymes in Cancer and Immunity. Cell Death Differ. 2010, 17, 616–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewen, C.L.; Kane, K.P.; Bleackley, R.C. A Quarter Century of Granzymes. Cell Death Differ. 2011, 19, 28–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holthof, L.C.; van der Horst, H.J.; Poels, R.; van der Schans, J.T.; Gelderloos, A.T.; Li, F.; Lokhorst, H.; Zweegman, S.; Themeli, M.; van de Donk, N.W.C.J.; et al. The Impact and Modulation of Microenvironment-Induced Immune Resistance Against CAR T Cell and Antibody Treatments in Multiple Myeloma. Blood 2019, 134, 137. [Google Scholar] [CrossRef]
- Frerichs, K.A.; Nagy, N.A.; Lindenbergh, P.L.; Bosman, P.; Marin Soto, J.; Broekmans, M.; Groen, R.W.J.; Themeli, M.; Nieuwenhuis, L.; Stege, C.; et al. CD38-Targeting Antibodies in Multiple Myeloma: Mechanisms of Action and Clinical Experience. Expert Rev. Clin. Immunol. 2018, 14, 197–206. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Study Name | Phase | Setting | Treatment | Number of Patients | ORR (%) | CR (%) | MRD Neg (%) | NCT Number | References |
|---|---|---|---|---|---|---|---|---|---|
| ALCYONE (MMY3007) | 3 | NTE NDMM | Dara-VMP vs. VMP | 706 | 90.9 vs. 73.9 | 42.6 vs. 24.4 | 22.3 vs. 6.2 | NCT03158688 | [36,37] |
| MAIA (MMY3008) | 3 | NTE NDMM | Dara-Rd vs. Rd | 737 | 92.9 vs. 81.3 | 47.6 vs. 24.9 | 24.2 vs. 7.3 | NTC02252172 | [38,39] |
| CASSIOPEIA (MMY3006) | 3 | TE NDMM | Dara-VTD vs. VTD | 1085 | 92.6 vs. 89.9 | 39 vs. 26 | 64 vs. 44 | NTC02541383 | [35,40] |
| POLLUX (MMY3003) | 3 | RRMM | Dara-Rd vs. Rd | 569 | 92.9 vs. 76.4 | 43.1 vs. 19.2 | 22.4 vs. 4.6 | NCT02076009 | [41] |
| CASTOR (MMY3004) | 3 | RRMM | Dara-Vd vs. Vd | 500 | 85 vs. 63 92 vs. 74 (1 prior line treatment) | 30 vs. 10 43 vs. 15 | 14 vs. 2 20 vs. 3 | NCT02136134 | [42] |
| CANDOR | 3 | RRMM | Dara-Kd vs. Kd | 466 | 84.3 vs. 74.7 | 29 vs. 10 | 14 vs. 3 | NCT03158688 | [35,43] |
| IKEMA | 3 | RRMM | Isa-Kd vs. Kd | 302 | 87 vs. 83 | 40 vs. 28 | 30 vs. 13 | NCT03275285 | [35,45] |
| ICARIA-MM | 3 | RRMM | Isa-Pd vs. Pd | 307 | 60 vs. 35 | 5 vs. 2 | 5 vs. 0 | NCT02990338 | [44,46] |
| CAR Construct | Study Name | Antigen | Number of Patients | High Risk/EMD (%) | CR (%) | CRS (%) | Neurotoxicity (%) | NCT Number | References |
|---|---|---|---|---|---|---|---|---|---|
| Ide-cel (bb2121) | CRB-401, Phase 1 | BCMA | 62 | 27/37 | 39 | 76 | 36 | NTC02658929 | [71] |
| Ide-cel (bb2121) | KarMMa, Phase 2 | BCMA | 128 | 35/39 | 33 | 84 | 18 | NCT03361748 | [74,75] |
| Cilta-cel (LCAR-B38M) | LEGEND-2, Phase 1/2 | Biepitope to BCMA (VHH1 and VHH2) | 57 | NR | 74 | 90 | 1 | NCT03090659 | [76] |
| Cilta-cel (JNJ-4528) | CARTITUDE-1, Phase 1b/2 | Biepitope to BCMA (VHH1 and VHH2) | 97 | 24/13 | 80 | 95 | 21 | NCT03548207 | [77,78] |
| Orva-cel (JCARH125) | EVOLVE, Phase 1/2 | BCMA | 62 | 41/23 | 36 | 89 | 13 | NCT03430011 | [79] |
| bb21217 | CRB-402, Phase 1 | BCMA | 69 | 33/NR | 29 | 70 | 16 | NCT03274219 | [80] |
| CAR-BCMA | Phase 1 | BCMA | 24 | 46/NR | 8 | 71 | NR | NCT02215967 | [81,82] |
| UPenn-CART-BCMA | Phase 1 | BCMA | 25 | 96/28 | 8 | 88 | 32 | NCT02546167 | [83] |
| CT053 | LUMMICAR STUDY 2, Phase 1b/2 | BCMA | 20 | 55/25 | 25 | 79 | 16 | NCT03915184 | [84] |
| ALLO-715 | UNIVERSAL, Phase 1 | BCMA | 31 | 48/23 | VGPR: 40 | 45 | 0 | NCT04093596 | [73] |
| C-CAR088 | Phase 1 | BCMA | 23 | 81/NR | 44 | 91 | 4 | NCT03751293 NCT03815383 NCT04322292 NCT048295018 | [85] |
| Agents | Type | Phase | Target | Number of Patients | ORR (%) | CRS (%) | NCT Number | References |
|---|---|---|---|---|---|---|---|---|
| AMG420 | BiTE | 1 | BCMAxCD3 | 42 | 70 | 38 | NCT03836053 | [94] |
| AMG701 | BiTE | 1/2 | BCMAxCD3 | 75 | 83 | 61 | NCT03287908 | [95] |
| Teclistamab (JNJ-64007957) | BsAb | 1/2 | BCMAxCD3 | 149 | 69 | 55 | NCT04557098 NCT03145181 | [96,97,98] |
| REGN5458 | BsAb | 1/2 | BCMAxCD3 | 49 | 62.5 | 39 | NCT03761108 | [99] |
| TNB-383B | BsAb | 1 | BCMAxCD3 | 58 | 80 | 45 | NCT03933735 | [100] |
| Elranatamab (PF-06863135) | BsAb | 2 | BCMAxCD3 | 30 | 83.3 | 73 | NCT04649359 | [101] |
| CC-93269 | BsAb | 1 | BCMAxCD3 | 30 | 89 | 77 | NCT03486067 | [102] |
| GBR1342 | BiTE | 1 | CD38xCD3 | 19 | NR | NR | NCT03309111 | [103] |
| AMG424 | BsAb | 1 | CD38xCD3 | NR | NR | NR | NCT03445663 | [104] |
| Talquetamab (JNJ-64407564) | BsAbs | 1 | GPRC5dxCD3 | NR | NR | NR | NCT04108195 NCT03399799 NCT04773522 | [105,106] |
| Cevostamab (BFCR4350A) | BiTE | 1 | FCRH5xCD3 | 160 | 54.8% | 80.7% | NCT03275103 | [107] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neumeister, P.; Schulz, E.; Pansy, K.; Szmyra, M.; Deutsch, A.J. Targeting the Microenvironment for Treating Multiple Myeloma. Int. J. Mol. Sci. 2022, 23, 7627. https://doi.org/10.3390/ijms23147627
Neumeister P, Schulz E, Pansy K, Szmyra M, Deutsch AJ. Targeting the Microenvironment for Treating Multiple Myeloma. International Journal of Molecular Sciences. 2022; 23(14):7627. https://doi.org/10.3390/ijms23147627
Chicago/Turabian StyleNeumeister, Peter, Eduard Schulz, Katrin Pansy, Marta Szmyra, and Alexander JA Deutsch. 2022. "Targeting the Microenvironment for Treating Multiple Myeloma" International Journal of Molecular Sciences 23, no. 14: 7627. https://doi.org/10.3390/ijms23147627