
Role of the Pangolin in Origin of SARS-CoV-2: An Evolutionary Perspective

 , and
, and {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Current Understanding on the Intermediate Host of SARS-CoV-2 and Our Perspective
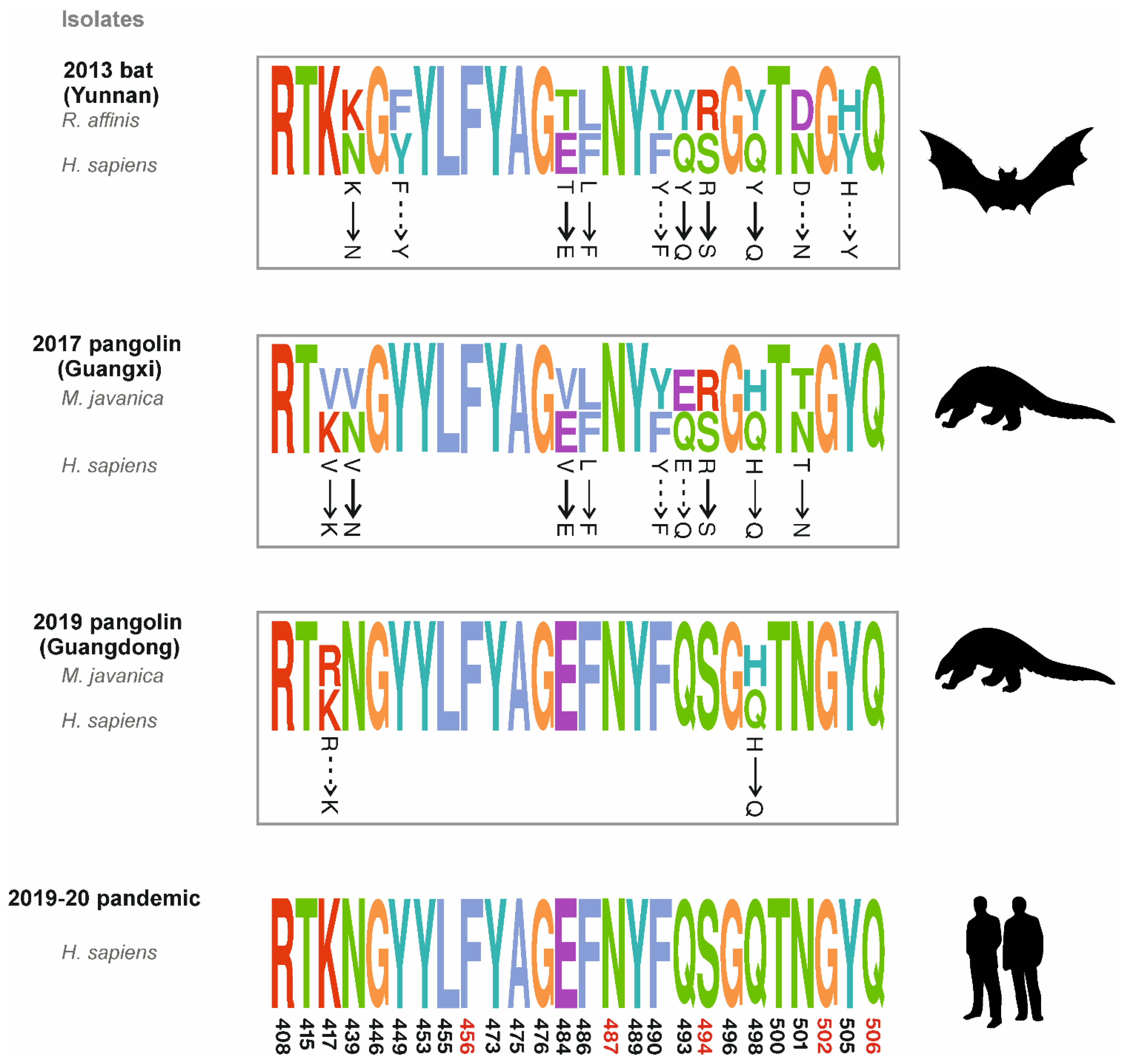
3. Sequence Comparative Analysis of Coronavirus Isolates from Bat and Pangolins
4. Role of Genomic Recombination between Bat and Pangolin Coronaviruses
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Manjunath, K.; Ranjan, R.K.; Kaushik, S.; Kumar, S.; Verma, V. COVID-19 pandemic: Insights into structure, function, and hACE2 receptor recognition by SARS-CoV-2. PLoS Pathog. 2020, 16, e1008762. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Zhang, S.; Qiao, S.; Yu, J.; Zeng, J.; Shan, S.; Tian, L.; Lan, J.; Zhang, L.; Wang, X. Bat and pangolin coronavirus spike glycoprotein structures provide insights into SARS-CoV-2 evolution. Nat. Commun. 2021, 12, 1607. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Friend, T.; Stebbing, J. What is the intermediate host species of SARS-CoV-2? Future Virol. 2021, 16, 153–156. [Google Scholar] [CrossRef]
- Wang, L.F.; Shi, Z.; Zhang, S.; Field, H.; Daszak, P.; Eaton, B.T. Review of bats and SARS. Emerg. Infect. Dis. 2006, 12, 1834–1840. [Google Scholar] [CrossRef]
- Zhao, J.; Cui, W.; Tian, B.P. The Potential Intermediate Hosts for SARS-CoV-2. Front. Microbiol. 2020, 11, 580137. [Google Scholar] [CrossRef] [PubMed]
- Lam, T.T.; Jia, N.; Zhang, Y.W.; Shum, M.H.; Jiang, J.F.; Zhu, H.C.; Tong, Y.G.; Shi, Y.X.; Ni, X.B.; Liao, Y.S.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef] [PubMed]
- Xiao, K.; Zhai, J.; Feng, Y.; Zhou, N.; Zhang, X.; Zou, J.J.; Li, N.; Guo, Y.; Li, X.; Shen, X.; et al. Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature 2020, 583, 286–289. [Google Scholar] [CrossRef]
- Wang, S.; Sundaram, J.P.; Spiro, D. VIGOR, an annotation program for small viral genomes. BMC Bioinform. 2010, 11, 451. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e9. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zhang, Y.; Qu, Y.; Zhang, C.; Liu, X.W.; Zhao, M.; Mu, Y.; Li, W. Key residues of the receptor binding domain in the spike protein of SARS-CoV-2 mediating the interactions with ACE2: A molecular dynamics study. Nanoscale 2021, 13, 9364–9370. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef]
- Liu, P.; Jiang, J.Z.; Wan, X.F.; Hua, Y.; Li, L.; Zhou, J.; Wang, X.; Hou, F.; Chen, J.; Zou, J.; et al. Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)? PLoS Pathog. 2020, 16, e1008421. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Niu, S.; Wang, J.; Bai, B.; Wu, L.; Zheng, A.; Chen, Q.; Du, P.; Han, P.; Zhang, Y.; Jia, Y.; et al. Molecular basis of cross-species ACE2 interactions with SARS-CoV-2-like viruses of pangolin origin. EMBO J. 2021, 40, e107786. [Google Scholar] [CrossRef]
- Nie, J.; Li, Q.; Zhang, L.; Cao, Y.; Zhang, Y.; Li, T.; Wu, J.; Liu, S.; Zhang, M.; Zhao, C.; et al. Functional comparison of SARS-CoV-2 with closely related pangolin and bat coronaviruses. Cell Discov. 2021, 7, 21. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- Temmam, S.; Vongphayloth, K.; Baquero, E.; Munier, S.; Bonomi, M.; Regnault, B.; Douangboubpha, B.; Karami, Y.; Chrétien, D.; Sanamxay, D.; et al. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature 2022, 604, 330–336. [Google Scholar] [CrossRef]
- Mallapaty, S. Closest known relatives of virus behind COVID-19 found in Laos. Nature 2021, 597, 603. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Barnes, C.O.; Weisblum, Y.; Schmidt, F.; Caskey, M.; Gaebler, C.; Cho, A.; Agudelo, M.; Finkin, S.; et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 2021, 12, 4196. [Google Scholar] [CrossRef]
- Cheng, M.H.; Krieger, J.M.; Banerjee, A.; Xiang, Y.; Kaynak, B.; Shi, Y.; Arditi, M.; Bahar, I. Impact of new variants on SARS-CoV-2 infectivity and neutralization: A molecular assessment of the alterations in the spike-host protein interactions. iScience 2022, 25, 103939. [Google Scholar] [CrossRef]
- Li, X.; Giorgi, E.E.; Marichannegowda, M.H.; Foley, B.; Xiao, C.; Kong, X.P.; Chen, Y.; Gnanakaran, S.; Korber, B.; Gao, F. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 2020, 6, eabb9153. [Google Scholar] [CrossRef]
- Patino-Galindo, J.A.; Filip, I.; Rabadan, R. Global Patterns of Recombination across Human Viruses. Mol. Biol. Evol. 2021, 38, 2520–2531. [Google Scholar] [CrossRef]
- Perez-Losada, M.; Arenas, M.; Galan, J.C.; Palero, F.; Gonzalez-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef]
- Zhu, Z.; Meng, K.; Meng, G. Genomic recombination events may reveal the evolution of coronavirus and the origin of SARS-CoV-2. Sci. Rep. 2020, 10, 21617. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.; Keck, J.G.; Stohlman, S.A.; Lai, M.M. High-frequency RNA recombination of murine coronaviruses. J. Virol. 1986, 57, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Makarenkov, V.; Mazoure, B.; Rabusseau, G.; Legendre, P. Horizontal gene transfer and recombination analysis of SARS-CoV-2 genes helps discover its close relatives and shed light on its origin. BMC Ecol. Evol. 2021, 21, 5. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Alizon, S.; Hurford, A.; Mideo, N.; Van Baalen, M. Virulence evolution and the trade-off hypothesis: History, current state of affairs and the future. J. Evol. Biol. 2009, 22, 245–259. [Google Scholar] [CrossRef]
- He, X.; Lau, E.H.Y.; Wu, P.; Deng, X.; Wang, J.; Hao, X.; Lau, Y.C.; Wong, J.Y.; Guan, Y.; Tan, X.; et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 2020, 26, 672–675. [Google Scholar] [CrossRef]
- Oran, D.P.; Topol, E.J. Prevalence of Asymptomatic SARS-CoV-2 Infection: A Narrative Review. Ann. Intern. Med. 2020, 173, 362–367. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein. Curr. Biol. 2020, 30, 2196–2203.e3. [Google Scholar] [CrossRef]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef]
- Weigang, S.; Fuchs, J.; Zimmer, G.; Schnepf, D.; Kern, L.; Beer, J.; Luxenburger, H.; Ankerhold, J.; Falcone, V.; Kemming, J.; et al. Within-host evolution of SARS-CoV-2 in an immunosuppressed COVID-19 patient as a source of immune escape variants. Nat. Commun. 2021, 12, 6405. [Google Scholar] [CrossRef]
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef]
- Corey, L.; Beyrer, C.; Cohen, M.S.; Michael, N.L.; Bedford, T.; Rolland, M. SARS-CoV-2 Variants in Patients with Immunosuppression. N. Engl. J. Med. 2021, 385, 562–566. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 evolution during treatment of chronic infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef]
- Choudhary, M.C.; Crain, C.R.; Qiu, X.; Hanage, W.; Li, J.Z. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Sequence Characteristics of Coronavirus Disease 2019 (COVID-19) Persistence and Reinfection. Clin. Infect. Dis. 2022, 74, 237–245. [Google Scholar] [CrossRef]
- Jung, C.; Kmiec, D.; Koepke, L.; Zech, F.; Jacob, T.; Sparrer, K.M.J.; Kirchhoff, F. Omicron: What Makes the Latest SARS-CoV-2 Variant of Concern So Concerning? J. Virol. 2022, 96, e0207721. [Google Scholar] [CrossRef]
- Callaway, E. Heavily mutated Omicron variant puts scientists on alert. Nature 2021, 600, 21. [Google Scholar] [CrossRef]
- Kupferschmidt, K. Where did ‘weird’ Omicron come from? Science 2021, 374, 1179. [Google Scholar] [CrossRef]
- Wei, C.; Shan, K.J.; Wang, W.; Zhang, S.; Huan, Q.; Qian, W. Evidence for a mouse origin of the SARS-CoV-2 Omicron variant. J. Genet. Genom. 2021, 48, 1111–1121. [Google Scholar] [CrossRef]
- Bull, J.J.; Lauring, A.S. Theory and empiricism in virulence evolution. PLoS Pathog. 2014, 10, e1004387. [Google Scholar] [CrossRef]
- Lytras, S.; Hughes, J.; Martin, D.; Swanepoel, P.; de Klerk, A.; Lourens, R.; Kosakovsky Pond, S.L.; Xia, W.; Jiang, X.; Robertson, D.L. Exploring the Natural Origins of SARS-CoV-2 in the Light of Recombination. Genome Biol. Evol. 2022, 14, evac018. [Google Scholar] [CrossRef]
- Pollett, S.; Conte, M.A.; Sanborn, M.; Jarman, R.G.; Lidl, G.M.; Modjarrad, K.; Maljkovic Berry, I. A comparative recombination analysis of human coronaviruses and implications for the SARS-CoV-2 pandemic. Sci. Rep. 2021, 11, 17365. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, S.K.; Minocha, R.; Thapa, P.J.; Srivastava, M.; Dandekar, T. Role of the Pangolin in Origin of SARS-CoV-2: An Evolutionary Perspective. Int. J. Mol. Sci. 2022, 23, 9115. https://doi.org/10.3390/ijms23169115
Gupta SK, Minocha R, Thapa PJ, Srivastava M, Dandekar T. Role of the Pangolin in Origin of SARS-CoV-2: An Evolutionary Perspective. International Journal of Molecular Sciences. 2022; 23(16):9115. https://doi.org/10.3390/ijms23169115
Chicago/Turabian StyleGupta, Shishir K., Rashmi Minocha, Prithivi Jung Thapa, Mugdha Srivastava, and Thomas Dandekar. 2022. "Role of the Pangolin in Origin of SARS-CoV-2: An Evolutionary Perspective" International Journal of Molecular Sciences 23, no. 16: 9115. https://doi.org/10.3390/ijms23169115