
Role of Histone Deacetylases in T-Cell Development and Function


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Chromatin and Post-Translational Modifications of Histones
1.2. Histone Deacetylases (HDACs)
1.3. T-Cells Maturation and Differentiation
2. HDACs in T-Cells
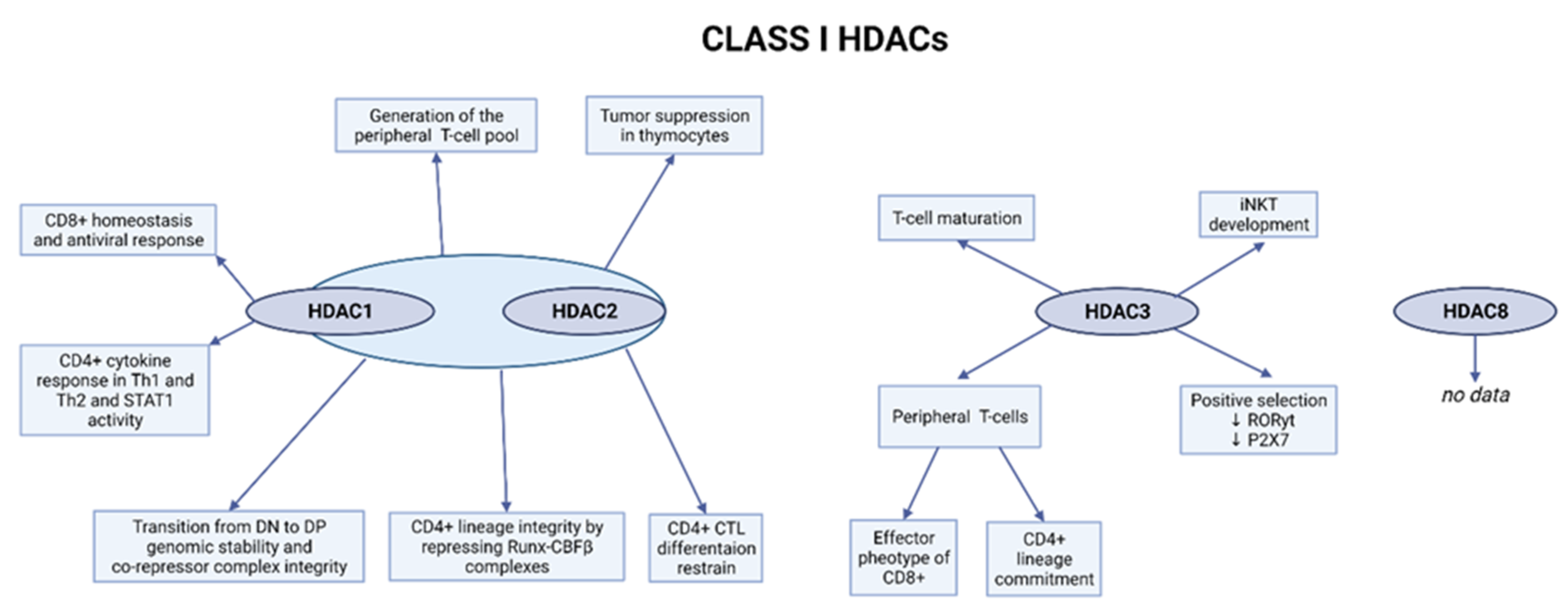
2.1. Class I HDACs
2.1.1. Role of HDAC1 and HDAC2 in the Early Stages of T-Cell Development
2.1.2. HDAC3 Is Involved in the Positive Selection Process and the Function of Peripheral T-Cells
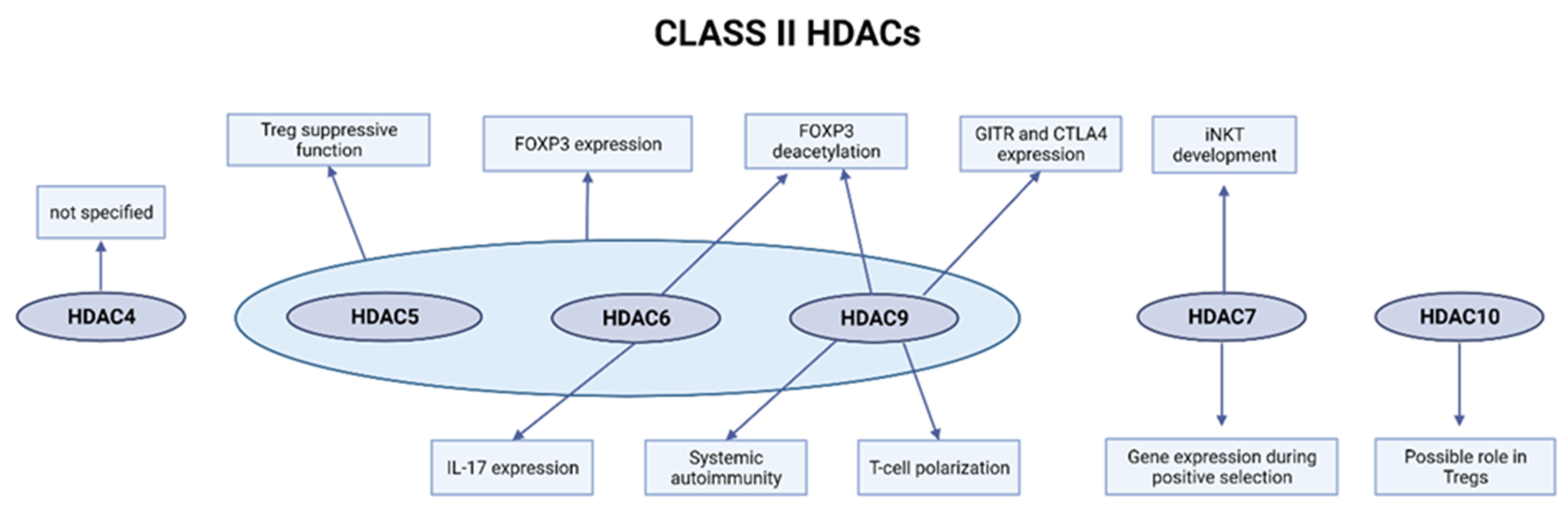
2.2. Class II HDACs
2.2.1. HDAC4 Is Expressed in the Multiple T-Cell Lineages but Is Not a Key Regulator of T-Cell Biology
2.2.2. Loss of HDAC5 Weakens the Tregs and Tconv Function
2.2.3. HDAC6 Is Involved in Treg Suppressive Function and FOXP3 Expression and Deacetylation
2.2.4. HDAC7 Regulates Gene Expression during Positive Selection and iNKT Development
2.2.5. Role of HDAC9 in Treg Function, T-Cell Polarization, and Systemic Autoimmunity
2.2.6. Role of HDAC10 in Treg Functionality and Immunosuppression
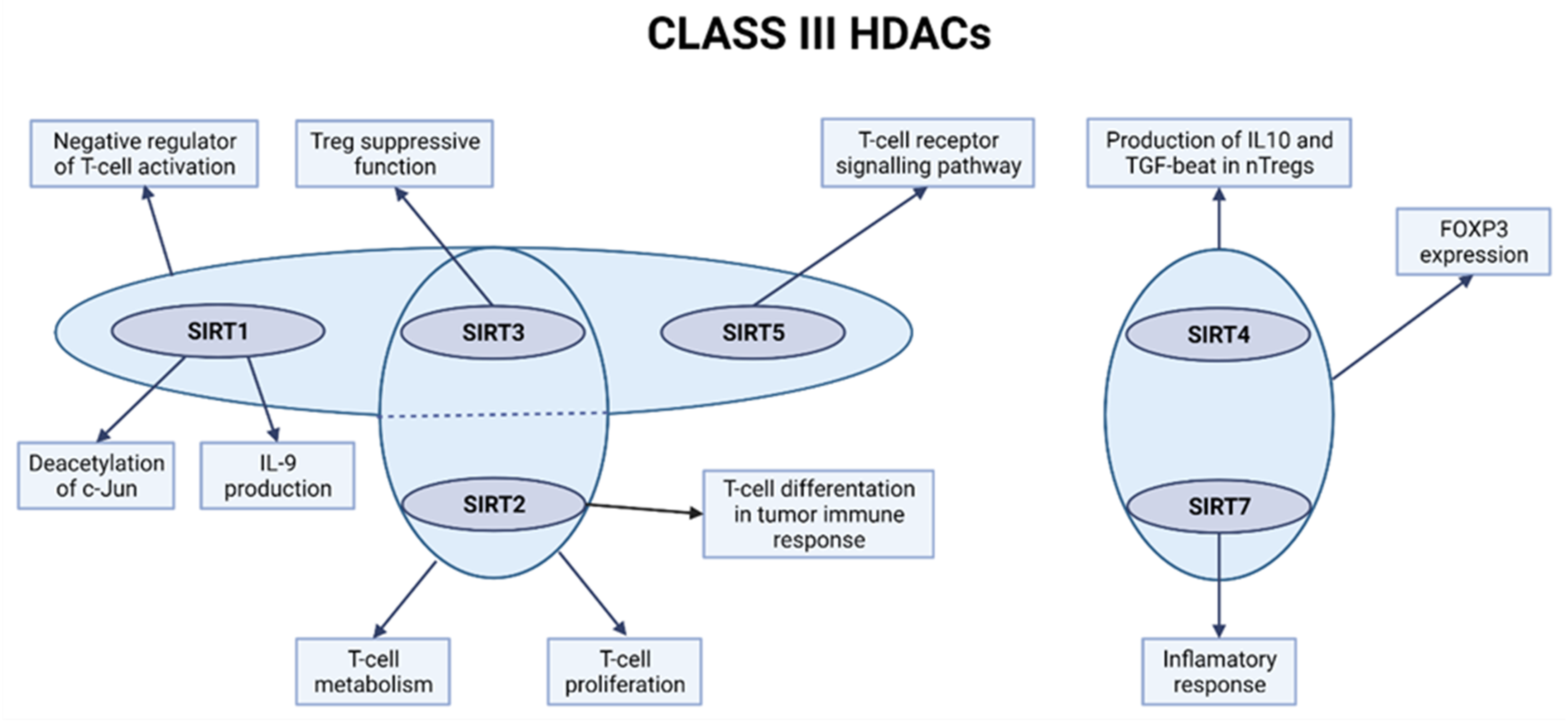
2.3. Class III HDACs
2.3.1. SIRT1 Is a Negative Regulator of T-Cell Activation
2.3.2. SIRT2 Regulates T-Cell Metabolism and Tumor T-Cell Immune Response
2.3.3. Role of SIRT3 in Promoting T-Cell Responses and Reducing Transplant Rejection
2.3.4. Role of SIRT4 and SIRT6 in Treg Regulations in Case of Traumatic Spinal Cord Injury
2.3.5. The Potential Involvement of SIRT5 in the T-Cell Receptor Signaling Pathway and SIRT7 in Inflammation Processes
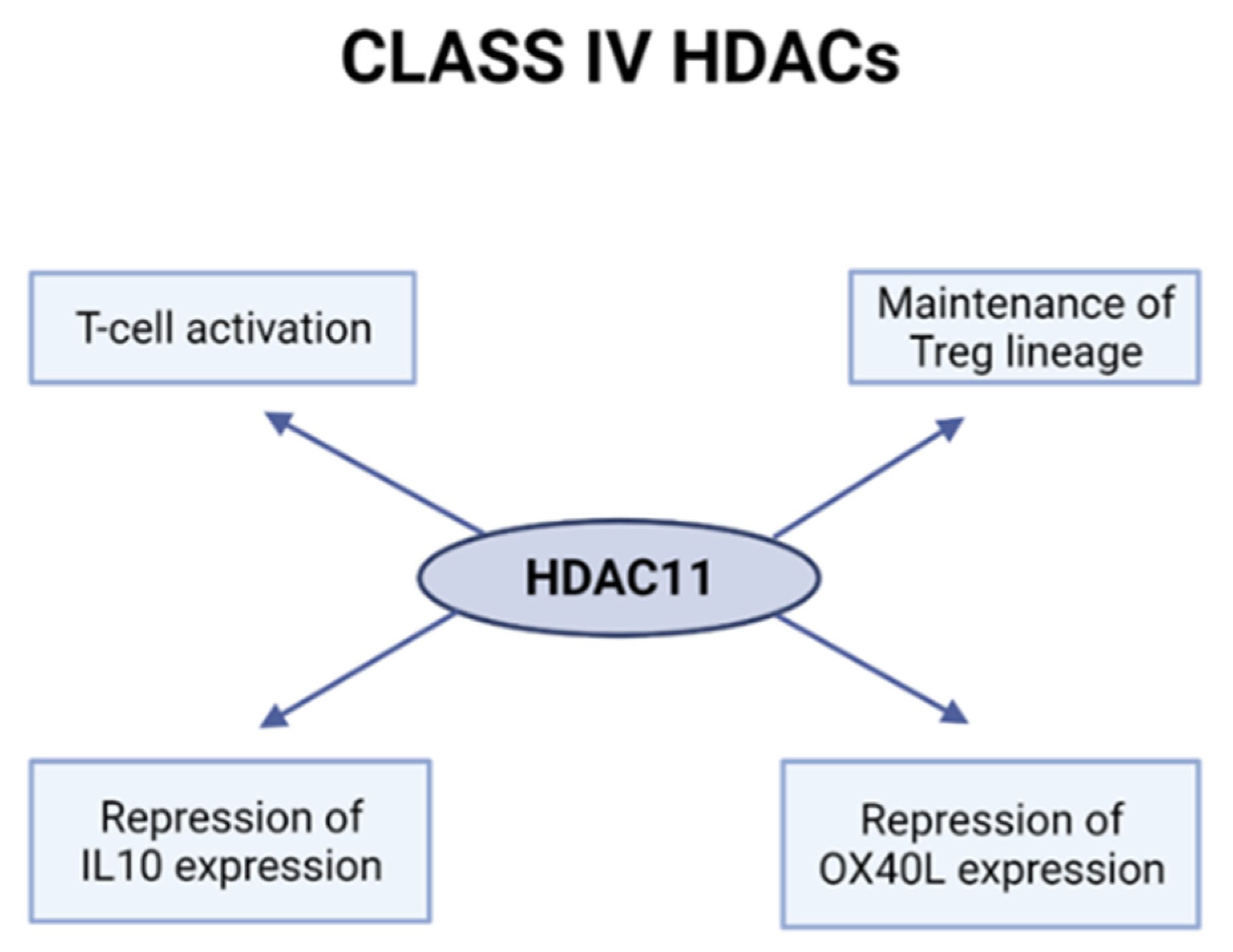
2.4. Class IV HDACs
HDAC11 Is Involved in T-Cell Activation and Treg Function
3. The Action of HDAC Inhibitors in the Maturation and Activation of T-Cells
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Woodcock, C.L.; Ghosh, R. Chromatin higher-order structure and dynamics. Cold Spring Harb. Perspect. Biol. 2010, 2, a000596. [Google Scholar] [CrossRef] [PubMed]
- Mariño-Ramírez, L.; Kann, M.G.; Shoemaker, B.A.; Landsman, D. Histone structure and nucleosome stability. Expert Rev. Proteom. 2005, 2, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Becker, P.B.; Workman, J.L. Nucleosome remodeling and epigenetics. Cold Spring Harb. Perspect. Biol. 2013, 5, a017905. [Google Scholar] [CrossRef] [Green Version]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [Green Version]
- Ramazi, S.; Allahverdi, A.; Zahiri, J. Evaluation of post-translational modifications in histone proteins: A review on histone modification defects in developmental and neurological disorders. J. Biosci. 2020, 45, 135. [Google Scholar] [CrossRef] [PubMed]
- Evertts, A.G.; Zee, B.M.; DiMaggio, P.A.; Gonzales-Cope, M.; Coller, H.A.; Garcia, B.A. Quantitative dynamics of the link between cellular metabolism and histone acetylation. J. Biol. Chem. 2013, 288, 12142–12151. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Coradin, M.; Porter, E.G.; Garcia, B.A. Accelerating the Field of Epigenetic Histone Modification Through Mass Spectrometry-Based Approaches. Mol. Cell. Proteom. 2021, 20, 100006. [Google Scholar] [CrossRef] [PubMed]
- Kschonsak, M.; Haering, C.H. Shaping mitotic chromosomes: From classical concepts to molecular mechanisms. Bioessays 2015, 37, 755–766. [Google Scholar] [CrossRef]
- Gujral, P.; Mahajan, V.; Lissaman, A.C.; Ponnampalam, A.P. Histone acetylation and the role of histone deacetylases in normal cyclic endometrium. Reprod. Biol. Endocrinol. 2020, 18, 84. [Google Scholar] [CrossRef]
- Bannister, A.J.; Schneider, R.; Kouzarides, T. Histone methylation: Dynamic or static? Cell 2002, 109, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Hyun, K.; Jeon, J.; Park, K.; Kim, J. Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 2017, 49, e324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greer, E.L.; Shi, Y. Histone methylation: A dynamic mark in health, disease and inheritance. Nat. Rev. Genet. 2012, 13, 343–357. [Google Scholar] [CrossRef] [Green Version]
- Rossetto, D.; Avvakumov, N.; Cote, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, T.G.; Poncet, S.; Derouiche, A.; Shi, L.; Mijakovic, I.; Noirot-Gros, M.-F. Role of Protein Phosphorylation in the Regulation of Cell Cycle and DNA-Related Processes in Bacteria. Front. Microbiol. 2016, 7, 184. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Chakravarti, D. A peek into the complex realm of histone phosphorylation. Mol. Cell. Biol. 2011, 31, 4858–4873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lu, Q.; Chang, C. Epigenetics in Health and Disease. Adv. Exp. Med. Biol. 2020, 1253, 3–55. [Google Scholar]
- Biswas, S.; Rao, C.M. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 2018, 837, 8–24. [Google Scholar] [CrossRef]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [Green Version]
- Erler, J.; Zhang, R.; Petridis, L.; Cheng, X.; Smith, J.C.; Langowski, J. The role of histone tails in the nucleosome: A computational study. Biophys. J. 2014, 107, 2911–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parbin, S.; Kar, S.; Shilpi, A.; Sengupta, D.; Deb, M.; Rath, S.K.; Patra, S.K. Histone deacetylases: A saga of perturbed acetylation homeostasis in cancer. J. Histochem. Cytochem. 2014, 62, 11–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartl, S.; Taplick, J.; Lagger, G.; Khier, H.; Kuchler, K.; Seiser, C. Identification of mouse histone deacetylase 1 as a growth factor-inducible gene. Mol. Cell. Biol. 1997, 17, 5033–5043. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.P.; Zhao, Y.T.; Zhao, T.C. Histone deacetylases and mechanisms of regulation of gene expression. Crit. Rev. Oncogenes. 2015, 20, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; He, C.; Wang, M.; Ma, X.; Mo, F.; Yang, S.; Han, J.; Wei, X. Targeting epigenetic regulators for cancer therapy: Mechanisms and advances in clinical trials. Signal Transduct. Target. Ther. 2019, 4, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitacre, J.M.; Lin, J.; Harding, A. T Cell Adaptive Immunity Proceeds through Environment-Induced Adaptation from the Exposure of Cryptic Genetic Variation. Front. Genet. 2012, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Meyer, C.; Zhu, C. T cell antigen recognition at the cell membrane. Mol. Immunol. 2012, 52, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Cho, S.; Spangrude, G.J. Hematopoietic stem cells: Generation and self-renewal. Cell Death Differ. 2007, 14, 1851–1859. [Google Scholar] [CrossRef]
- Muro, R.; Takayanagi, H.; Nitta, T. T cell receptor signaling for gammadeltaT cell development. Inflamm. Regen. 2019, 39, 6. [Google Scholar] [CrossRef]
- Ghaedi, M.; Steer, C.A.; Martinez-Gonzalez, I.; Halim, T.; Abraham, N.; Takei, F. Common-Lymphoid-Progenitor-Independent Pathways of Innate and T Lymphocyte Development. Cell Rep. 2016, 15, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Zarnitsyna, V.I.; Evavold, B.D.; Schoettle, L.N.; Blattman, J.N.; Antia, R. Estimating the diversity, completeness, and cross-reactivity of the T cell repertoire. Front. Immunol. 2013, 4, 485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, E.G.; Balk, S.P.; Aupeix, K.; Colonna, M.; Strominger, J.L.; Groh-Spies, V. Human T-cell receptor (TCR) alpha/beta + CD4-CD8- T cells express oligoclonal TCRs, share junctional motifs across TCR V beta-gene families, and phenotypically resemble memory T cells. Proc. Natl. Acad. Sci. USA 1993, 90, 11787–11791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Obaldia, M.E.; Bell, J.J.; Bhandoola, A. Early T-cell progenitors are the major granulocyte precursors in the adult mouse thymus. Blood 2013, 121, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, W.; Taniuchi, I. Transcriptional regulation of early T-cell development in the thymus. Eur. J. Immunol. 2016, 46, 531–538. [Google Scholar] [CrossRef] [Green Version]
- Robert, P.; Kunze-Schumacher, H.; Greiff, V.; Krueger, A. Modeling the Dynamics of T-Cell Development in the Thymus. Entropy 2021, 23, 437. [Google Scholar] [CrossRef]
- Zhao, Q.; Dai, H.; Liu, X.; Jiang, H.; Liu, W.; Feng, Z.; Zhang, N.; Gao, Y.; Dong, Z.; Zhou, X.; et al. Helper T Cells in Idiopathic Membranous Nephropathy. Front. Immunol. 2021, 12, 665629. [Google Scholar] [CrossRef]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Swain, S.L.; McKinstry, K.K.; Strutt, T.M. Expanding roles for CD4(+) T cells in immunity to viruses. Nat. Rev. Immunol. 2012, 12, 136–148. [Google Scholar] [CrossRef]
- Zhu, X.; Zhu, J. CD4 T Helper Cell Subsets and Related Human Immunological Disorders. Int. J. Mol. Sci. 2020, 21, 8011. [Google Scholar] [CrossRef]
- Rosendahl Huber, S.; van Beek, J.; de Jonge, J.; Luytjes, W.; van Baarle, D. T cell responses to viral infections-opportunities for Peptide vaccination. Front. Immunol. 2014, 5, 171. [Google Scholar] [CrossRef]
- Trapani, J.A. Target cell apoptosis induced by cytotoxic T cells and natural killer cells involves synergy between the pore-forming protein, perforin, and the serine protease, granzyme B. Aust. N. Z. J. Med. 1995, 25, 793–799. [Google Scholar] [CrossRef] [PubMed]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workman, C.J.; Szymczak-Workman, A.L.; Collison, L.W.; Pillai, M.R.; Vignali, D.A.A. The development and function of regulatory T cells. Cell. Mol. Life Sci. 2009, 66, 2603–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sojka, D.K.; Huang, Y.-H.; Fowell, D.J. Mechanisms of regulatory T-cell suppression-a diverse arsenal for a moving target. Immunology 2008, 124, 13–22. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Gol-Ara, M.; Jadidi-Niaragh, F.; Sadria, R.; Azizi, G.; Mirshafiey, A. The role of different subsets of regulatory T cells in immunopathogenesis of rheumatoid arthritis. Arthritis 2012, 2012, 805875. [Google Scholar] [CrossRef]
- Park, S.-Y.; Kim, J.-S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Torchy, M.P.; Hamiche, A.; Klaholz, B.P. Structure and function insights into the NuRD chromatin remodeling complex. Cell. Mol. Life Sci. 2015, 72, 2491–2507. [Google Scholar] [CrossRef]
- Tiana, M.; Acosta-Iborra, B.; Puente-Santamaría, L.; Hernansanz-Agustin, P.; Worsley-Hunt, R.; Masson, N.; García-Rio, F.; Mole, D.; Ratcliffe, P.; Wasserman, W.W.; et al. The SIN3A histone deacetylase complex is required for a complete transcriptional response to hypoxia. Nucleic Acids Res. 2018, 46, 120–133. [Google Scholar] [CrossRef] [Green Version]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1454–1458. [Google Scholar] [CrossRef] [Green Version]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.-M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Oberoi, J.; Fairall, L.; Watson, P.J.; Yang, J.-C.; Czimmerer, Z.; Kampmann, T.; Goult, B.T.; Greenwood, A.J.; Gooch, J.T.; Kallenberger, B.C.; et al. Structural basis for the assembly of the SMRT/NCoR core transcriptional repression machinery. Nat. Struct. Mol. Biol. 2011, 18, 177–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millard, C.J.; Watson, P.J.; Fairall, L.; Schwabe, J.W. Targeting Class I Histone Deacetylases in a “Complex” Environment. Trends Pharmacol. Sci. 2017, 38, 363–377. [Google Scholar] [CrossRef] [PubMed]
- Grausenburger, R.; Bilic, I.; Boucheron, N.; Zupkovitz, G.; El-Housseiny, L.; Tschismarov, R.; Zhang, Y.; Rembold, M.; Gaisberger, M.; Hartl, A.; et al. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J. Immunol. 2010, 185, 3489–3497. [Google Scholar] [CrossRef]
- schismarov, R.; Firner, S.; Gil-Cruz, C.; Göschl, L.; Boucheron, N.; Steiner, G.; Matthias, P.; Seiser, C.; Ludewig, B.; Ellmeier, W. HDAC1 controls CD8+ T cell homeostasis and antiviral response. PLoS ONE 2014, 9, e110576. [Google Scholar]
- Göschl, L.; Preglej, T.; Hamminger, P.; Bonelli, M.; Andersen, L.; Boucheron, N.; Gülich, A.F.; Müller, L.; Saferding, V.; Mufazalov, I.A.; et al. A T cell-specific deletion of HDAC1 protects against experimental autoimmune encephalomyelitis. J. Autoimmun. 2018, 86, 51–61. [Google Scholar] [CrossRef]
- Göschl, L.; Preglej, T.; Boucheron, N.; Saferding, V.; Müller, L.; Platzer, A.; Hirahara, K.; Shih, H.-Y.; Backlund, J.; Matthias, P.; et al. Histone deacetylase 1 (HDAC1): A key player of T cell-mediated arthritis. J. Autoimmun. 2020, 108, 102379. [Google Scholar] [CrossRef]
- Dovey, O.M.; Foster, C.T.; Conte, N.; Edwards, S.A.; Edwards, J.M.; Singh, R.; Vassiliou, G.; Bradley, A.; Cowley, S.M. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 2013, 121, 1335–1344. [Google Scholar] [CrossRef] [Green Version]
- Boucheron, N.; Tschismarov, R.; Goeschl, L.; Moser, M.A.; Lagger, S.; Sakaguchi, S.; Winter, M.; Lenz, F.; Vitko, D.; Breitwieser, F.P.; et al. CD4(+) T cell lineage integrity is controlled by the histone deacetylases HDAC1 and HDAC2. Nat. Immunol. 2014, 15, 439–448. [Google Scholar] [CrossRef] [Green Version]
- Heideman, M.R.; Wilting, R.H.; Yanover, E.; Velds, A.; de Jong, J.; Kerkhoven, R.M.; Jacobs, H.; Wessels, L.F.; Dannenberg, J.-H. Dosage-dependent tumor suppression by histone deacetylases 1 and 2 through regulation of c-Myc collaborating genes and p53 function. Blood 2013, 121, 2038–2050. [Google Scholar] [CrossRef]
- Preglej, T.; Hamminger, P.; Luu, M.; Bulat, T.; Andersen, L.; Göschl, L.; Stolz, V.; Rica, R.; Sandner, L.; Waltenberger, D.; et al. Histone deacetylases 1 and 2 restrain CD4+ cytotoxic T lymphocyte differentiation. JCI Insight 2020, 5, e133393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, D.; Gao, J.; Su, L. Foxp3 inhibits HDAC1 activity to modulate gene expression in human T cells. Virology 2011, 421, 12–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daams, R.; Sime, W.; Leandersson, K.; Sitnicka, E.; Massoumi, R. Deletion of Nemo-like Kinase in T Cells Reduces Single-Positive CD8(+) Thymocyte Population. J. Immunol. 2020, 205, 1830–1841. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.; La Perle, K.; Kwiatkowski, S.; Sullivan, L.A.; Sams, G.H.; Johns, J.; Curphey, D.P.; Wen, J.; McConnell, K.; Qi, J.; et al. Mechanism, Consequences, and Therapeutic Targeting of Abnormal IL15 Signaling in Cutaneous T-cell Lymphoma. Cancer Discov. 2016, 6, 986–1005. [Google Scholar] [CrossRef] [Green Version]
- Hsu, F.-C.; Belmonte, P.; Constans, M.M.; Chen, M.W.; McWilliams, D.C.; Hiebert, S.W.; Shapiro, V.S. Histone Deacetylase 3 Is Required for T Cell Maturation. J. Immunol. 2015, 195, 1578–1590. [Google Scholar] [CrossRef]
- Dash, B.; Shapiro, M.J.; Thapa, P.; Arocha, S.R.; Chung, J.-Y.; Schwab, A.D.; Shaylene, A.M.; Rajcula, M.J.; Shapiro, V.S.; McCue, S.A. The Interaction between NKAP and HDAC3 Is Critical for T Cell Maturation. Immunohorizons 2019, 3, 352–367. [Google Scholar] [CrossRef]
- Thapa, P.; Arocha, S.R.; Chung, J.Y.; Sant’Angelo, D.B.; Shapiro, V.S. Histone deacetylase 3 is required for iNKT cell development. Sci. Rep. 2017, 7, 5784. [Google Scholar] [CrossRef]
- Philips, R.L.; Chen, M.W.; McWilliams, D.C.; Belmonte, P.J.; Constans, M.M.; Shapiro, V.S. HDAC3 Is Required for the Downregulation of RORgammat during Thymocyte Positive Selection. J. Immunol. 2016, 197, 541–554. [Google Scholar] [CrossRef] [Green Version]
- Stengel, K.; Zhao, Y.; Klus, N.J.; Kaiser, J.F.; Gordy, L.E.; Joyce, S.; Hiebert, S.W.; Summers, A.R. Histone Deacetylase 3 Is Required for Efficient T Cell Development. Mol. Cell. Biol. 2015, 35, 3854–3865. [Google Scholar] [CrossRef] [Green Version]
- Philips, R.L.; McCue, S.A.; Rajcula, M.J.; Shapiro, V.S. Cutting Edge: HDAC3 Protects Double-Positive Thymocytes from P2X7 Receptor-Induced Cell Death. J. Immunol. 2019, 202, 1033–1038. [Google Scholar] [CrossRef]
- Müller, L.; Hainberger, D.; Stolz, V.; Ellmeier, W. NCOR1-a new player on the field of T cell development. J. Leukoc. Biol. 2018, 104, 1061–1068. [Google Scholar] [CrossRef] [Green Version]
- Tay, R.E.; Olawoyin, O.; Cejas, P.; Xie, Y.; Meyer, C.A.; Ito, Y.; Weng, Q.Y.; Fisher, D.E.; Long, H.W.; Brown, M.; et al. Hdac3 is an epigenetic inhibitor of the cytotoxicity program in CD8 T cells. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Philips, R.L.; Lee, J.H.; Gaonkar, K.; Chanana, P.; Chung, J.Y.; Romero Arocha, S.R.; Schwab, A.; Ordog, T.; Shapiro, V.S. HDAC3 restrains CD8-lineage genes to maintain a bi-potential state in CD4(+)CD8(+) thymocytes for CD4-lineage commitment. eLife 2019, 8, e43821. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-J.; Greégoire, S. Class II histone deacetylases: From sequence to function, regulation, and clinical implication. Mol. Cell. Biol. 2005, 25, 2873–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Muslin, A.J.; Xing, H. 14-3-3 proteins: Regulation of subcellular localization by molecular interference. Cell. Signal. 2000, 12, 703–709. [Google Scholar] [CrossRef]
- Tong, J.J. Identification of HDAC10, a novel class II human histone deacetylase containing a leucine-rich domain. Nucleic Acids Res. 2002, 30, 1114–1123. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Peng, L.; Seto, E.; Huang, S.; Qiu, Y. Modulation of histone deacetylase 6 (HDAC6) nuclear import and tubulin deacetylase activity through acetylation. J. Biol. Chem. 2012, 287, 29168–29174. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, X.; Yin, C.; Chen, X.; Zhang, Z.; Brown, S.; Xie, H.; Zhou, L.; Mi, Q.-S. HDAC4 is expressed on multiple T cell lineages but dispensable for their development and function. Oncotarget 2017, 8, 17562–17572. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Zhu, Q.; Jiang, T.; Wang, R.; Shen, Y.; Zhu, X.; Wang, Y.; Bai, F.; Ding, Q.; Zhou, X.; et al. Genome-wide DNA methylation patterns in CD4+ T cells from Chinese Han patients with rheumatoid arthritis. Mod. Rheumatol. 2017, 27, 441–447. [Google Scholar] [CrossRef]
- Nijhuis, L.; Peeters, J.G.C.; Vastert, S.J.; Van Loosdregt, J. Restoring T Cell Tolerance, Exploring the Potential of Histone Deacetylase Inhibitors for the Treatment of Juvenile Idiopathic Arthritis. Front. Immunol. 2019, 10, 151. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Jiao, J.; Wang, L.; O’Brien, S.; Newick, K.; Wang, L.C.S.; Falkensammer, E.; Liu, Y.; Han, R.; Kapoor, V.; et al. HDAC5 controls the functions of Foxp3(+) T-regulatory and CD8(+) T cells. Int. J. Cancer 2016, 138, 2477–2486. [Google Scholar] [CrossRef] [Green Version]
- Beier, U.H.; Wang, L.; Han, R.; Akimova, T.; Liu, Y.; Hancock, W.W. Histone deacetylases 6 and 9 and sirtuin-1 control Foxp3+ regulatory T cell function through shared and isoform-specific mechanisms. Sci. Signal. 2012, 5, ra45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Zoeten, E.F.; Wang, L.; Butler, K.; Beier, U.H.; Akimova, T.; Sai, H.; Bradner, J.E.; Mazitschek, R.; Kozikowski, A.P.; Matthias, P.; et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol. Cell. Biol. 2011, 31, 2066–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellmeier, W.; Seiser, C. Histone deacetylase function in CD4(+) T cells. Nat. Rev. Immunol. 2018, 18, 617–634. [Google Scholar] [CrossRef]
- Yan, B.; Liu, Y.; Bai, H.; Chen, M.; Xie, S.; Li, D.; Liu, M.; Zhou, J. HDAC6 regulates IL-17 expression in T lymphocytes: Implications for HDAC6-targeted therapies. Theranostics 2017, 7, 1002–1009. [Google Scholar] [CrossRef] [Green Version]
- Dequiedt, F.; Kasler, H.; Fischle, W.; Kiermer, V.; Weinstein, M.; Herndier, B.G.; Verdin, E. HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity 2003, 18, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Kasler, H.G.; Young, B.D.; Mottet, D.; Lim, H.W.; Collins, A.M.; Olson, E.N.; Verdin, E. Histone deacetylase 7 regulates cell survival and TCR signaling in CD4/CD8 double-positive thymocytes. J. Immunol. 2011, 186, 4782–4793. [Google Scholar] [CrossRef]
- Kasler, H.G.; Lee, I.S.; Lim, H.W.; Verdin, E. Histone Deacetylase 7 mediates tissue-specific autoimmunity via control of innate effector function in invariant Natural Killer T Cells. eLife 2018, 7, e32109. [Google Scholar] [CrossRef]
- Kasler, H.G.; Lim, H.W.; Mottet, D.; Collins, A.M.; Lee, I.; Verdin, E. Nuclear export of histone deacetylase 7 during thymic selection is required for immune self-tolerance. EMBO J. 2012, 31, 4453–4465. [Google Scholar] [CrossRef] [Green Version]
- Myers, D.R.; Lau, T.; Markegard, E.; Lim, H.W.; Kasler, H.; Zhu, M.; Barczak, A.; Huizar, J.P.; Zikherman, J.; Erle, D.J.; et al. Tonic LAT-HDAC7 Signals Sustain Nur77 and Irf4 Expression to Tune Naive CD4 T Cells. Cell Rep. 2017, 19, 1558–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, R.; de Zoeten, E.F.; Özkaynak, E.; Chen, C.; Wang, L.; Porrett, P.M.; Li, B.; Turka, L.A.; Olson, E.N.; Greene, M.I.; et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 2007, 13, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Cao, Q.; Reilly, C.M.; Young, N.L.; Garcia, B.A.; Mishra, N. Histone deacetylase 9 deficiency protects against effector T cell-mediated systemic autoimmunity. J. Biol. Chem. 2011, 286, 28833–28843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Zoeten, E.F.; Wang, L.; Sai, H.; Dillmann, W.H.; Hancock, W.W. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology 2010, 138, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Beier, U.H.; Angelin, A.; Akimova, T.; Wang, L.; Liu, Y.; Xiao, H.; Koike, M.A.; Hancock, S.A.; Bhatti, T.R.; Han, R.; et al. Essential role of mitochondrial energy metabolism in Foxp3(+) T-regulatory cell function and allograft survival. FASEB J. 2015, 29, 2315–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahiya, S.; Beier, U.H.; Wang, L.; Han, R.; Jiao, J.; Akimova, T.; Angelin, A.; Wallace, D.C.; Hancock, W.W. HDAC10 deletion promotes Foxp3(+) T-regulatory cell function. Sci. Rep. 2020, 10, 424. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Faller, D.V. Transcription Regulation by Class III Histone Deacetylases (HDACs)-Sirtuins. Transl. Oncogenom. 2008, 3, 53–65. [Google Scholar]
- Zhang, J.; Lee, S.-M.; Shannon, S.; Gao, B.; Chen, W.; Chen, A.; Divekar, R.; McBurney, M.W.; Braley-Mullen, H.; Zaghouani, H.; et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J. Clin. Investig. 2009, 119, 3048–3058. [Google Scholar] [CrossRef] [Green Version]
- Kong, S.; Yeung, P.; Fang, D. The class III histone deacetylase sirtuin 1 in immune suppression and its therapeutic potential in rheumatoid arthritis. J. Genet. Genom. 2013, 40, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Bi, Y.; Chen, X.; Li, C.; Li, Y.; Zhang, Z.; Wang, J.; Lu, Y.; Yu, Q.; Su, H.; et al. Histone Deacetylase SIRT1 Negatively Regulates the Differentiation of Interleukin-9-Producing CD4(+) T Cells. Immunity 2016, 44, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, C.; Hirota, K.; Stieglitz, B.; Van Snick, J.; Tolaini, M.; Lahl, K.; Sparwasser, T.; Helmby, H.; Stockinger, B. An IL-9 fate reporter demonstrates the induction of an innate IL-9 response in lung inflammation. Nat. Immunol. 2011, 12, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Purwar, R.; Schlapbach, C.; Xiao, S.; Kang, H.S.; Elyaman, W.; Jiang, X.; Jetten, A.; Khoury, S.; Fuhlbrigge, R.C.; Kuchroo, V.K.; et al. Robust tumor immunity to melanoma mediated by interleukin-9-producing T cells. Nat. Med. 2012, 18, 1248–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequeira, J.; Boily, G.; Bazinet, S.; Saliba, S.; He, X.; Jardine, K.; Kennedy, C.; Staines, W.; Rousseaux, C.; Mueller, R.; et al. sirt1-null mice develop an autoimmune-like condition. Exp. Cell Res. 2008, 314, 3069–3074. [Google Scholar] [CrossRef]
- Hamaidi, I.; Zhang, L.; Kim, N.; Wang, M.H.; Iclozan, C.; Fang, B.; Liu, M.; Koomen, J.M.; Berglund, A.E.; Yoder, S.J.; et al. Sirt2 Inhibition En.nhances Metabolic Fitness and Effector Functions of Tumor-Reactive T Cells. Cell Metab. 2020, 32, 420–436. [Google Scholar] [CrossRef] [PubMed]
- Hamaidi, I.; Kim, S. Sirtuins are crucial regulators of T cell metabolism and functions. Exp. Mol. Med. 2022, 54, 207–215. [Google Scholar] [CrossRef]
- Jiang, C.; Liu, J.; Guo, M.; Gao, X.; Wu, X.; Bai, N.; Guo, W.; Li, N.; Yi, F.; Cheng, R.; et al. The NAD-dependent deacetylase SIRT2 regulates T cell differentiation involved in tumor immune response. Int. J. Biol. Sci. 2020, 16, 3075–3084. [Google Scholar] [CrossRef]
- Toubai, T.; Tamaki, H.; Peltier, D.C.; Rossi, C.; Oravecz-Wilson, K.; Liu, C.; Zajac, C.; Wu, J.; Sun, Y.; Fujiwara, H.; et al. Mitochondrial Deacetylase SIRT3 Plays an Important Role in Donor T Cell Responses after Experimental Allogeneic Hematopoietic Transplantation. J. Immunol. 2018, 201, 3443–3455. [Google Scholar] [CrossRef]
- Lin, W.; Chen, W.; Liu, W.; Xu, Z.; Zhang, L. Sirtuin4 suppresses the anti-neuroinflammatory activity of infiltrating regulatory T cells in the traumatically injured spinal cord. Immunology 2019, 158, 362–374. [Google Scholar] [CrossRef]
- Heinonen, T.; Ciarlo, E.; Théroude, C.; Pelekanou, A.; Herderschee, J.; Le Roy, D.; Roger, T. Sirtuin 5 Deficiency Does Not Compromise Innate Immune Responses to Bacterial Infections. Front. Immunol. 2018, 9, 2675. [Google Scholar] [CrossRef]
- Wang, K.; Hu, Z.; Zhang, C.; Yang, L.; Feng, L.; Yang, P.; Yu, H. SIRT5 Contributes to Colorectal Cancer Growth by Regulating T Cell Activity. J. Immunol. Res. 2020, 2020, 3792409. [Google Scholar] [CrossRef]
- Kim, W.; Kim, E.J. SIRT7 an emerging sirtuin: Deciphering newer roles. J. Physiol. Pharmacol. 2013, 64, 531–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez-Navarro, A.; Martínez-Rojas, M.; Albarrán-Godinez, A.; Pérez-Villalva, R.; Auwerx, J.; de la Cruz, A.; Noriega, L.G.; Rosetti, F.; Bobadilla, N.A. Sirtuin 7 Deficiency Reduces Inflammation and Tubular Damage Induced by an Episode of Acute Kidney Injury. Int. J. Mol. Sci. 2022, 23, 2573. [Google Scholar] [CrossRef] [PubMed]
- Vakhrusheva, O.; Smolka, C.; Gajawada, P.; Kostin, S.; Boettger, T.; Kubin, T.; Braun, T.; Bober, E. Sirt7 increases stress resistance of cardiomyocytes and prevents apoptosis and inflammatory cardiomyopathy in mice. Circ. Res. 2008, 102, 703–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanginlar, C.; Logie, C. HDAC11 is a regulator of diverse immune functions. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Cueto, M.A.; Asselbergs, F.; Atadja, P. Cloning and functional characterization of HDAC11, a novel member of the human histone deacetylase family. J. Biol. Chem. 2002, 277, 25748–25755. [Google Scholar] [CrossRef] [Green Version]
- Núñez-Álvarez, Y.; Suelves, M. HDAC11: A multifaceted histone deacetylase with proficient fatty deacylase activity and its roles in physiological processes. FEBS J. 2021, 289, 2771–2792. [Google Scholar] [CrossRef]
- Villagra, A.; Cheng, F.; Wang, H.-W.; Suarez, I.; Glozak, M.; Maurin, M.; Nguyen, D.; Wright, K.L.; Atadja, P.W.; Bhalla, K.; et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2009, 10, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Woods, D.M.; Woan, K.V.; Cheng, F.; Sodré, A.L.; Wang, D.; Wu, Y.; Wang, Z.; Chen, J.; Powers, J.; Pinilla-Ibarz, J.; et al. T cells lacking HDAC11 have increased effector functions and mediate enhanced alloreactivity in a murine model. Blood 2017, 130, 146–155. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Wang, L.; Dahiya, S.; Beier, U.; Han, R.; Samanta, A.; Bergman, J.; Sotomayor, E.M.; Seto, E.; Kozikowski, A.P.; et al. Histone/protein deacetylase 11 targeting promotes Foxp3+ Treg function. Sci. Rep. 2017, 7, 8626. [Google Scholar] [CrossRef] [Green Version]
- Buglio, D.; Khaskhely, N.M.; Voo, K.S.; Martinez-Valdez, H.; Liu, Y.-J.; Younes, A. HDAC11 plays an essential role in regulating OX40 ligand expression in Hodgkin lymphoma. Blood 2011, 117, 2910–2917. [Google Scholar] [CrossRef] [Green Version]
- Akimova, T.; Beier, U.; Liu, Y.; Wang, L.; Hancock, W.W. Histone/protein deacetylases and T-cell immune responses. Blood 2012, 119, 2443–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Tai, Y.-T.; Tian, Z.; Hideshima, T.; Chauhan, D.; Nanjappa, P.; Exley, A.M.; Anderson, K.C.; Munshi, N.C. HDAC inhibition by LBH589 affects the phenotype and function of human myeloid dendritic cells. Leukemia 2011, 25, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhao, D.; Kirschbaum, M.; Zhang, C.; Lin, C.-L.; Todorov, I.; Kandeel, F.; Forman, S.; Zeng, D. HDAC inhibitor reduces cytokine storm and facilitates induction of chimerism that reverses lupus in anti-CD3 conditioning regimen. Proc. Natl. Acad. Sci. USA 2008, 105, 4796–4801. [Google Scholar] [CrossRef] [Green Version]
- Dagtas, A.S.; Edens, R.E.; Gilbert, K.M. Histone deacetylase inhibitor uses p21(Cip1) to maintain anergy in CD4+ T cells. Int. Immunopharmacol. 2009, 9, 1289–1297. [Google Scholar] [CrossRef]
- McCaw, T.R.; Randall, T.D.; Forero, A.; Buchsbaum, D.J. Modulation of antitumor immunity with histone deacetylase inhibitors. Immunotherapy 2017, 9, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lv, H.; Jia, X.; Hu, G.; Kong, L.; Zhang, T.; Li, L.; Pan, Y.; Zhai, Q.; Meng, B.; et al. Clinical significance of enhancer of zeste homolog 2 and histone deacetylases 1 and 2 expression in peripheral T-cell lymphoma. Oncol. Lett. 2019, 18, 1415–1423. [Google Scholar] [CrossRef] [Green Version]
- Higuchi, T.; Nakayama, T.; Arao, T.; Nishio, K.; Yoshie, O. SOX4 is a direct target gene of FRA-2 and induces expression of HDAC8 in adult T-cell leukemia/lymphoma. Blood 2013, 121, 3640–3649. [Google Scholar] [CrossRef]
- Kozako, T.; Aikawa, A.; Shoji, T.; Fujimoto, T.; Yoshimitsu, M.; Shirasawa, S.; Tanaka, H.; Honda, S.-I.; Shimeno, H.; Arima, N.; et al. High expression of the longevity gene product SIRT1 and apoptosis induction by sirtinol in adult T-cell leukemia cells. Int. J. Cancer 2012, 131, 2044–2055. [Google Scholar] [CrossRef]
- Iżykowska, K.; Rassek, K.; Korsak, D.; Przybylski, G.K. Novel targeted therapies of T cell lymphomas. J. Hematol. Oncol. 2020, 13, 176. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pieniawska, M.; Iżykowska, K. Role of Histone Deacetylases in T-Cell Development and Function. Int. J. Mol. Sci. 2022, 23, 7828. https://doi.org/10.3390/ijms23147828
Pieniawska M, Iżykowska K. Role of Histone Deacetylases in T-Cell Development and Function. International Journal of Molecular Sciences. 2022; 23(14):7828. https://doi.org/10.3390/ijms23147828
Chicago/Turabian StylePieniawska, Monika, and Katarzyna Iżykowska. 2022. "Role of Histone Deacetylases in T-Cell Development and Function" International Journal of Molecular Sciences 23, no. 14: 7828. https://doi.org/10.3390/ijms23147828
APA StylePieniawska, M., & Iżykowska, K. (2022). Role of Histone Deacetylases in T-Cell Development and Function. International Journal of Molecular Sciences, 23(14), 7828. https://doi.org/10.3390/ijms23147828