
UDP-Glucose: A Cereblon-Dependent Glucokinase Protein Degrader

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
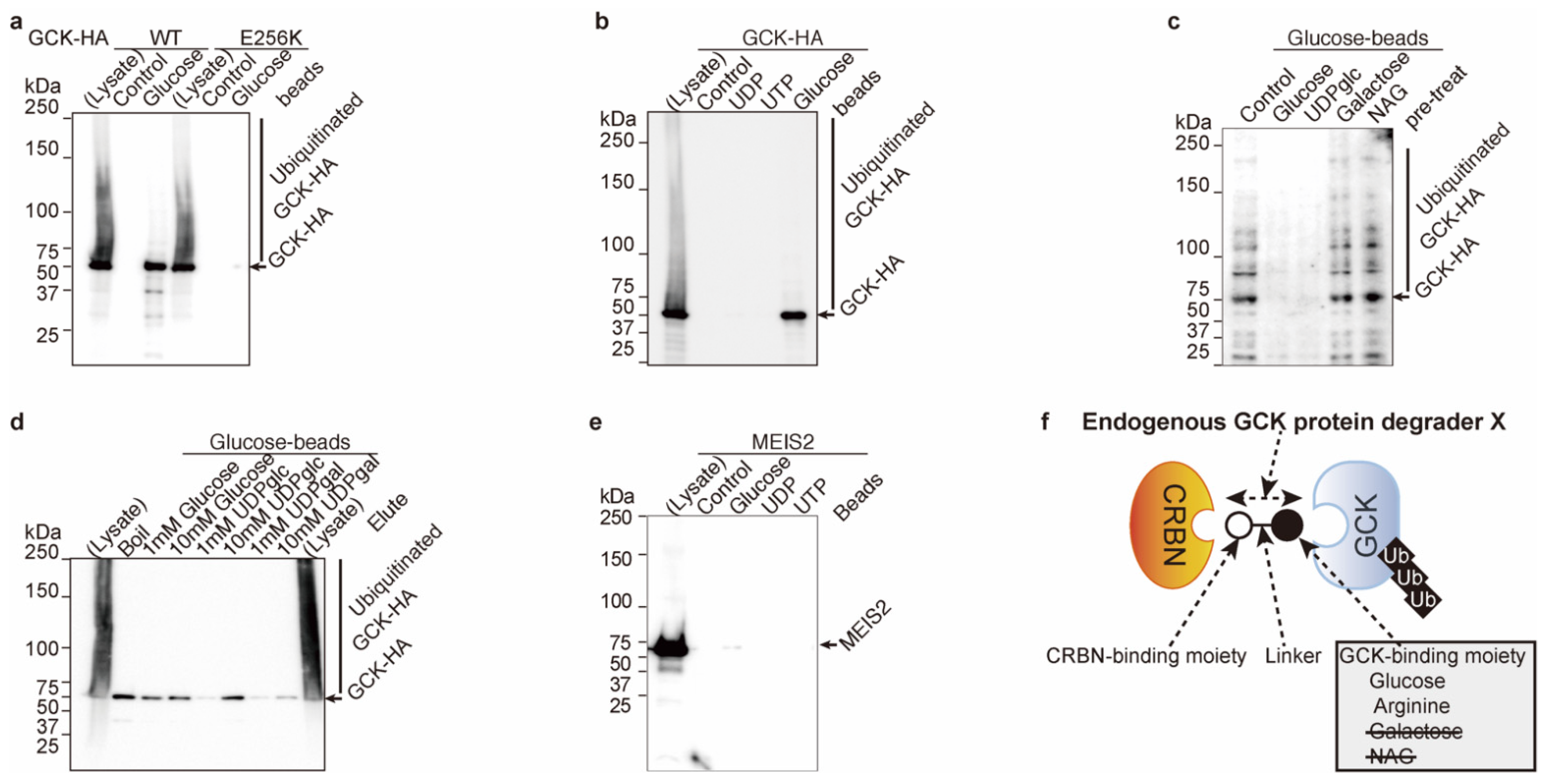
2.1. Glucose Inhibits Glucokinase Ubiquitination and Degradation
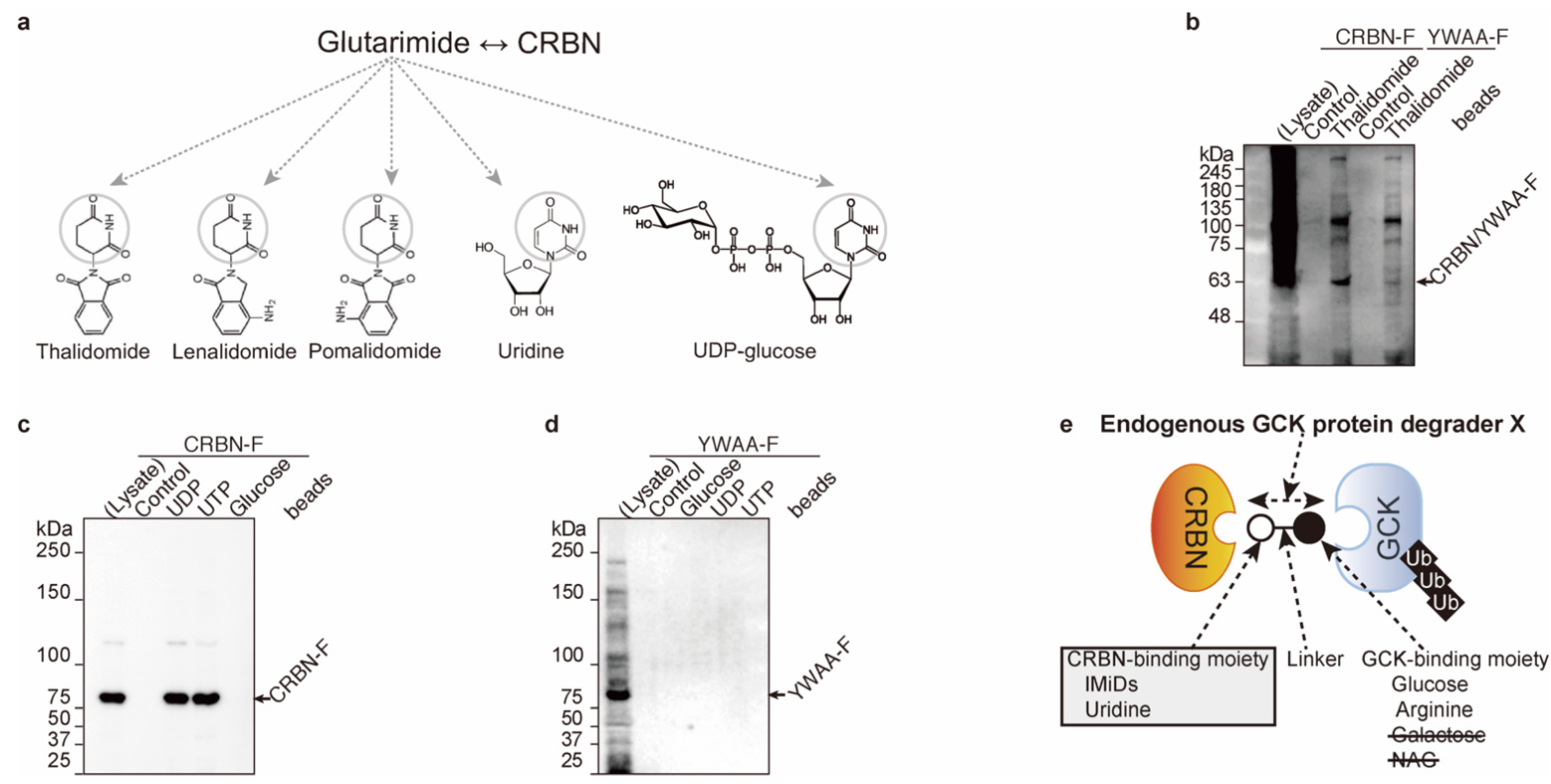
2.2. Uridine Binds to Cereblon
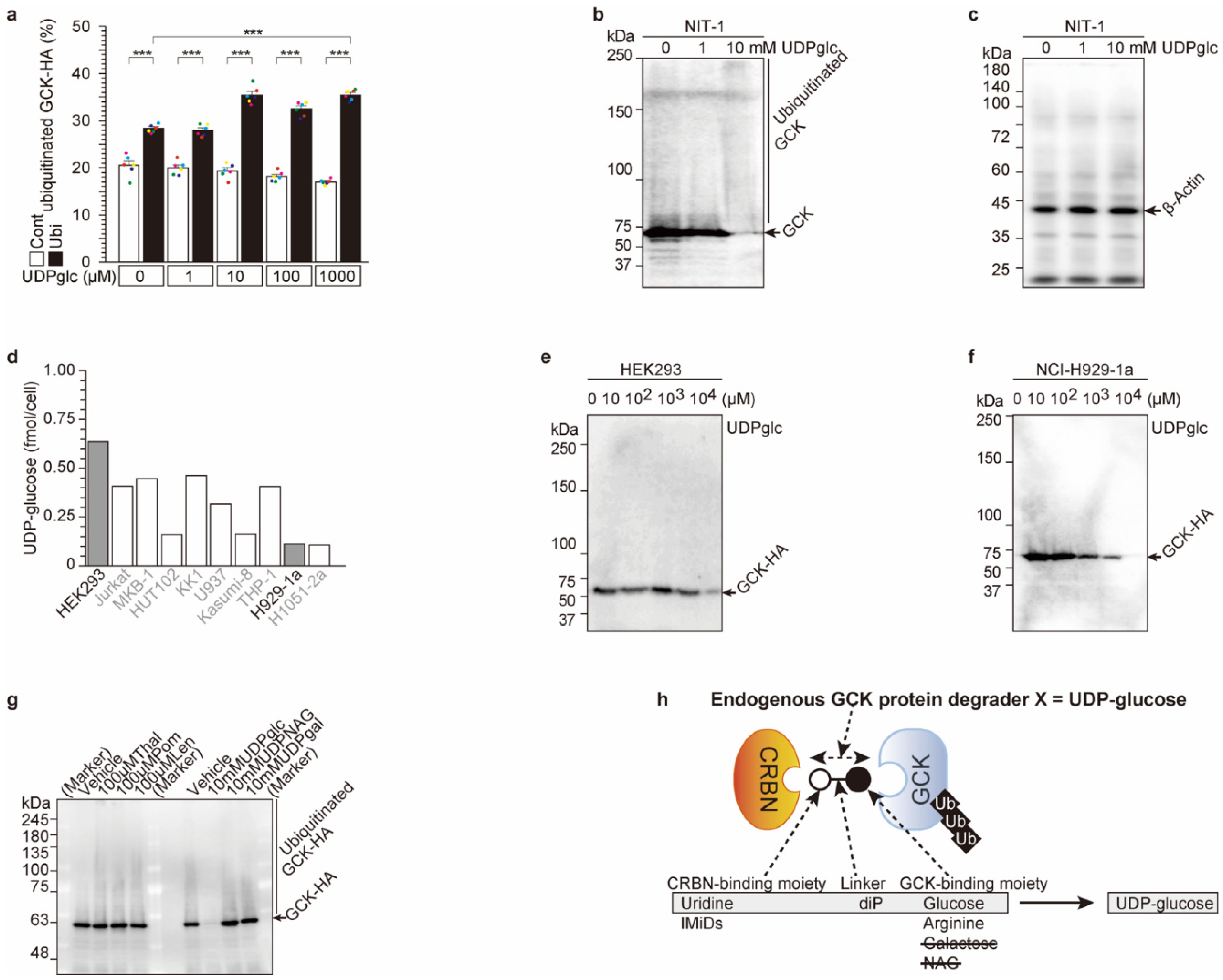
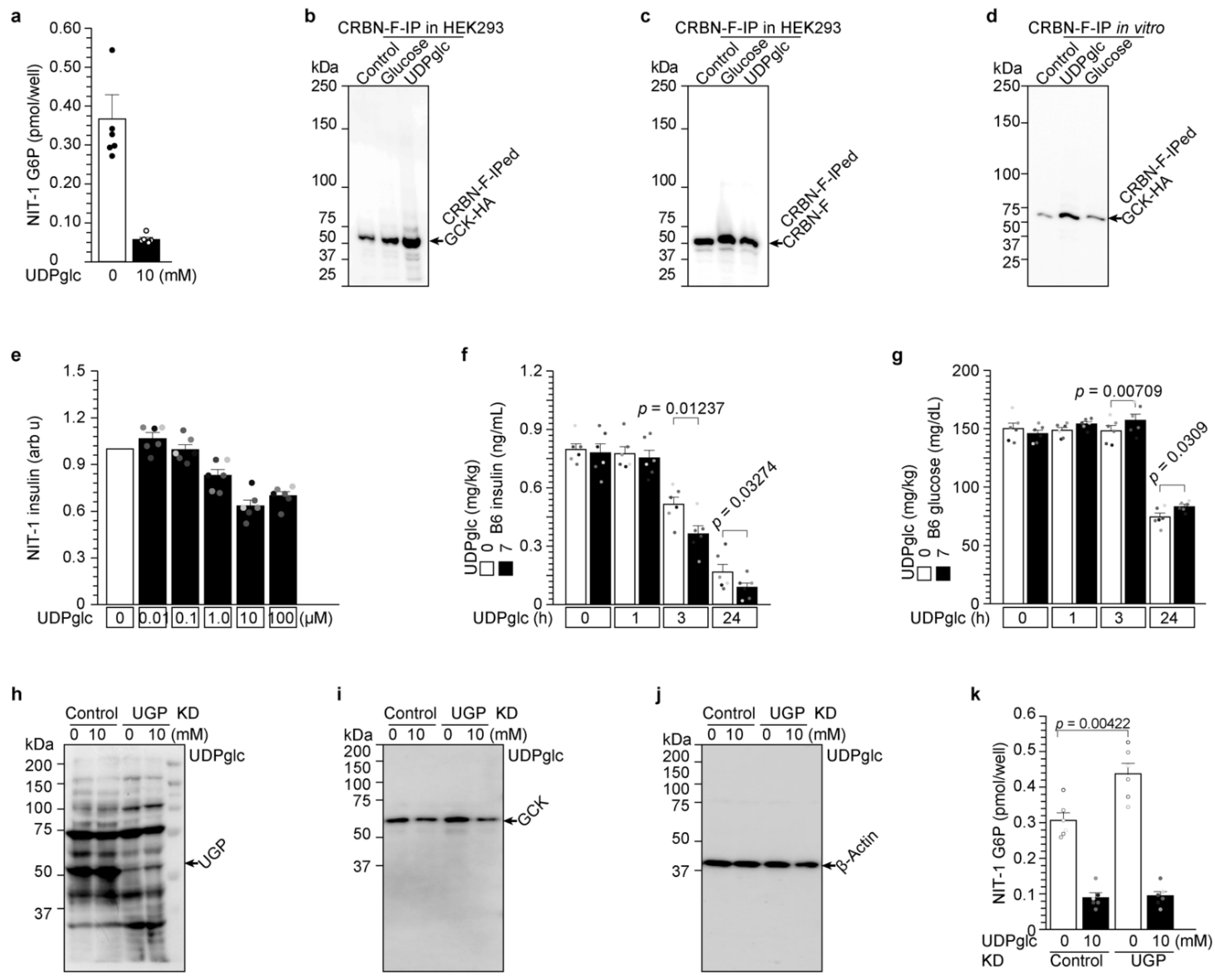
2.3. UDP-Glucose Induces Cereblon-Dependent Glucokinase Ubiquitination and Degradation
2.4. UDP-Glucose Induces Glucokinase Degradation and Reduces Insulin Secretion from 3 h to 24 h
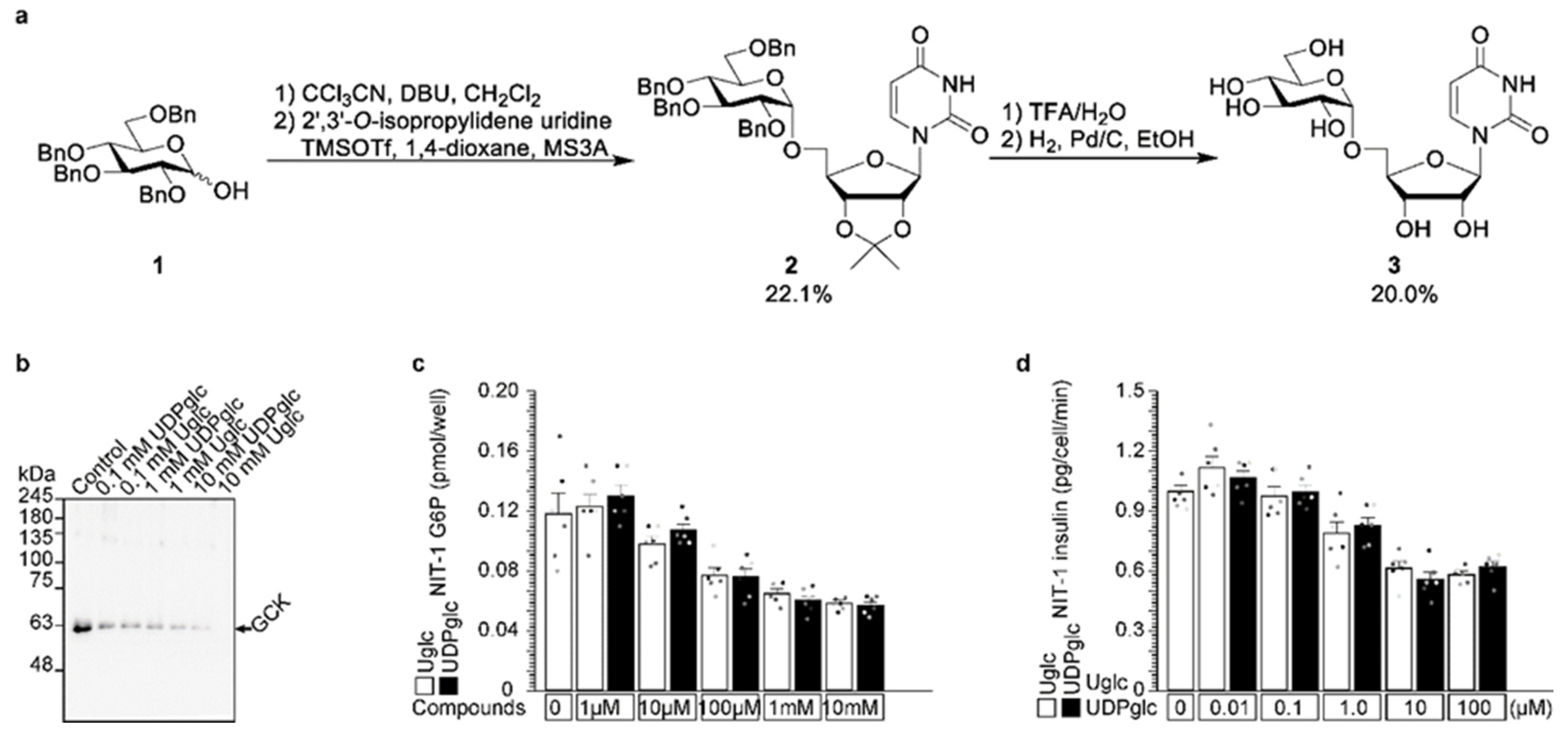
2.5. Uridine-Glucose Degrades Glucokinase and Reduces Insulin Secretion
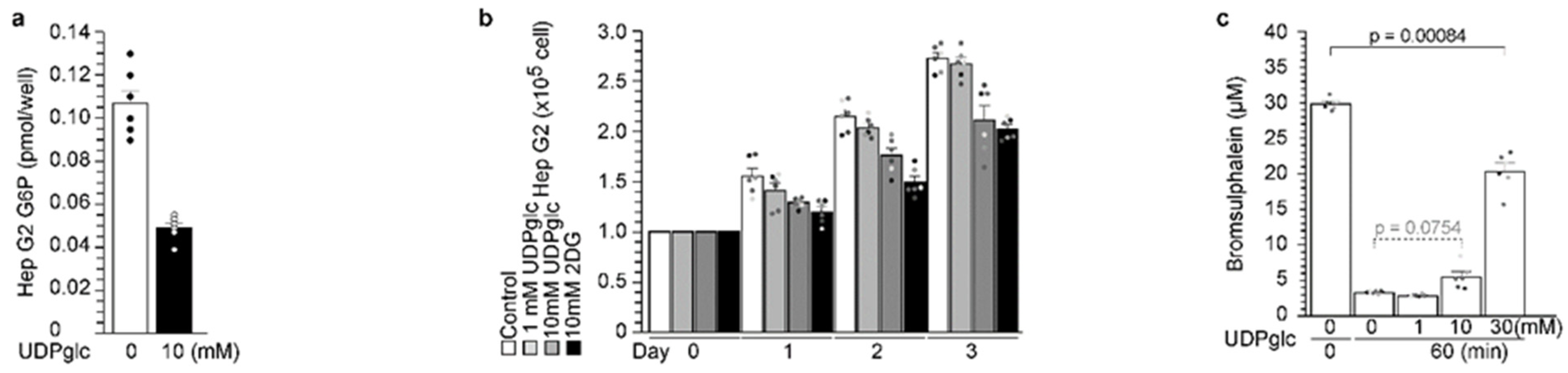
2.6. UDP-Glucose Reduced G6P Production in Hepatocytes
3. Discussion
4. Materials and Methods
4.1. Antibodies and Cell Culture
4.2. Analysis of Insulin Secretion from Cells
4.3. Immunoprecipitation and Western Blot
4.4. Chemical Detection
4.5. Chemical Synthesis
4.5.1. General
4.5.2. 2-Bromoethyl 2,3,4,6-Tetra-O-acetyl-β-d-glucopyranoside (2)
4.5.3. 6-Bromohexyl 2,3,4,6-Tetra-O-acetyl-β-d-glucopyranoside (3)
4.5.4. 2-S-Thioacetylethyl 2,3,4,6-Tetra-O-acetyl-β-d-glucopyranoside (4)
4.5.5. 6-S-Thioacetylhexyl 2,3,4,6-Tetra-O-acetyl-β-d-glucopyranoside (5)
4.5.6. 2-S-Ethyl-β-d-glucopyranose S-Linked Maleimide (6)
4.5.7. 6-S-Hexyl-β-d-glucopyranose S-Linked Maleimide (7)
4.6. Statistical Analysis
4.7. Ethical Approval
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cho, J.; Horikawa, Y.; Enya, M.; Takeda, J.; Imai, Y.; Imai, Y.; Handa, H.; Imai, T. L-Arginine prevents cereblon-mediated ubiquitination of glucokinase and stimulates glucose-6-phosphate production in pancreatic β-cells. Commun. Biol. 2020, 3, 497. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, Y.; Enya, M.; Mabe, H.; Fukushima, K.; Takubo, N.; Ohashi, M.; Ikeda, F.; Hashimoto, K.; Watada, H.; Takeda, J. NEUROD1-deficient diabetes (MODY6): Identification of the first cases in Japanese and the clinical features. Pediatr. Diabetes 2018, 19, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Horikawa, Y. Maturity-onset diabetes of the young as a model for elucidating the multifactorial origin of type 2 diabetes mellitus. J. Diabetes Investig. 2018, 9, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Bekes, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Dis. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Fischer, E.S.; Böhm, K.; Lydeard, J.R.; Yang, H.; Stadler, M.B.; Cavadini, S.; Nagel, J.; Serluca, F.; Acker, V.; Lingaraju, G.M.; et al. Structure of the DDB1-CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 2014, 512, 49–53. [Google Scholar] [CrossRef]
- Chen, Y.-A.; Peng, Y.-J.; Hu, M.-C.; Huang, J.-J.; Chien, Y.-C.; Wu, J.-T.; Chen, T.-Y.; Tang, C.-Y. The Cullin 4A/B-DDB1-Cereblon E3 Ubiquitin Ligase Complex Mediates the Degradation of CLC-1 Chloride Channels. Sci. Rep. 2015, 29, 10667. [Google Scholar] [CrossRef]
- Fu, S.J.; Hu, M.C.; Peng, Y.J.; Fang, H.Y.; Hsiao, C.T.; Chen, T.Y.; Jeng, C.J.; Tang, C.Y. CUL4-DDB1-CRBN E3 Ubiquitin Ligase Regulates Proteostasis of ClC-2 Chloride Channels: Implication for Aldosteronism and Leukodystrophy. Cells 2020, 9, 1332. [Google Scholar] [CrossRef]
- Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K.; Imamura, Y.; Yamaguchi, Y.; Handa, H. Identification of a primary target of thalidomide teratogenicity. Science 2005, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Girona, A.; Mendy, D.; Ito, T.; Miller, K.; Gandhi, A.K.; Kang, J.; Karasawa, S.; Carmel, G.; Jackson, P.; Abbasian, M.; et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia 2012, 26, 2326–2335. [Google Scholar] [CrossRef]
- Rosano, C.; Sabini, E.; Rizzi, M.; Deriu, D.; Murshudov, G.; Bianchi, M.; Serafini, G.; Magnani, M.; Bolognesi, M. Binding of non-catalytic ATP to human hexokinase I highlights the structural components for enzyme-membrane association control. Structure 1999, 7, 1427–1437. [Google Scholar] [CrossRef]
- Devlin, T.M. Chapter 15, Carbohydrate metabolism I: Major metabolic pathways and their control. In Textbook of Biochemistry, 6th ed.; Wiley-Liss: Hoboken, NJ, USA, 2005; pp. 595–600. [Google Scholar]
- Heim, C.; Pliatsika, D.; Mousavizadeh, F.; Bär, K.; Alvarez, B.H.; Giannis, A.; Hartmann, M.D. De-Novo Design of Cereblon (CRBN) Effectors Guided by Natural Hydrolysis Products of Thalidomide Derivatives. J. Med. Chem. 2019, 62, 6615–6629. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.; Ito, T.; Yamaguchi, Y.; Handa, H. Discovery of CRBN as a target of thalidomide: A breakthrough for progress in the development of protein degraders. Chem. Soc. Rev. 2022, 51, 6234–6250. [Google Scholar] [CrossRef]
- Umeda, M.; Hiramoto, M.; Watanabe, A.; Tsunoda, N.; Imai, T. Arginine-induced insulin secretion in endoplasmic reticulum. Biochem. Biophys. Res. Commun. 2015, 466, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Uebi, T.; Umeda, M.; Imai, T. Estrogen induces Estrogen Receptor alpha expression and hepatocyte proliferation in the livers of the male mice. Genes Cells 2015, 20, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Umeda, M.; Hiramoto, M.; Imai, T. Partial hepatectomy induces delayed hepatocyte proliferation and normal liver regeneration in ovariectomized mice. Clin. Exp. Gastroenterol. 2015, 8, 175–182. [Google Scholar]
- Tsugawa, Y.; Natori, M.; Handa, H.; Imai, T. Estradiol accelerates liver regeneration through estrogen receptor α. Clin. Exp. Gastroenterol. 2019, 12, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, Y.; Hiramoto, M.; Imai, T. Estrogen induces estrogen receptor α expression and hepatocyte proliferation in late pregnancy. Biochem. Biophys. Res. Commun. 2019, 511, 592–596. [Google Scholar] [CrossRef]
- Cho, J.; Hiramoto, M.; Masaike, Y.; Sakamoto, S.; Imai, Y.; Imai, Y.; Handa, H.; Imai, T. UGGT1 retains proinsulin in the endoplasmic reticulum in an arginine dependent manner. Biochem. Biophys. Res. Commun. 2020, 527, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Tsugawa, Y.; Imai, Y.; Imai, T. Chorionic gonadotropin stimulates maternal hepatocyte proliferation during pregnancy. Biochem. Biophys. Res. Commun. 2021, 579, 110–115. [Google Scholar] [CrossRef]
- Cho, J.; Tsugawa, Y.; Imai, T. R1526 residue in arginine/proinsulin binding domain of UGGT1 is involved in proinsulin binding. Biochem. Biophys. Res. Commun. 2022, 615, 131–135. [Google Scholar] [CrossRef]
- Soga, T.; Ohashi, Y.; Ueno, Y.; Naraoka, H.; Tomita, M.; Nishioka, T. Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J. Proteome Res. 2003, 2, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Soga, T.; Baran, R.; Suematsu, M.; Ueno, Y.; Ikeda, S.; Sakurakawa, T.; Kakazu, Y.; Ishikawa, T.; Robert, M.; Nishioka, T.; et al. Differential metabolomics reveals ophthalmic acid as an oxidative stress biomarker indicating hepatic glutathione consumption. J. Biol. Chem. 2006, 281, 16768–16776. [Google Scholar] [CrossRef] [PubMed]
- Stolz, R.M.; Northrop, B.H. Experimental and Theoretical Studies of Selective Thiol–Ene and Thiol–Yne Click Reactions Involving N-Substituted Maleimides. J. Org. Chem 2013, 78, 8105–8116. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, J.; Miyagawa, A.; Yamaguchi, K.; Abe, W.; Tsugawa, Y.; Yamamura, H.; Imai, T. UDP-Glucose: A Cereblon-Dependent Glucokinase Protein Degrader. Int. J. Mol. Sci. 2022, 23, 9094. https://doi.org/10.3390/ijms23169094
Cho J, Miyagawa A, Yamaguchi K, Abe W, Tsugawa Y, Yamamura H, Imai T. UDP-Glucose: A Cereblon-Dependent Glucokinase Protein Degrader. International Journal of Molecular Sciences. 2022; 23(16):9094. https://doi.org/10.3390/ijms23169094
Chicago/Turabian StyleCho, Jaeyong, Atsushi Miyagawa, Kazuki Yamaguchi, Wakana Abe, Yoji Tsugawa, Hatsuo Yamamura, and Takeshi Imai. 2022. "UDP-Glucose: A Cereblon-Dependent Glucokinase Protein Degrader" International Journal of Molecular Sciences 23, no. 16: 9094. https://doi.org/10.3390/ijms23169094