1. Introduction
The domestic silkworm,
Bombyx mori, is an important economic insect and model organism of lepidoptera, having important economic and scientific research value [
1]. For instance, silk from the silkworm is a biomaterial with excellent biocompatibility [
2]. Moreover, the silkworm model is suitable for drug discovery due to its low cost, few ethical concerns, and similar pharmacokinetic and pharmacodynamic properties [
3,
4].
B. mori nucleopolyhedrovirus (BmNPV) is an obligate virus that specially infects insects; the disease caused by BmNPV is extremely contagious and seriously endangers sericulture production around the world [
5]. However, the molecular mechanism of BmNPV infection is not yet fully understood. In recent years, studies on the infection mechanism of BmNPV has been in full swing, and many effectors involved in viral invasion have been identified and verified. For instance, several studies have been devoted to elucidating the viral invasion mechanism from transcriptomic or proteomic perspective [
6,
7]. For example,
B. mori E3 ubiquitin–protein ligase SINA-like 10 (SINAL10) could initiate viral proliferation, which serves as a binding protein for envelope fusion protein GP64 of BmNPV [
8]. Additionally, it has been reported that
B. mori membrane protein BmREEPa is required for BmNPV to enter into cells by interacting with GP64 [
9].
Insects’ innate immune system is evolutionarily highly conserved, defending the host from invading pathogenic microorganisms, such as bacteria, fungi, and viruses. The innate immunity is triggered by the recognition of pathogen-associated molecular patterns (PAMPs) by the host itself [
10]. In general, the components located on the surface of microbial cell walls, such as lipopolysaccharide, lipoteichoic acid, and peptidoglycan, all belong to the category of PAMPs [
11]. In insects, pattern recognition receptors (PRRs) are crucial in the recognition of PAMPs, which include C-type lectins (CTLs), peptidoglycan recognition proteins (PGRPs), β-1,3 glucan recognition protein (βGRP), Gram-negative-bacteria-binding proteins (GNBPs), integrins, hemolin, and apolipophorin Ⅲ (ApoLp-Ⅲ), among others [
11,
12]. The recognition of invading pathogens by the host itself activates downstream signal transduction pathways and thereby a series of innate immune responses, including the synthesis of antimicrobial peptides (AMPs), and here the melanization is initiated, finally killing and eliminating the pathogens. In
Drosophila melanogaster, the mutants of IMD pathway are defective for PGRP-LC, and other genes displayed more sensitivity to cricket paralysis virus (CrPV) infection [
13]. The expression of
BmPGRP2-2 could be induced by BmNPV, wherein the protein inhibits PTEN-phosphoinositide 3-kinase (PI3K)/Akt signaling pathway to promote BmNPV replication [
14].
C-type lectins, as highly conserved calcium-dependent receptors, are defined by one or more C-type lectin-like domains (CTLDs). CTLDs that contain characteristic motifs, such as Glu-Pro-Asn (EPN) motif or Gln-Pro-Asp (QPD) motif, are closely linked with the specificity of carbohydrate binding of CTLs [
15]. Up until now, C-type lectins have been identified in many mammals, plants, insects, mollusks, and other multicellular organisms [
16]. Mammalian CTLs are known to play important roles in the homeostasis of the immune system, allergic responses, recognition of apoptotic cells and tumors, and complement activation [
17,
18]. Among the arthropods, CTLs specifically recognize the PAMPs on the surface of microbes, and then trigger the aforementioned immune responses to eliminate the invading pathogens. For instance,
B. mori CTL5 may be involved in the JAK/STAT pathway and slightly stimulated by
B. mori cytoplasmic polyhedrosis virus (BmCPV) [
19]. In mosquitos, mosGCTL-7 interacts with the envelope protein of the Japanese encephalitis virus (JEV) via N-glycan at N154, and then promotes virus entry into cells [
20].
Aedes aegypti mosGCTL-1 collaborates with the human CD45 homolog mosPTP-1 to enable attachment and entry of the West Nile virus (WNV) [
21]. In
Musca domestica L, MdCTL1 and MdCTL2 could effectively reduce the infection of Sf 9 cells by
Autographa californica multicapsid nucleopolyhedrovirus (AcMNPV) and dramatically inhibit the proliferation of influenza H1 N1 virus [
22]. In short, the roles of insect C-type lectins in viral infection have been increasingly investigated and reported. However, little is known about the potential internal relationship between C-type lectins with BmNPV infection and replication.
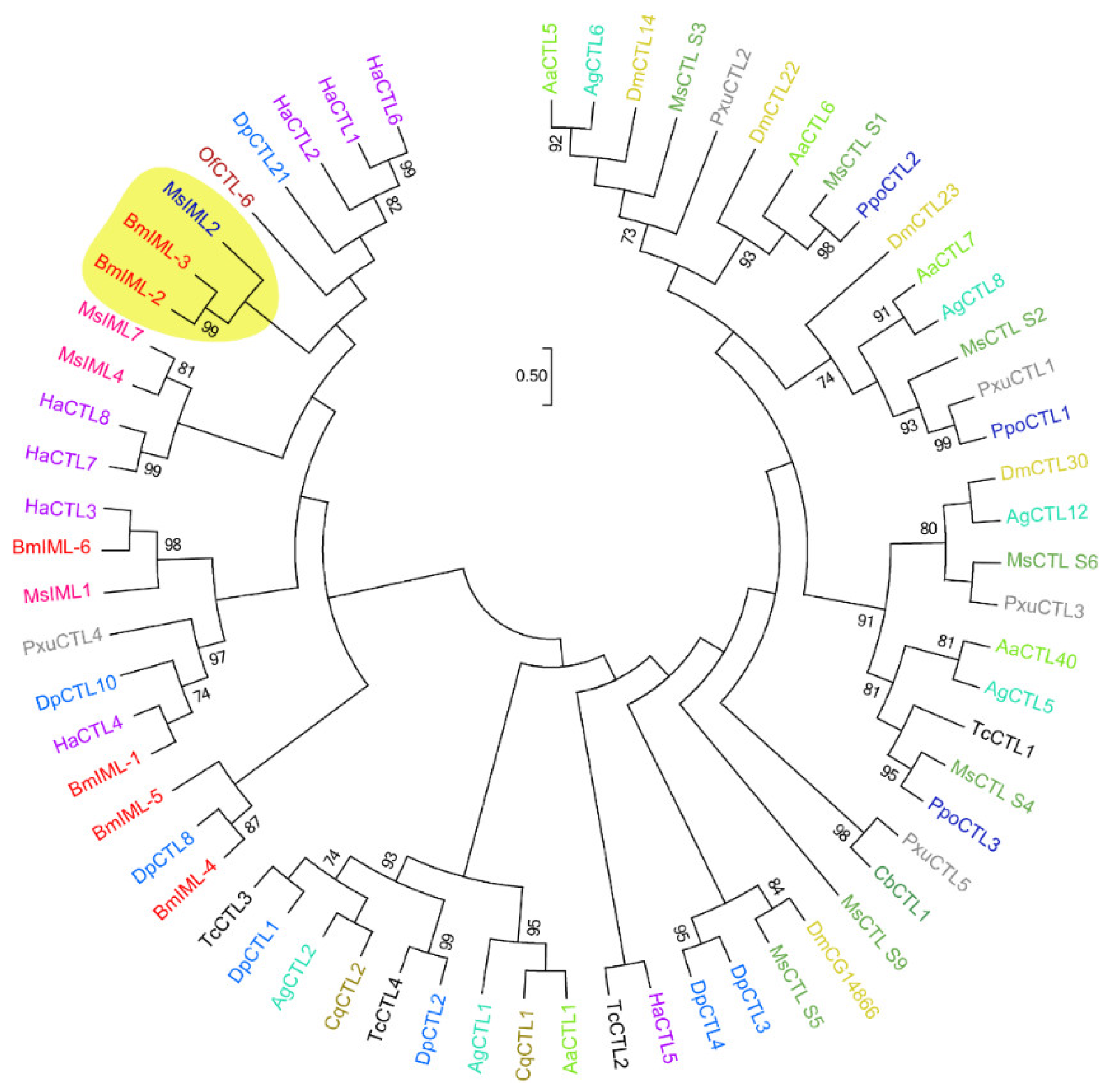
In the present study, we investigated the functional roles of C-type lectins BmIML-2 in viral infection and replication. At first, bioinformatics analysis indicated that BmIML-2 possesses two tandem CTLD that defined a canonical CTL in insects. The phylogenetic tree analysis showed that BmIML-2 has the closest homologous evolutionary relationship with B. mori IML-3 and Manduca sexta IML-2. In addition, overexpression assay and siRNA interference-mediated knockdown were conducted. The results of overexpression and knockdown demonstrated that BmIML-2 acts as a negative regulator in the process of viral infection and replication. Moreover, the results of qRT-PCR revealed that BmIML-2 may take part in the initiation of apoptosis, thus regulating the expression of several apoptosis-related genes. Furthermore, immunofluorescence assay was performed to analyze the subcellular localization of BmIML-2. In sum, our findings demonstrated that BmIML-2 could inhibit the infection and replication of BmNPV, which may be due to its involvement in the regulation of the apoptotic signal pathway.
3. Discussion
BmNPV is an important infectious pathogen that endangers sericulture production, causing great economic losses to sericulture. Insect CTLs are widely known as important PRRs that contribute to triggering and regulation of host immune responses, as well as in defense against pathogens [
23,
24]. In the present study, we selected a putative CTL gene from the genome database of silkworm, called
BmIML-2, to investigate its potential functions during the infection and replication of BmNPV. Our results demonstrated that
BmIML-2 could significantly inhibit the proliferation of BmNPV, which might play key roles in viral infection. Bioinformatic analysis results indicated that mature BmIML-2 contain an EPD and an EPN motif. The characteristic motif was critical for the determination of carbohydrate-binding capacity. The CTLD-containing QPD motif known as galactose-type sugar binding exhibits an affinity to galactose specifically, while CTLD with EPN motif would prefer to bind mannose-type sugar [
15]. Hence, we speculated that BmIML-2 may have the ability to specifically recognize and bind to several viral envelope proteins in the early stages of BmNPV infection.
The mRNA of
BmIML-2 was detected in the selected tissues, and it exhibited the highest expression level in fat bodies, followed by malpighian tubes. Interestingly, its transcript level significantly increased at 24 h after silkworms received BmNPV infection, while it was dramatically downregulated 48 h and 72 h after viral infection compared with the control group. Previous studies suggested that the expression level of
B. mori βGRP4 dramatically decreased from 12 h to 72 h after BmNPV infection [
25]. It was likely that more BmIML-2 were induced and were secreted to recognize the invading BmNPV within 24 h of infection, and BmNPV might suppress the host’s own immune defense mechanisms after 48 h to 72 h. We considered that BmIML-2 might respond to the BmNPV infection and possess multiple functions in regulating innate immune responses.
To further confirm the involvement of
BmIML-2 in the infection and replication of BmNPV, we performed overexpression and knockdown assays to determine the roles and regulation of
BmIML-2. Our results suggested that the overexpression of
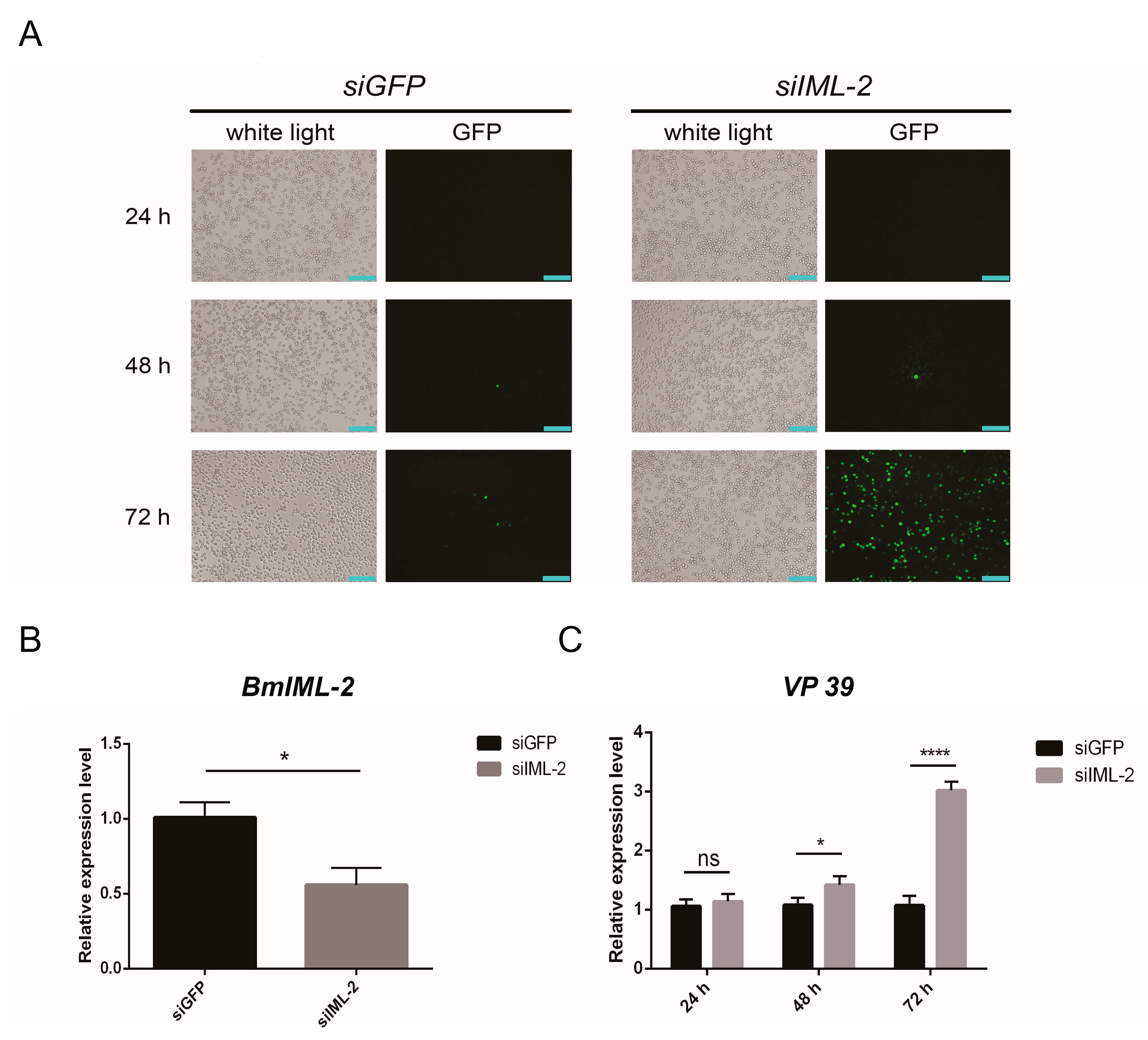
BmIML-2 led to a significant reduction of viral proliferation in BmN cells. Conversely, the increased proliferation of BmNPV was observed after the knockdown of
BmIML-2 via siRNA. Moreover, several other PRRs identified previously have also shown that up- or downregulation of these genes would significantly modulate the proliferation of BmNPV, such as BmβGRP4 [
25] and BmPGRP2-2 [
14]. Thus, we conceived that BmNPV, acting as a typical PPR, was provided with the effective capacity to inhibit the infection and replication of BmNPV.
Apoptosis acts as an extremely powerful response to the infection of virus, which integrates with other innate antiviral defenses in insects, then reduces the viral proliferation and spread [
26,
27]. More importantly, the antiviral defense mediated by apoptosis is critical for insects lacking the vertebrate-specific acquired immune system [
28]. Apoptosis signal pathway in several insect cell lines seems to be initiated following the challenge of NPVs [
29]. Then, the signal of apoptosis initiated by NPV challenge ultimately stimulates the caspase cascade, which consists of upstream promoters and downstream effector caspases [
30,
31,
32]. In silkworm, the
Bm-p53 gene was able to regulate apoptosis via the activation of caspase cascade, and overexpression of
Bm-p53 in BmN cells promoted apoptosis [
33]. To further confirm whether
BmIML-2 regulates the infection and replication of BmNPV by involving the signal pathway of apoptosis,
BmIML-2 was overexpressed in the BmN cells and then infected with BmNPV to induce apoptosis. The results showed that the relative transcript level of apoptosis-related genes such as
BmCaspase1,
BmDredd,
BmPkc,
BmApaf1,
BmPTEN, and
Bmp53 was upregulated when compared with the control group. At the same time, the expression levels of several genes that are unrelated to apoptosis did not change significantly in virally infected
BmIML-2-overexpressing cells (
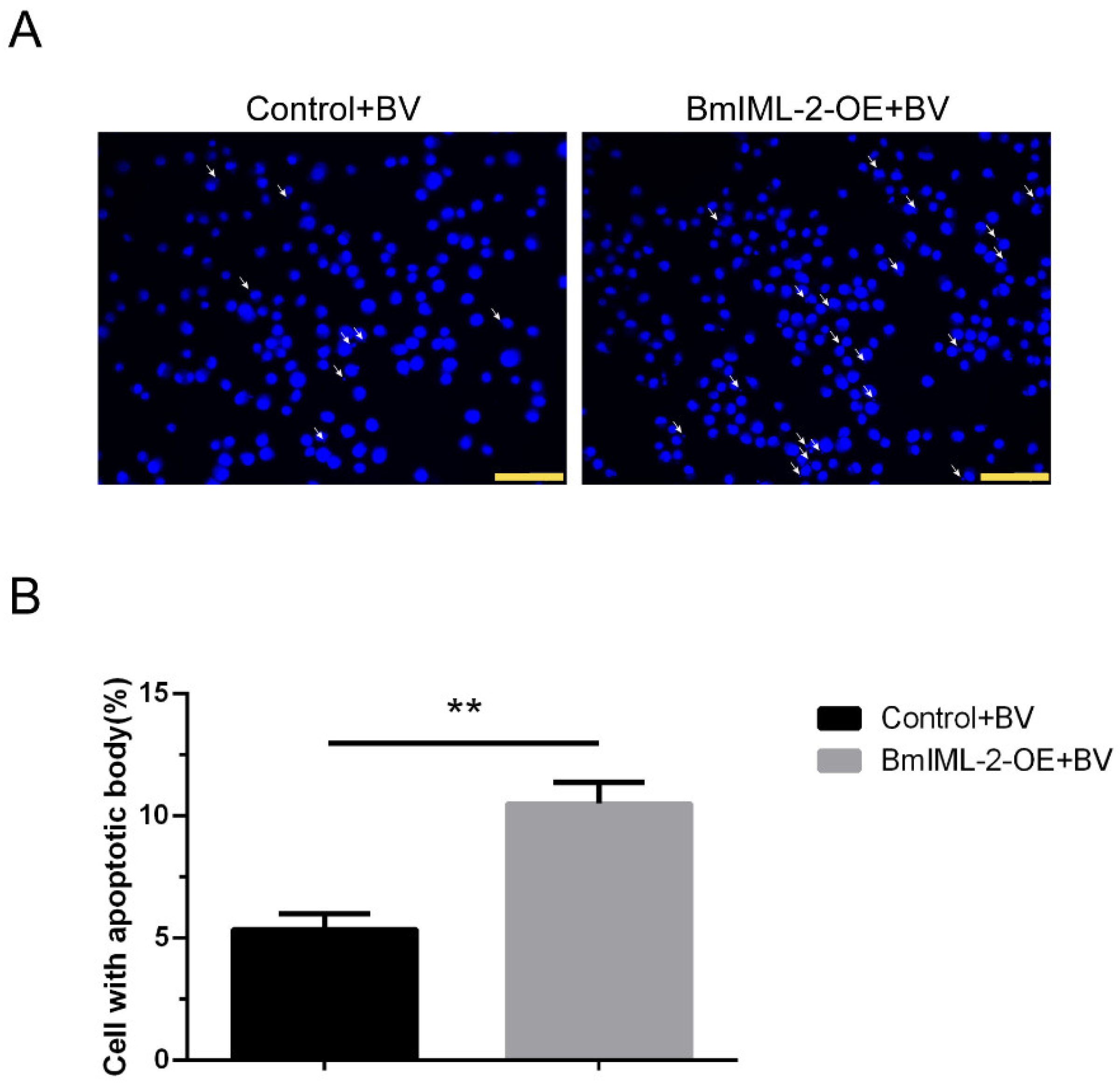
Figure S3). Moreover, cells overexpressing
BmIML-2 with apoptotic body formation were observed under a microscope (
Figure 7). The above results provided evidence that
BmIML-2 could positively regulate the signal pathway that induces the occurrence of apoptosis in BmN cells. PRRs in insects that are involved in the regulation of apoptosis signal pathway were reported in other investigations. For example,
Bombyx mori peptidoglycan recognition protein BmPGRP2-2 negatively regulated the expression of BmPTEN and then inhibited the apoptosis through repressing PTEN-phosphoinositide 3-kinase (PI3K)/Akt signaling [
14]. Additionally, another important PRR, BmβGRP4, could positively regulate the transcript level of
BmPTEN and repress the expression of an apoptosis suppressor gene named
BmIAP [
25].
In conclusion, a C-type lectin gene called BmIML-2 was characterized and identified from the genome database of B. mori. The significant change of BmIML-2 transcript level was detected in fat bodies and malpighian tubes upon BmNPV challenge. Afterwards, in vitro overexpression and knockdown assays suggested that BmIML-2 exhibited dramatical inhibitory ability for the proliferation of BmNPV. Moreover, we demonstrated that BmIML-2 suppressed BmNPV proliferation by participating in the regulation of the expression of apoptosis-related genes. Finally, we found that BmIML-2 was mainly localized in the cytoplasm with the help of immunofluorescence analysis. These results imply that BmIML-2 plays key roles in the process of BmNPV proliferation, possibly via the apoptosis signal pathway.
4. Materials and Methods
4.1. Preparation of Silkworm, BmN Cells, and Virus
The silkworm strain p50 was provided in the Sericulture Research Institute, Jiangsu University of Science and Technology University, Zhenjiang, China. Briefly, the larvae were raised with fresh mulberry leaves under a rearing environment of 26 ± 1 °C, 70–85% relative humidity, and a 12 h light/12 h dark photoperiod. The BmN cell line from Bombyx mori ovarian was cultured in TC-100 (Livning Biological Technology Co., Ltd., Beijing) medium supplemented with 10% (v/v) fetal bovine serum (FBS) (Gibco) contained at 28 °C. The BmNPV particles used for oral infection were suspended in sterile water (1.0 × 107 OB/mL). The recombinant EGFP-tagged budded virus of BmNPV was maintained in our laboratory, which used to assess the viral infection and reproduction.
4.2. Bioinformatics Analysis of BmIML-2
The EXPASY (Expert Protein Analysis System) websites (
http://www.expasy.org (accessed on 3 February 2022)) were used to predict the deduced amino acids sequences, molecular weight, and isoelectric point. The SignalP-5.0 server (
http://www.cbs.dtu.dk/services/SignalP/ (accessed on 4 February 2022)) was used to predict the putative signal peptides. The prediction of conserved domains and motifs was performed in the SMART website (
http://smart.embl-heidelberg.de/smart/set_mode.cgi (accessed on 4 February 2022)). The phylogenetic tree analysis of BmIML-2 was generated by MEGA 6.0 software with a neighbor-joining method (bootstrap = 1000 replications, Poisson model, uniform rates).
4.3. RNA Extraction and cDNA Synthesis
The first day of fifth instar silkworm larvae were anesthetized on ice and dissected; then, tissue samples including heads, midguts, fat bodies, malpighian tubes, and hemolymph were collected separately. In addition, each first day of fifth instar larvae from the same batch were infected by orally fed BmNPV suspension (1.0 × 107 OB/mL). After 48 h infection, the larvae were dissected, and tissues were harvested. Then, the total RNA samples from the aforementioned tissues were extracted separately with TRIzol Reagent (TIANGEN, Biotech Co., Ltd., Beijing, China). In addition, the first strand cDNA for subsequent experiments was synthesized by using a FastKing RT Kit following the manufacturer’s recommendations (TIANGEN, China).
4.4. Overexpression Vector Construction and Small Interfering RNA (siRNA) Synthesis
The ORF region of the
BmIML-2 was cloned using the specific primers listed in
Table S1 that provided the restriction sites
Kpn I and
Xba I. The amplified PCR products were ligated with pMD-19T vector for sequencing verification. The digested fragment was constructed into the pIZT/V5-His-mCherry vector that had undergone the same double-digestion treatment. Then, the constructed vector was verified by double digestion and sequencing, and the empty vector was used as a control. Moreover, the siRNA targeting
BmIML-2 were synthesized by GenePharma (Suzhou, China). The oligonucleotide sequence designed for
BmIML-2 was
siIML-2 (5′-GCGCUGAUCAAUGACCUUUTT-3′), and a sequence designed for
siEGFP (5′-GCGAUGCCACCUACGGCAATT-3′) was used as a negative control.
4.5. Cell Transient Transfection and BmNPV Infection
BmN cells were seeded into 12-well culture plates for 24 h and transfected with the overexpression vector or siRNA by using GP-transfect-Mate transfection reagent (GenePharma, Suzhou, China) according to the manufacturer’s instructions. Briefly, 1.6 µg overexpression vector or 80 pm siRNA oligo was added to 100 µL serum-free TC-100 medium and then mixed with an equal volume of medium containing 5 µL of transfection reagent for the preparation of transfection complexes. After incubation at room temperature for 20 min, the complex was finally added to the culture medium for transfection. Afterwards, the transfected BmN cells in each well were infected with BmNPV-EGFP with a multiplicity of infection (MOI) of 3. Then, cells were harvested 24, 48, and 72 h after viral infection, and genomic DNA or total RNA were extracted for subsequent qRT-PCR analysis. Moreover, the fluorescence intensity in the wells was observed and photographed at different points in time under an inverted fluorescence microscope (Ti-E, Nikon, Tokyo, Japan).
4.6. Genomic DNA Extraction
Genomic DNA of BmN cells was extracted with appropriate volume DNA extraction buffer (10 mM Tris-HCl, 100 mM EDTA, 100 mM NaCl, 0.5% SDS; pH = 8.0). After adding an equal volume of saturated phenol to mix thoroughly, we centrifuged the mixture for 15 min, aspirated the supernatant, and purified the mixture with chloroform isoamyl alcohol and absolute ethanol containing 0.1 M sodium acetate (NaAc), followed by RNase treatment. The purity and concentration of DNA were measured by a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, NewYork, NY, USA) and were stored at −80 °C for further use.
4.7. Quantitative Real-Time PCR (qRT-PCR) Analysis
The transcript levels of
BmIML-2 or other genes were quantified using qRT-PCR assay. Specific primers listed in
Table S1 were used. The qRT-PCR solution was prepared with UltraSYBR Mixture (ComWin Biotech Co.,Ltd., Beijing, China) according to the manufacturer’s recommendations. In addition, the qRT-PCR assay was carried out on a LightCycler
® 96 PCR instrument (Roche, Basel, Switzerland). The amplification conditions consisted of a pre-denaturation at 95 °C 10 min, followed by 40 additional cycles of denaturation at 95 °C for 15 s, and annealing and extension at 60 °C for 1 min. Each reaction was performed in 3 biological and 2 technical replicates. The relative expression level of each mRNA was quantified with the 2
−ΔΔCt method.
B. mori glyceraldehyde-3-phosphate dehydrogenase (
BmGAPDH) gene was used as an internal reference to calibrate the total amount of RNA.
4.8. Observation of Apoptosis Morphology of Cells after BmIML-2 Overexpression
Briefly, the BmN cells overexpressing BmIML-2 were infected with BmNPV-EGFP with a MOI of 3. After 48 h infection, cells were fixed with 4% paraformaldehyde for 20 min after culture medium was removed completely. After that, cells were washed three times with sterile PBS, and the cell nuclei were stained with 4,6-diamidino-2-phenylindole (DAPI) (Sangon Biotech Co., Ltd., Shanghai, China) (0.5 ug/mL) for 15 min. Then, the treated cells were observed by using a fluorescence microscope. The cells transfected with empty vector were used as a control.
4.9. Immunofluorescence Analysis of BmIML-2
Firstly, the BmN cells overexpressing BmIML-2 (polyhistidine tag) cultured overnight for 24 h were suspended and placed on the cell round coverslip for 4 h. Then, the cells were rinsed with PBS for three times and fixed with 4% paraformaldehyde for 20 min. After being washed three times, cells were permeated with 1% Triton X-100 (PBS preparation) for 20 min at room temperature and washed with PBS as mentioned above. Then, the permeabilized cells were blocked with 5% BSA prepared by PBST for 1 h. After that, the cells were incubated with mouse monoclonal anti-polyhistidine antibody (1:800 in 5% BSA) at 4 °C overnight. After being rinsed three times, the cells were incubated with goat anti-mouse secondary antibody conjugated with Alexa Flour 488 (1:500 in 5% BSA) for 2 h at room temperature. Subsequently, cells were rinsed with PBS, and the nuclei were stained with DAPI (0.5 ug/mL) for 15 min; then, we repeated the washing above for the last time. Finally, the stained cells were mounted and photographed under a fluorescence microscope.
4.10. Statistical Analysis
All treatments were carried out in triplicate, and results were represented as means ± standard deviation (S.D.). Data analysis and comparison were performed by using Graph Pad Prism 6.0 software. The difference between the two groups of data was compared with Student’s t-test, while analysis of variance (ANOVA) was used to test the significance of the difference between the means of two or more samples. A significant difference was considered when p-value < 0.05.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







