
Identify Bitter Peptides by Using Deep Representation Learning Features


Abstract
:1. Introduction
2. Result and Discussion
2.1. Results of Preliminary Optimization
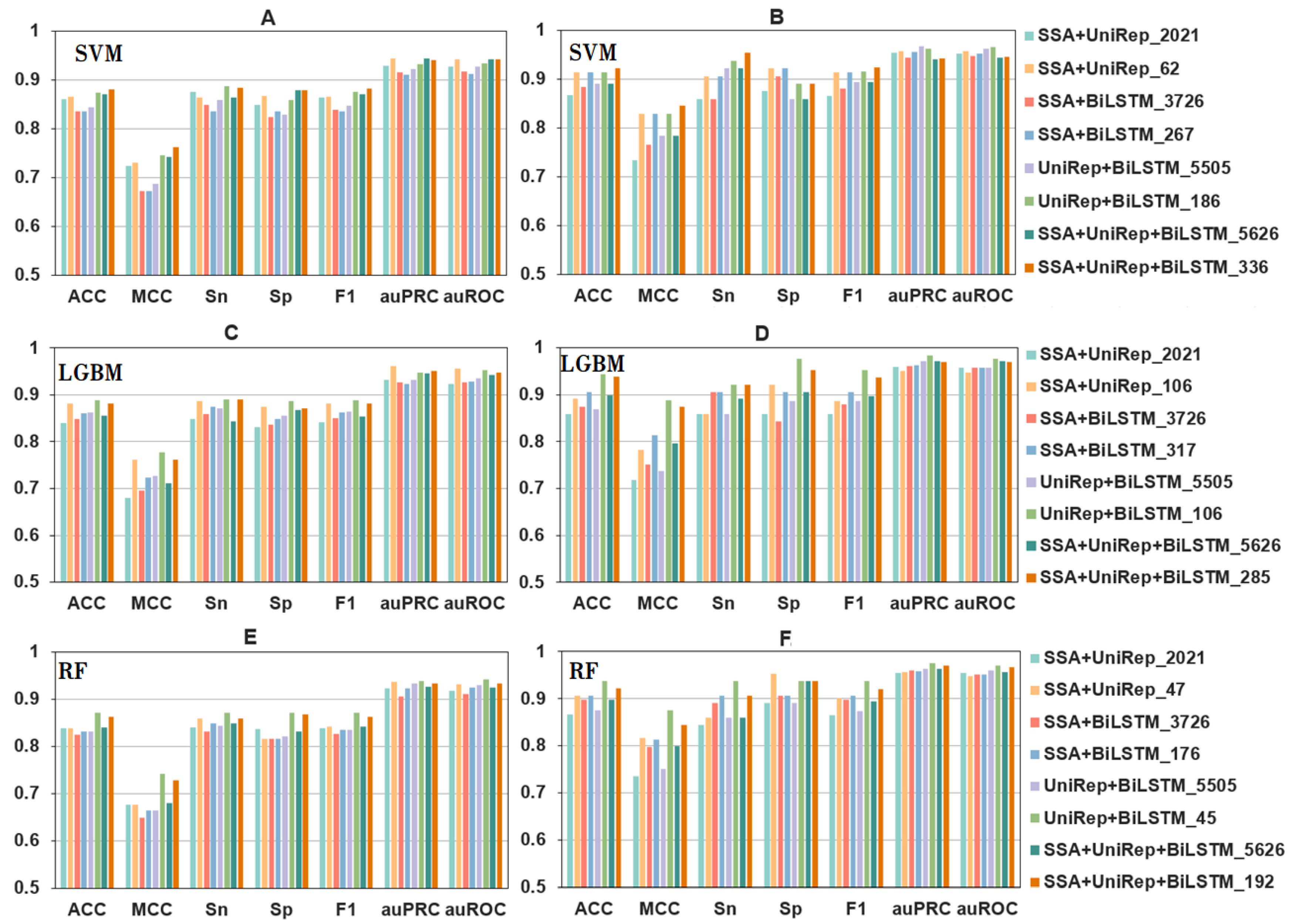
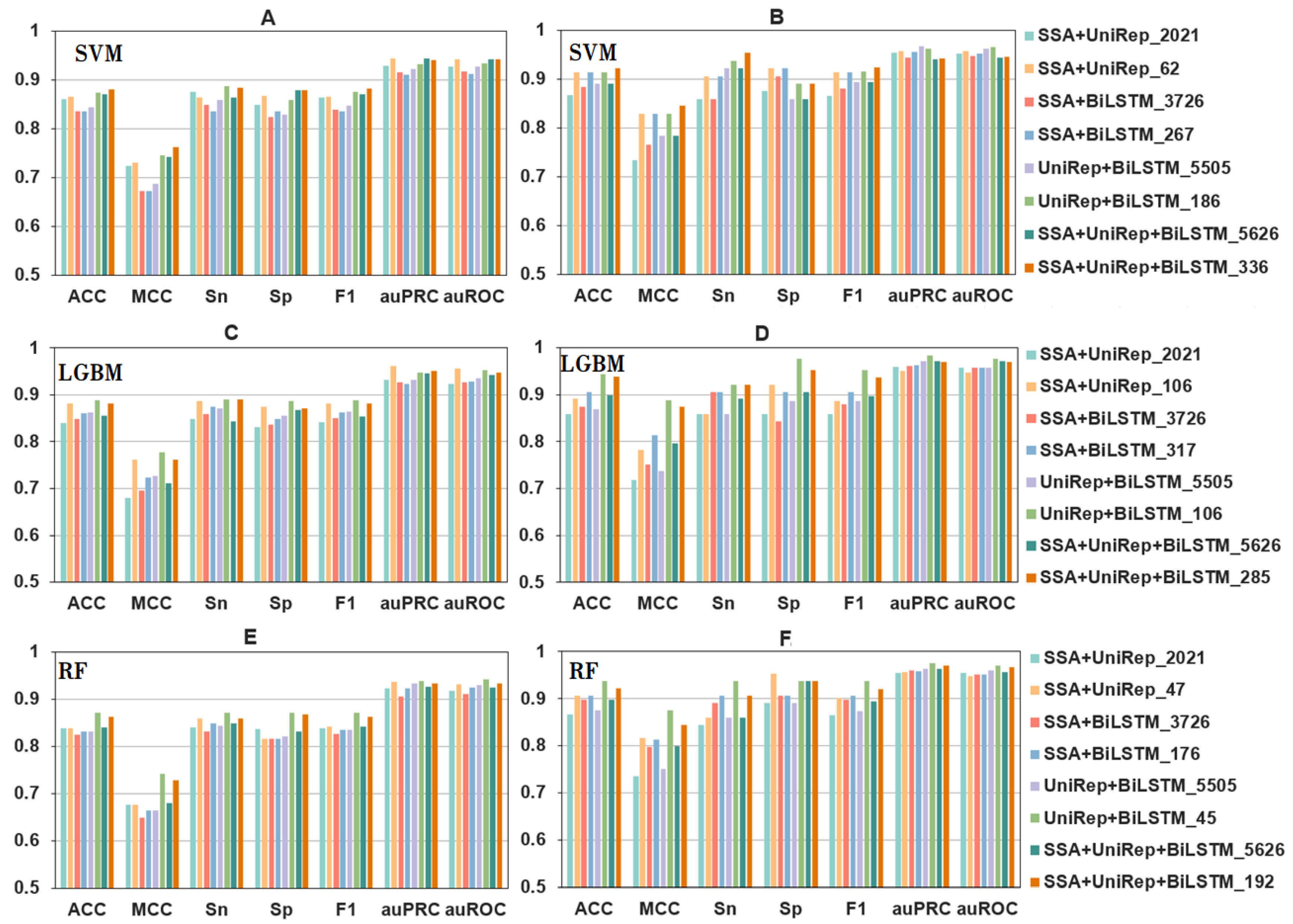
2.2. The Effects of Feature Fusion on the Automatic Identification of Bitter Peptides
2.3. The Effects of Feature Selection on the Automatic Identification of Bitter Peptides
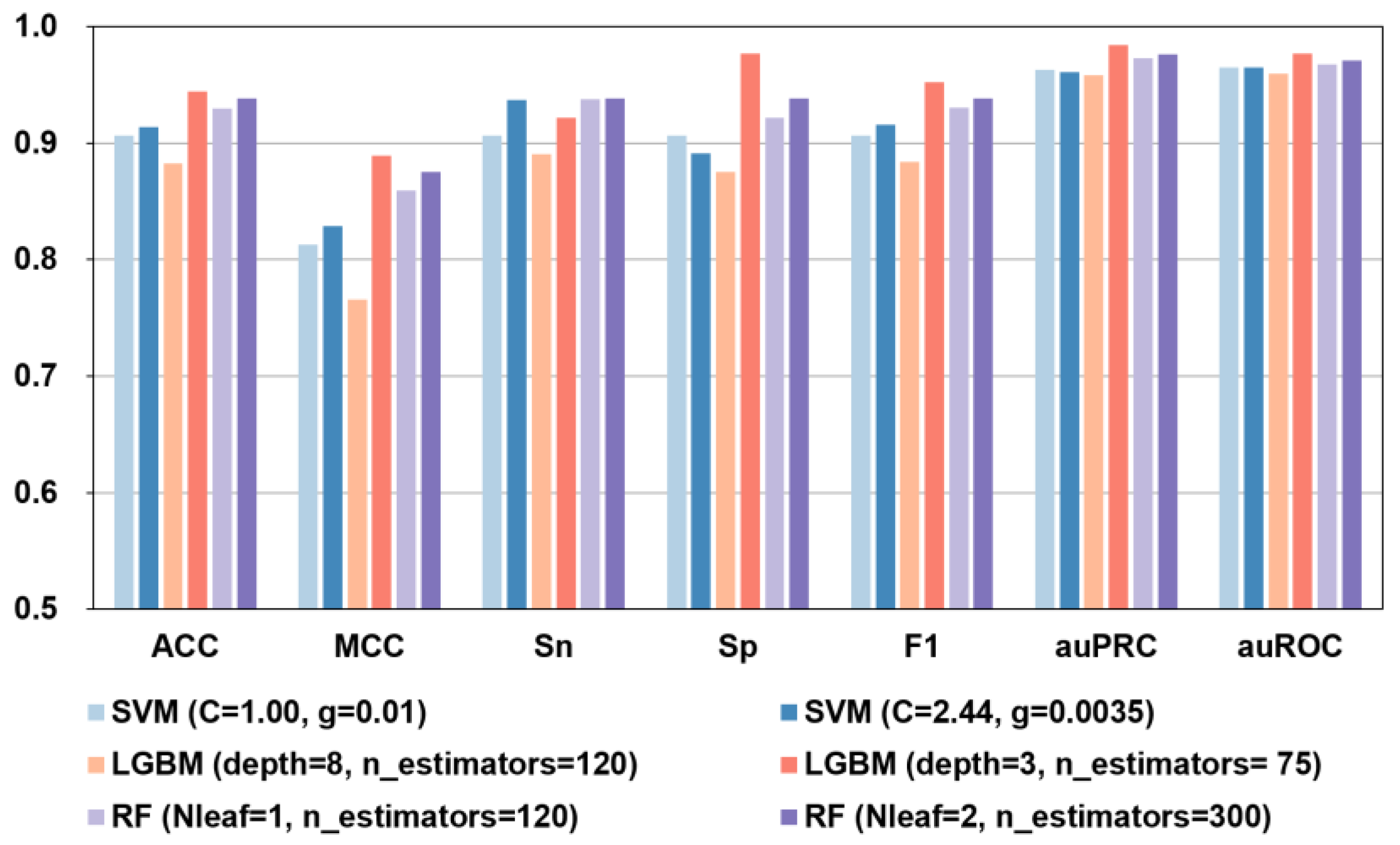
2.4. The Effect of Machine Learning Model Parameter Optimization on the Automated Identification of Bitter Peptides
2.5. Comparison with Existing Methods
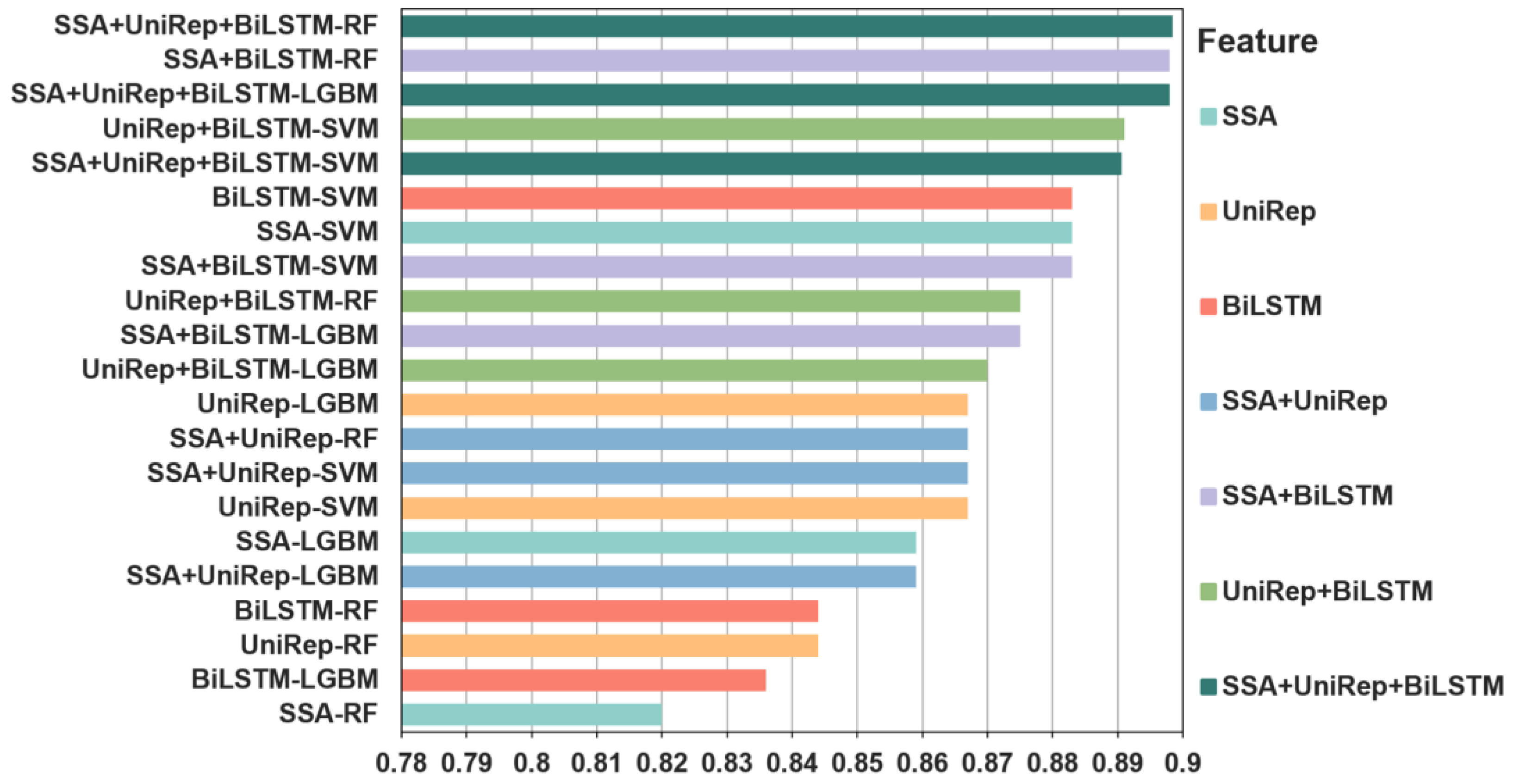
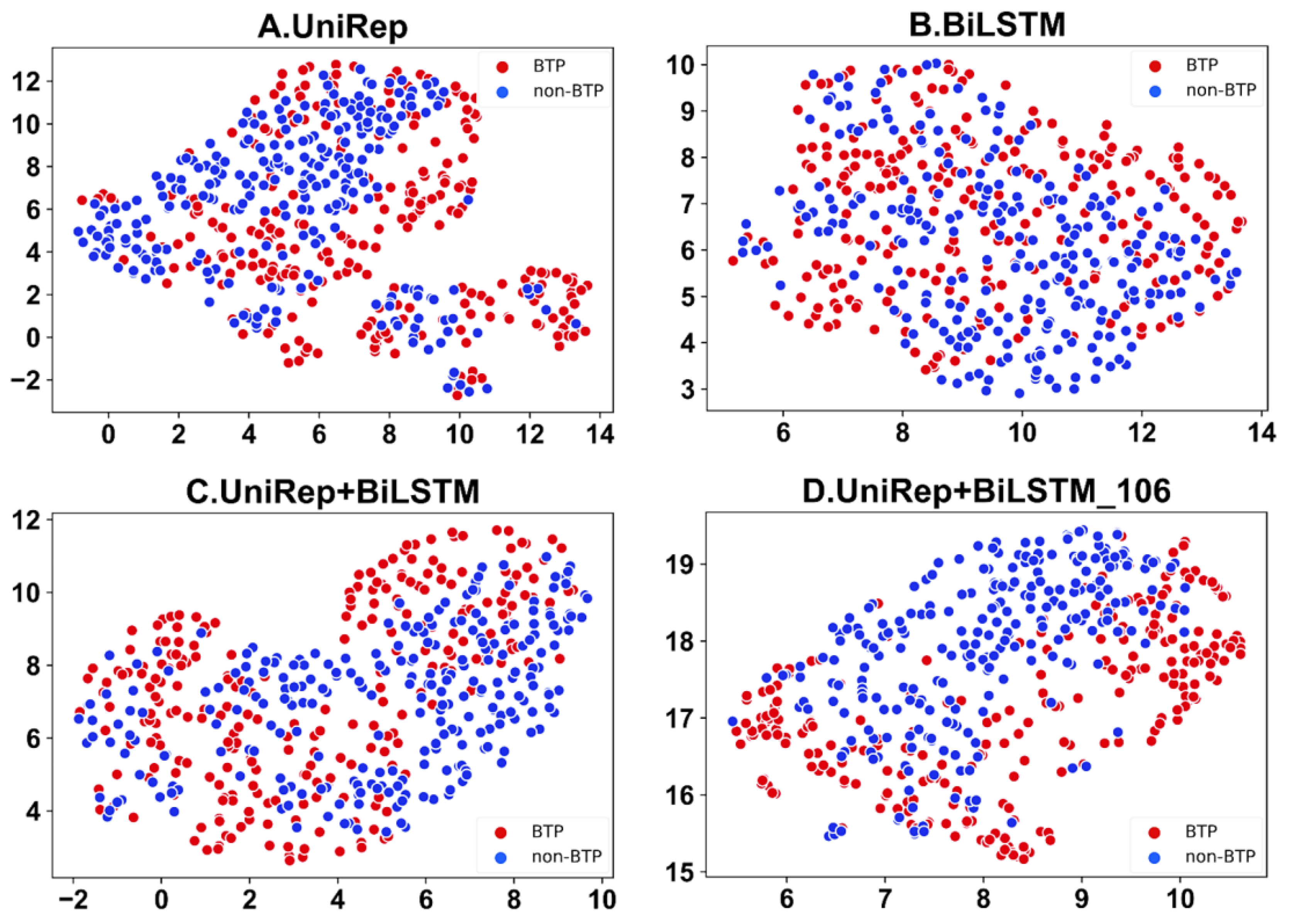
2.6. Feature Visualization of the Picric Peptide Automatic Recognition Effect
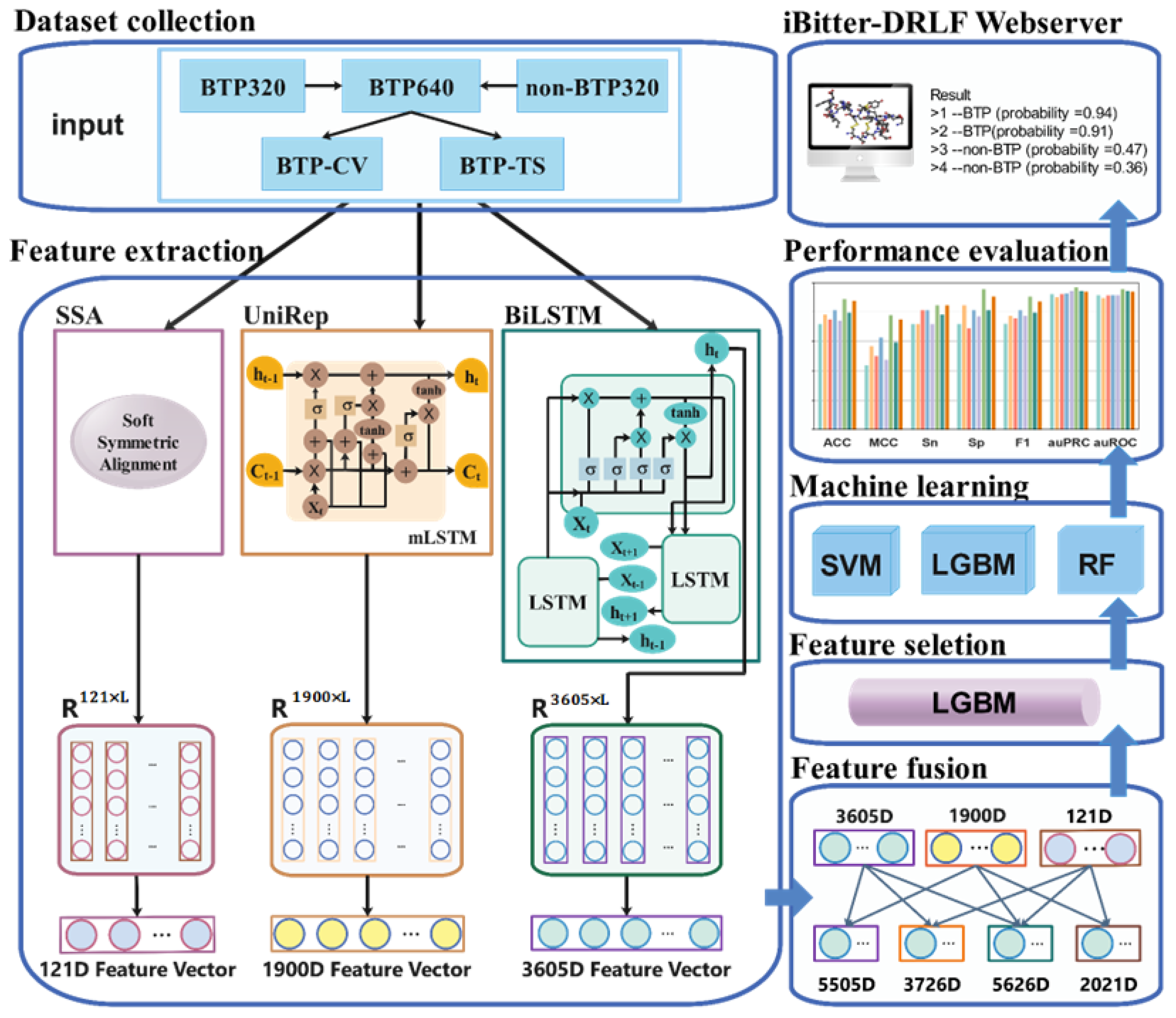
2.7. iBitter-DRLF Webserver
3. Materials and Methods
3.1. Benchmark Dataset
3.2. Feature Extraction
3.2.1. Pre-Trained SSA Embedding Model
3.2.2. Pre-Trained UniRep Embedding Model
3.2.3. Pre-Trained BiLSTM Embedding Model
3.2.4. Feature Fusion
3.3. Feature Selection Method
3.4. Machine Learning Methods
3.5. Evaluation Metrics and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Maehashi, K.; Huang, L. Bitter peptides and bitter taste receptors. Cell. Mol. Life Sci. 2009, 66, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Puying, Z.; Xiaoying, Z.; Rui, Q.; Jing, W.; Xionghai, R. Research Progress on Flavour Protease for Debittering. Food Nutr. China 2021, 27, 29–34. [Google Scholar]
- Maehashi, K.; Matsuzaki, M.; Yamamoto, Y.; Udaka, S. Isolation of Peptides from an Enzymatic Hydrolysate of Food Proteins and Characterization of Their Taste Properties. Biosci. Biotechnol. Biochem. 1999, 63, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Ayana, W.; Marina, S.; Anat, L.; Niv, M.Y. BitterDB: A database of bitter compounds. Nucleic Acids Res. 2012, 40, D413–D419. [Google Scholar]
- Cao, C.; Wang, J.; Kwok, D.; Cui, F.; Zhang, Z.; Zhao, D.; Li, M.J.; Zou, Q. webTWAS: A resource for disease candidate susceptibility genes identified by transcriptome-wide association study. Nucleic Acids Res. 2021, 50, D1123–D1130. [Google Scholar] [CrossRef]
- Le, N.Q.K.; Hung, T.N.K.; Do, D.T.; Lam, L.H.T.; Dang, L.H.; Huynh, T.-T. Radiomics-based machine learning model for efficiently classifying transcriptome subtypes in glioblastoma patients from MRI. Comput. Biol. Med. 2021, 132, 104320. [Google Scholar] [CrossRef]
- Ramzan, Z.; Hassan, M.A.; Asif, H.M.S.; Farooq, A. A Machine Learning-based Self-risk Assessment Technique for Cervical Cancer. Curr. Bioinform. 2021, 16, 315–332. [Google Scholar] [CrossRef]
- Su, Q.; Chen, Q. Application of Machine Learning in Animal Disease Analysis and Prediction. Curr. Bioinform. 2021, 16, 972–982. [Google Scholar] [CrossRef]
- Hyun-Ock, K.; Li-Chan, E.C.Y. Quantitative Structure—Activity Relationship Study of Bitter Peptides. J. Agric. Food Chem. 2006, 54, 10102–10111. [Google Scholar]
- Wu, J.; Aluko, R.E. Quantitative structure-activity relationship study of bitter di- and tri-peptides including relationship with angiotensin I-converting enzyme inhibitory activity. J. Pept. Sci. 2006, 13, 63–69. [Google Scholar] [CrossRef]
- Yin, J.; Diao, Y.; Wen, Z.; Wang, Z.; Li, M. Studying Peptides Biological Activities Based on Multidimensional Descriptors (E) Using Support Vector Regression. Int. J. Pept. Res. Ther. 2010, 16, 111–121. [Google Scholar] [CrossRef]
- Tong, J.; Liu, S.; Zhou, P.; Wu, B.; Li, Z. A novel descriptor of amino acids and its application in peptide QSAR. J. Theor. Biol. 2008, 253, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.-H.; Long, H.-X.; Bo, Z.; Wang, Y.-Q.; Wu, Y.-Z. New descriptors of amino acids and their application to peptide QSAR study. Peptides 2008, 29, 1798–1805. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Yang, L.; Kang, L.; Mei, H.; Li, Z. Using multidimensional patterns of amino acid attributes for QSAR analysis of peptides. Amino Acids 2008, 37, 583–591. [Google Scholar] [CrossRef]
- Huang, W.; Shen, Q.; Su, X.; Ji, M.; Liu, X.; Chen, Y.; Lu, S.; Zhuang, H.; Zhang, J. BitterX: A tool for understanding bitter taste in humans. Sci. Rep. 2016, 6, 23450. [Google Scholar] [CrossRef] [Green Version]
- Dagan-Wiener, A.; Nissim, I.; Ben Abu, N.; Borgonovo, G.; Bassoli, A.; Niv, M.Y. Bitter or not? BitterPredict, a tool for predicting taste from chemical structure. Sci. Rep. 2017, 7, 12074. [Google Scholar] [CrossRef] [Green Version]
- Charoenkwan, P.; Yana, J.; Schaduangrat, N.; Nantasenamat, C.; Hasan, M.M.; Shoombuatong, W. iBitter-SCM: Identification and characterization of bitter peptides using a scoring card method with propensity scores of dipeptides. Genomics 2020, 112, 2813–2822. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Nantasenamat, C.; Hasan, M.M.; Moni, M.A.; Lio, P.; Shoombuatong, W. iBitter-Fuse: A Novel Sequence-Based Bitter Peptide Predictor by Fusing Multi-View Features. Int. J. Mol. Sci. 2021, 22, 8958. [Google Scholar] [CrossRef]
- Charoenkwan, P.; Nantasenamat, C.; Hasan, M.; Manavalan, B.; Shoombuatong, W. BERT4Bitter: A bidirectional encoder representations from transformers (BERT)-based model for improving the prediction of bitter peptides. Bioinformatics 2021, 37, 2556–2562. [Google Scholar] [CrossRef]
- He, W.; Jiang, Y.; Jin, J.; Li, Z.; Zhao, J.; Manavalan, B.; Su, R.; Gao, X.; Wei, L. Accelerating bioactive peptide discovery via mutual information-based meta-learning. Brief. Bioinform. 2022, 23, bbab499. [Google Scholar] [CrossRef]
- Chen, X.; Li, C.; Bernards, M.T.; Shi, Y.; Shao, Q.; He, Y. Sequence-based peptide identification, generation, and property prediction with deep learning: A review. Mol. Syst. Des. Eng. 2021, 6, 406–428. [Google Scholar] [CrossRef]
- Arif, M.; Kabir, M.; Ahmad, S.; Khan, A.; Ge, F.; Khelifi, A.; Yu, D.-J. DeepCPPred: A deep learning framework for the discrimination of cell-penetrating peptides and their uptake efficiencies. IEEE/ACM Trans. Comput. Biol. Bioinform. 2021, 1. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Cui, F.; Zou, Q.; Zhang, L.; Xu, L. Anticancer peptides prediction with deep representation learning features. Brief. Bioinform. 2021, 22, bbab008. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-Y.; Lin, T.-T.; Cheng, W.-C.; Lu, I.-H.; Lin, C.-Y.; Chen, S.-H. Peptide-Based Drug Predictions for Cancer Therapy Using Deep Learning. Pharmaceuticals 2022, 15, 422. [Google Scholar] [CrossRef]
- Alley, E.C.; Khimulya, G.; Biswas, S.; AlQuraishi, M.; Church, G.M. Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods 2019, 16, 1315–1322. [Google Scholar] [CrossRef]
- Rao, R.; Bhattacharya, N.; Thomas, N.; Duan, Y.; Chen, X.; Canny, J.; Abbeel, P.; Song, Y.S. Evaluating Protein Transfer Learning with TAPE. Adv. Neural Inf. Process. Syst. 2019, 32, 9689–9701. [Google Scholar]
- Lv, Z.; Wang, P.; Zou, Q.; Jiang, Q. Identification of sub-Golgi protein localization by use of deep representation learning features. Bioinformatics 2020, 36, 5600–5609. [Google Scholar] [CrossRef]
- Zhao, Q.; Ma, J.; Wang, Y.; Xie, F.; Lv, Z.; Xu, Y.; Shi, H.; Han, K. Mul-SNO: A Novel Prediction Tool for S-Nitrosylation Sites Based on Deep Learning Methods. IEEE J. Biomed. Health Inform. 2021, 26, 2379–2387. [Google Scholar] [CrossRef]
- Minkiewicz, P.; Dziuba, J.; Iwaniak, A.; Dziuba, M.; Darewicz, M. BIOPEP Database and Other Programs for Processing Bioactive Peptide Sequences. J. AOAC Int. 2019, 91, 965–980. [Google Scholar] [CrossRef] [Green Version]
- Bepler, T.; Berger, B. Learning protein sequence embeddings using information from structure. arXiv 2019, arXiv:1902.08661v2. [Google Scholar]
- Graves, A.; Schmidhuber, J. Framewise phoneme classification with bidirectional LSTM and other neural network architectures. Neural Netw. 2005, 18, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Wang, D.; Ding, H.; Zhong, B.; Xu, L. Escherichia coli DNA N-4-Methycytosine Site Prediction Accuracy Improved by Light Gradient Boosting Machine Feature Selection Technology. IEEE Access 2020, 8, 14851–14859. [Google Scholar] [CrossRef]
- Lv, Z.; Ding, H.; Wang, L.; Zou, Q. A Convolutional Neural Network Using Dinucleotide One-hot Encoder for identifying DNA N6-Methyladenine Sites in the Rice Genome. Neurocomputing 2021, 422, 214–221. [Google Scholar] [CrossRef]
- Lv, Z.; Jin, S.; Ding, H.; Zou, Q. A Random Forest Sub-Golgi Protein Classifier Optimized via Dipeptide and Amino Acid Composition Features. Front. Bioeng. Biotechnol. 2019, 7, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malebary, S.; Rahman, S.; Barukab, O.; Ash’ari, R.; Khan, S.A. iAcety-SmRF: Identification of Acetylation Protein by Using Statistical Moments and Random Forest. Membranes 2022, 12, 265. [Google Scholar] [CrossRef]
- Bao, W.; Cui, Q.; Chen, B.; Yang, B. Phage_UniR_LGBM: Phage Virion Proteins Classification with UniRep Features and LightGBM Model. Comput. Math. Methods Med. 2022, 2022, 9470683. [Google Scholar] [CrossRef]
- Jiao, Y.; Du, P. Performance measures in evaluating machine learning based bioinformatics predictors for classifications. Quant. Biol. 2016, 4, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Xing, P.; Zeng, J.; Chen, J.; Su, R.; Guo, F. Improved prediction of protein-protein interactions using novel negative samples, features, and an ensemble classifier. Artif. Intell. Med. 2017, 83, 67–74. [Google Scholar] [CrossRef]
- Cui, F.; Zhang, Z.; Zou, Q. Sequence representation approaches for sequence-based protein prediction tasks that use deep learning. Brief. Funct. Genom. 2021, 20, 61–73. [Google Scholar] [CrossRef]
- Naseer, S.; Hussain, W.; Khan, Y.D.; Rasool, N. NPalmitoylDeep-pseaac: A predictor of N-Palmitoylation Sites in Proteins Using Deep Representations of Proteins and PseAAC via Modified 5-Steps Rule. Curr. Bioinform. 2021, 16, 294–305. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Model | Dim | 10-Fold Cross-Validation | Independent Test | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | MCC | Sn | Sp | F1 | auPRC | auROC | ACC | MCC | Sn | Sp | F1 | auPRC | auROC | |||
| SSA b | SVM c | 121 | 0.826 | 0.652 | 0.836 | 0.816 | 0.828 | 0.89 | 0.898 | 0.883 a | 0.766 | 0.891 | 0.875 | 0.884 | 0.951 | 0.944 |
| LGBM c | 0.787 | 0.575 | 0.816 | 0.758 | 0.793 | 0.874 | 0.886 | 0.859 | 0.722 | 0.906 | 0.812 | 0.866 | 0.949 | 0.941 | ||
| RF c | 0.791 | 0.584 | 0.828 | 0.754 | 0.798 | 0.848 | 0.865 | 0.82 | 0.644 | 0.875 | 0.766 | 0.83 | 0.934 | 0.922 | ||
| UniRep b | SVM c | 1900 | 0.865 | 0.73 | 0.867 | 0.863 | 0.865 | 0.937 | 0.931 | 0.867 | 0.735 | 0.844 | 0.891 | 0.864 | 0.952 | 0.948 |
| LGBM c | 0.84 | 0.68 | 0.828 | 0.852 | 0.838 | 0.939 | 0.93 | 0.867 | 0.735 | 0.844 | 0.891 | 0.864 | 0.953 | 0.952 | ||
| RF c | 0.842 | 0.684 | 0.836 | 0.848 | 0.841 | 0.927 | 0.92 | 0.844 | 0.688 | 0.828 | 0.859 | 0.841 | 0.946 | 0.943 | ||
| BiLSTM b | SVM c | 3605 | 0.818 | 0.637 | 0.82 | 0.816 | 0.819 | 0.91 | 0.912 | 0.883 | 0.766 | 0.906 | 0.859 | 0.885 | 0.956 | 0.951 |
| LGBM c | 0.855 | 0.711 | 0.863 | 0.848 | 0.857 | 0.924 | 0.926 | 0.836 | 0.673 | 0.812 | 0.859 | 0.832 | 0.95 | 0.95 | ||
| RF c | 0.818 | 0.637 | 0.828 | 0.809 | 0.82 | 0.9 | 0.908 | 0.844 | 0.688 | 0.844 | 0.844 | 0.844 | 0.954 | 0.949 | ||
| Feature | Model | Dim | 10-Fold Cross-Validation | Independent Test | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | MCC | Sn | Sp | F1 | auPRC | auROC | ACC | MCC | Sn | Sp | F1 | auPRC | auROC | |||
| SSA + UniRep b | SVM c | 2021 | 0.861 | 0.723 | 0.875 a | 0.848 | 0.863 | 0.929 | 0.927 | 0.867 | 0.734 | 0.859 | 0.875 | 0.866 | 0.954 | 0.952 |
| LGBM c | 0.840 | 0.680 | 0.848 | 0.832 | 0.841 | 0.933 | 0.924 | 0.859 | 0.719 | 0.859 | 0.859 | 0.859 | 0.960 | 0.958 | ||
| RF c | 0.838 | 0.676 | 0.840 | 0.836 | 0.838 | 0.923 | 0.917 | 0.867 | 0.735 | 0.844 | 0.891 | 0.864 | 0.955 | 0.954 | ||
| SSA + BiLSTM b | SVM c | 3726 | 0.836 | 0.672 | 0.848 | 0.824 | 0.838 | 0.915 | 0.917 | 0.883 | 0.766 | 0.859 | 0.906 | 0.880 | 0.943 | 0.947 |
| LGBM c | 0.848 | 0.696 | 0.859 | 0.836 | 0.849 | 0.927 | 0.927 | 0.875 | 0.751 | 0.906 | 0.844 | 0.879 | 0.961 | 0.957 | ||
| RF c | 0.824 | 0.649 | 0.832 | 0.816 | 0.826 | 0.906 | 0.911 | 0.898 | 0.797 | 0.891 | 0.906 | 0.898 | 0.959 | 0.951 | ||
| UniRep + BiLSTM b | SVM c | 5505 | 0.844 | 0.688 | 0.859 | 0.828 | 0.846 | 0.921 | 0.926 | 0.891 | 0.783 | 0.922 | 0.859 | 0.894 | 0.966 | 0.962 |
| LGBM c | 0.863 | 0.727 | 0.871 | 0.855 | 0.864 | 0.932 | 0.935 | 0.870 | 0.737 | 0.859 | 0.886 | 0.887 | 0.972 | 0.958 | ||
| RF c | 0.832 | 0.664 | 0.844 | 0.820 | 0.834 | 0.932 | 0.930 | 0.875 | 0.750 | 0.859 | 0.891 | 0.873 | 0.963 | 0.960 | ||
| SSA + UniRep + BiLSTM b | SVM c | 5626 | 0.871 | 0.742 | 0.863 | 0.879 | 0.870 | 0.943 | 0.941 | 0.891 | 0.783 | 0.922 | 0.859 | 0.894 | 0.940 | 0.943 |
| LGBM c | 0.855 | 0.711 | 0.844 | 0.867 | 0.854 | 0.945 | 0.942 | 0.898 | 0.797 | 0.891 | 0.906 | 0.898 | 0.971 | 0.971 | ||
| RF c | 0.840 | 0.680 | 0.848 | 0.832 | 0.841 | 0.926 | 0.925 | 0.898 | 0.799 | 0.859 | 0.937 | 0.894 | 0.963 | 0.957 | ||
| Feature | Model | Dim | 10-Fold Cross-Validation | Independent Test | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ACC | MCC | Sn | Sp | F1 | auPRC | auROC | ACC | MCC | Sn | Sp | F1 | auPRC | auROC | |||
| SSA b | SVM c | 53 | 0.820 | 0.641 | 0.840 | 0.801 | 0.824 | 0.910 | 0.909 | 0.914 | 0.829 | 0.937 | 0.891 | 0.916 | 0.948 | 0.941 |
| LGBM c | 77 | 0.816 | 0.634 | 0.848 | 0.785 | 0.822 | 0.877 | 0.892 | 0.883 | 0.768 | 0.922 | 0.844 | 0.887 | 0.947 | 0.940 | |
| RF c | 16 | 0.805 | 0.610 | 0.820 | 0.789 | 0.808 | 0.860 | 0.881 | 0.867 | 0.734 | 0.875 | 0.859 | 0.868 | 0.888 | 0.894 | |
| UniRep b | SVM c | 65 | 0.875 | 0.750 | 0.875 | 0.875 | 0.875 | 0.946 | 0.943 | 0.906 | 0.813 | 0.891 | 0.922 | 0.905 | 0.952 | 0.952 |
| LGBM c | 313 | 0.854 | 0.707 | 0.855 | 0.852 | 0.854 | 0.946 | 0.938 | 0.914 | 0.829 | 0.891 | 0.937 | 0.912 | 0.954 | 0.948 | |
| RF c | 329 | 0.836 | 0.672 | 0.824 | 0.848 | 0.834 | 0.918 | 0.908 | 0.891 | 0.785 | 0.844 | 0.937 | 0.885 | 0.958 | 0.957 | |
| BiLSTM b | SVM c | 344 | 0.820 | 0.641 | 0.824 | 0.816 | 0.821 | 0.913 | 0.915 | 0.922 | 0.844 | 0.937 | 0.906 | 0.923 | 0.955 | 0.956 |
| LGBM c | 339 | 0.871 | 0.742 | 0.883 | 0.859 | 0.873 | 0.925 | 0.929 | 0.906 | 0.813 | 0.906 | 0.906 | 0.906 | 0.969 | 0.966 | |
| RF c | 434 | 0.830 | 0.660 | 0.836 | 0.824 | 0.831 | 0.906 | 0.914 | 0.898 | 0.797 | 0.906 | 0.891 | 0.899 | 0.957 | 0.950 | |
| SSA + UniRep b | SVM c | 62 | 0.865 | 0.730 | 0.863 | 0.867 | 0.865 | 0.944 | 0.942 | 0.914 | 0.828 | 0.906 | 0.922 | 0.913 | 0.958 | 0.957 |
| LGBM c | 106 | 0.881 | 0.762 | 0.887 | 0.875 | 0.882 | 0.961 | 0.957 | 0.891 | 0.783 | 0.859 | 0.922 | 0.887 | 0.952 | 0.947 | |
| RF c | 47 | 0.838 | 0.676 | 0.859 | 0.816 | 0.841 | 0.937 | 0.931 | 0.906 | 0.816 | 0.859 | 0.953 | 0.902 | 0.956 | 0.947 | |
| SSA + BiLSTM b | SVM c | 267 | 0.836 | 0.672 | 0.836 | 0.836 | 0.836 | 0.910 | 0.911 | 0.914 | 0.828 | 0.906 | 0.922 | 0.913 | 0.956 | 0.952 |
| LGBM c | 317 | 0.861 | 0.723 | 0.875 | 0.848 | 0.863 | 0.924 | 0.929 | 0.906 | 0.813 | 0.906 | 0.906 | 0.906 | 0.962 | 0.958 | |
| RF c | 176 | 0.832 | 0.664 | 0.848 | 0.816 | 0.835 | 0.922 | 0.925 | 0.906 | 0.813 | 0.906 | 0.906 | 0.906 | 0.959 | 0.952 | |
| UniRep + BiLSTM b | SVM c | 186 | 0.873 | 0.746 | 0.887 | 0.859 | 0.875 | 0.932 | 0.934 | 0.914 | 0.829 | 0.937 | 0.891 | 0.916 | 0.961 | 0.965 |
| LGBM c | 106 | 0.889a | 0.777 | 0.891 | 0.887 | 0.889 | 0.947 | 0.952 | 0.944 | 0.889 | 0.922 | 0.977 | 0.952 | 0.984 | 0.977 | |
| RF c | 45 | 0.871 | 0.742 | 0.871 | 0.871 | 0.871 | 0.937 | 0.941 | 0.938 | 0.875 | 0.938 | 0.938 | 0.938 | 0.976 | 0.971 | |
| SSA + UniRep + BiLSTM b | SVM c | 336 | 0.881 | 0.762 | 0.883 | 0.879 | 0.881 | 0.940 | 0.942 | 0.922 | 0.845 | 0.953 | 0.891 | 0.924 | 0.942 | 0.946 |
| LGBM c | 285 | 0.881 | 0.762 | 0.891 | 0.871 | 0.882 | 0.951 | 0.947 | 0.938 | 0.875 | 0.922 | 0.953 | 0.937 | 0.969 | 0.969 | |
| RF c | 192 | 0.863 | 0.727 | 0.859 | 0.867 | 0.863 | 0.932 | 0.932 | 0.922 | 0.844 | 0.906 | 0.937 | 0.921 | 0.970 | 0.967 | |
| Classifier | ACC | MCC | Sn | Sp | auROC |
|---|---|---|---|---|---|
| iBitter-DRLF | 0.944 a | 0.889 | 0.922 | 0.977 | 0.977 |
| iBitter-Fuse | 0.930 | 0.859 | 0.938 | 0.922 | 0.933 |
| BERT4Bitter | 0.922 | 0.844 | 0.938 | 0.906 | 0.964 |
| iBitter-SCM | 0.844 | 0.688 | 0.844 | 0.844 | 0.904 |
| MIMML | 0.938 | 0.875 | 0.938 | 0.938 | 0.955 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, J.; Lin, X.; Jiang, Y.; Jiang, L.; Lv, Z. Identify Bitter Peptides by Using Deep Representation Learning Features. Int. J. Mol. Sci. 2022, 23, 7877. https://doi.org/10.3390/ijms23147877
Jiang J, Lin X, Jiang Y, Jiang L, Lv Z. Identify Bitter Peptides by Using Deep Representation Learning Features. International Journal of Molecular Sciences. 2022; 23(14):7877. https://doi.org/10.3390/ijms23147877
Chicago/Turabian StyleJiang, Jici, Xinxu Lin, Yueqi Jiang, Liangzhen Jiang, and Zhibin Lv. 2022. "Identify Bitter Peptides by Using Deep Representation Learning Features" International Journal of Molecular Sciences 23, no. 14: 7877. https://doi.org/10.3390/ijms23147877
APA StyleJiang, J., Lin, X., Jiang, Y., Jiang, L., & Lv, Z. (2022). Identify Bitter Peptides by Using Deep Representation Learning Features. International Journal of Molecular Sciences, 23(14), 7877. https://doi.org/10.3390/ijms23147877