Abstract
Osteoclastogenesis is an ongoing rigorous course that includes osteoclast precursors fusion and bone resorption executed by degradative enzymes. Osteoclastogenesis is controlled by endogenous signaling and/or regulators or affected by exogenous conditions and can also be controlled both internally and externally. More evidence indicates that autophagy, inflammation, and immunity are closely related to osteoclastogenesis and involve multiple intracellular organelles (e.g., lysosomes and autophagosomes) and certain inflammatory or immunological factors. Based on the literature on osteoclastogenesis induced by different regulatory aspects, emerging basic cross-studies have reported the emerging disquisitive orientation for osteoclast differentiation and function. In this review, we summarize the partial potential therapeutic targets for osteoclast differentiation and function, including the signaling pathways and various cellular processes.
1. Introduction
Bone renewal and remodeling are dynamic courses that include old bone removal by osteoclasts and new bone generation by osteoblasts [1]. Bone also provides mechanical assistance to the body, locomotion, and protects the internal organs, such as the brain, lung, kidney, and bone marrow [2]. Typically, bone maintains a normal metabolic equilibrium. However, excessive osteoclast activity or osteoblast depletion causes degenerative joint- and bone-related diseases, such as osteoporosis and osteolytic Paget disease [3,4]. In contrast, attenuating osteoclast activity and/or osteoclast dysfunction or reinforcement osteoblast remodeling causes abnormal bone density-related diseases, such as osteopetrosis and osteogenesis imperfecta [5,6].
Osteoclasts are multinucleated terminal giant cells regulated by two essential cytokines: macrophage colony-stimulating factor (M-CSF or CSF-1) and receptor activator of nuclear factor κB (NF-κB) ligand (RANKL). M-CSF is commonly secreted by bone marrow stromal cells (BMSCs) and osteoblasts and is critical in osteoclastogenesis biological function. M-CSF binds to its cognate receptor c-Fms (on monocyte/macrophage and osteoclast surfaces) and promotes osteoclastogenesis [2,7]. M-CSF promoted RANKL-induced osteoclast survival and motility, whereas incomplete blockade of c-Fms attenuated bone loss and did not prevent ongoing osteoclastogenesis. Nevertheless, M-CSF cannot be considered as irreplaceable and indispensable as RANKL. RANKL combines with receptor activator of NF-κB (RANK) on the surface of osteoclast precursors (OCPs) or mature osteoclasts and simultaneously activates downstream cascade signals to accelerate osteoclastogenesis [8,9]. Osteoprotegerin (OPG) is a secretory glycoprotein produced by osteoblasts or BMSCs. OPG competitively combines with RANKL to block RANKL binding to RANK, thereby inhibiting osteoclastogenesis [10,11,12]. The RANKL–RANK–OPG system is a well-known key regulatory axis and is indispensable during osteoclastogenesis [9,13,14] (Figure 1).
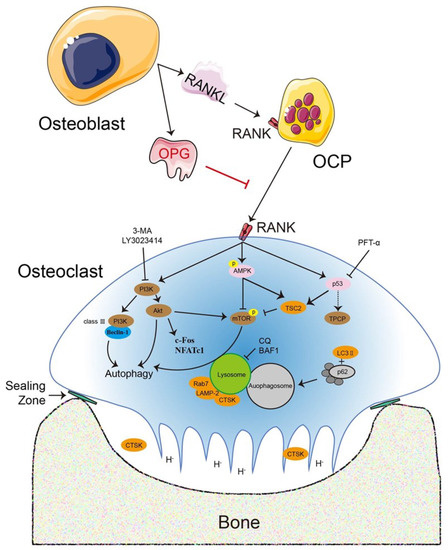
Figure 1.
The RANKL–RANK–OPG system is a key regulatory axis that is indispensable during osteoclastogenesis and is regulated by numerous signaling pathways and dependent or independent routes. RANKL and OPG are secreted by osteoblasts or other cells and maintain the dynamic equilibrium for bone metabolism, which includes bone matrix degradation and regeneration mediated by osteoclasts and osteoblasts, respectively. The association with osteoclastogenesis is required for RANKL to bind to its receptor RANK, whereas OPG suppresses osteoclastogenesis. Various signaling pathways or regulators directly or indirectly regulate osteoclastogenesis, such as the PI3K–Akt and AMPK–mTOR signaling pathways, p53, and the transcription factors c-Fos and NFATc1. An exogenous secreted factor, OPG inhibits osteoclast differentiation and function directly. Accordingly, the role of various critical regulators in osteoclastogenesis has been examined with PI3K inhibitors [3-methyladenine (3-MA) and LY3023414], the p53 inhibitor pifithrin-α (PFT-α), and lysosomal inhibitors [chloroquine (CQ) and bafilomycin A1 (BAF1)].
Although this regulatory mechanism was identified in previous osteoclastogenesis studies, understanding of the mechanism is limited. Generally, many aspects affect osteoclast differentiation and function, which involves the regulatory mechanism of OCP migration and fusion, osteoclast secretion, and the exogenous action of other cells. Therefore, this review highlights the role of osteoclastogenesis signaling transduction in the interaction with autophagy, inflammation, and immunity and proposes their relationship for future applications in bone cell differentiation and function.
2. Master Regulators of Osteoclastogenesis
Osteoclastogenesis requires a series of transcription factors and degrading enzymes including M-CSF, which is secreted by osteoblasts or BMSCs and is a survival and proliferative factor [15,16]. Similarly, RANKL indirectly and/or directly stimulates osteoclastogenesis by regulating the phosphatidylinositol 3-kinase (PI3K)–protein kinase B (Akt) signaling pathway, AMP-activated protein kinase (AMPK), and p53, lysosomal-associated regulators, the transcription factors nuclear factor of activated T cells c1 (NFATc1) and activator protein-1 (AP-1), and OPG [17].
PI3K is a lipid kinase that contains three subfamilies (class I, II, III). Class I PI3K is critical for cell survival, differentiation, proliferation, and metabolism [18]. A lack of class I PI3Kα (p85α) in mouse bone marrow monocytes/macrophages (BMMs) led to the reduction of activated Akt and phosphatidylinositol-3,4,5-trisphosphate (PIP3) levels, which is related to M-CSF binding to its tyrosine kinase receptor c-Fms [19,20]. In mice, class 1 PI3Kγ (p110γ) knockout resulted in augmented bone mass by decreasing osteoclast numbers and impairing osteoclast formation in vivo and in vitro, but did not affect bone formation [20].
The role of class II PI3K is unclear, especially in cell signaling transduction. Class III PI3K is mainly involved in regulating vesicle trafficking, including autophagy, phagocytosis, and endocytosis [21]. Class III PI3K combines with Beclin1 to form a complex that initiates autophagy, thereby affecting the regulation of downstream signaling [22,23]. These actions were suppressed by the PI3K inhibitor 3-methyladenine (3-MA), which caused the phosphorylation of mammalian target of rapamycin (mTOR) and p70S6K during osteoclastogenesis [8].
Akt has a positive regulatory function in RANKL-induced osteoclastogenesis by increasing NFATc1 expression but does not affect the expression of c-Fos and the inactive form of GSK-3β [24]. Akt contains three members: Akt1, Akt2, and Akt3. Akt1 and Akt2 are expressed in similar locations, such as bone, fat, liver, and muscle. Akt3 is limited to the testis and brain and was not expressed on BMMs and osteoclasts [25,26,27,28]. Mice with double-elimination of Akt1 and Akt2 exhibited loss of bone mass and development injury and even died shortly after birth [29]. The major Akt1 isoform is present in bone cells (osteoblasts, BMMs, osteoclasts), where its deficiency in mice caused decreased bone mass and formation by impairing osteoblast and osteoclast function [27]. Importantly, each isoform performs different functions that cannot be replaced and compensated by another [30,31]. Recent, a novel inhibitor, LY3023414, was examined in the PI3K–mTOR signaling pathway in a phase Ⅱ clinical study, where it suppressed Akt1 and Akt2 expression in osteoblastogenesis [25]. These data suggested that the PI3K–Akt signaling pathway is critical in bone cells, especially RANKL-induced osteoclastogenesis.
AMPK is a critical sensor in regulating cellular energy metabolism in eukaryotes. AMPK contains three subunits: α, β, and γ. AMPKα subunit phosphorylation at Thr172 is induced by decreased ATP/AMP and the activation of calcium/calmodulin-dependent protein kinase kinase β (CaMKKβ) and liver kinase (LKB1), which are the two upstream kinases [32,33,34]. An important monitor in response to the balance of cellular ATP level, AMPK is coupled with its functional homeostasis in regulating mitochondrial function, autophagy, and glucose metabolism [35]. In the presence of a physiological concentration of ATP (5 mmol/L) in mammalian cells, AMP led to the increased allosteric activation of both the γ1 and γ2 complexes by approximately 10-fold, whereas γ3 complex activation became almost negligible (1.4-fold higher) [36]. CaMKKβ and LKB1 are potential activators in bone cells. Phosphorylation of CaMKKβ promotes osteoclast differentiation and function. Otherwise, it has an inhibitory effect. In this section, we mainly focus on the relationship between AMPK and osteoclast differentiation and function.
AMPK plays a negative role in osteoclastogenesis by regulating autophagy [32,37,38,39]. The AMPKα isoform has a catalytic action in osteoclast differentiation and function via signaling cascades. The activation of AMPKα stimulated OPG secretion from osteoblasts and bone mineral density and indirectly attenuated the differentiation and function of osteoclasts induced from RAW264.7 cells to alleviate bone loss [40]. Continuous activation of the AMPKα2 isoform enhanced osteogenesis while attenuating M-CSF and RANKL secretion and the RANKL/OPG ratio to result in lower osteoclastogenesis [39]. In contrast, inactivation of AMPKα at Thr172 promoted osteoclast differentiation and function regulated by CaMKK and TGF-β-activated kinase 1 (TAK1) in vitro [41]. An in vivo study reported results consistent with those previous results, where osteoclasts caused decreased bone mass [42]. The AMPK regulatory β and γ subunits are critical in bone cells. Deletion of the AMPKβ isoform did not affect the osteoclast number but reduced trabecular bone mass and density [43,44]. In MC3T3 cells (osteoblastic cell line), activation of the AMPKβ isoform at Ser108 by prostaglandin E2 (PGE2) was an important mediator of the arachidonic acid derivative and promoted OPG synthesis [43]. The AMPKγ subunit consists of isoforms γ1, γ2, and γ3, which have different functions in osteoclastogenesis. Silencing of AMPKγ activity using knockdown or inhibitor (dorsomorphin) decreased its expression and osteoclast size but had no effect on osteoclast number [45,46].
More studies have focused on osteoblast mediation by p53, which indirectly controls osteoclastogenesis. p53 is a primary tumor suppressor that is also a negative regulator of bone remodeling with multiple regulatory mechanisms in osteoclastogenesis and osteoblast-dependent osteoclastogenesis [47]. p53 is related to osteoblast differentiation and has a dual role in regulating bone mass, where p53 deficiency in mice enhanced bone mass by increasing osteoblast differentiation while accelerating osteoclastogenesis that may have been induced by M-CSF secretion [47,48,49]. p53 is also regulated by the tumor suppressor ARF that controls ribosomal biogenesis and is involved in bone remodeling, including osteoblast function and osteoclast activity independently of p53 in vivo [50]. Interestingly, ARF inhibited cell proliferation in a p53-dependent or -independent manner, while its deletion in vivo enhanced osteoclastogenesis and bone resorption [51].
Previous data demonstrated that p53 accelerates osteoblastic differentiation driven by the transcription factors runt-related transcription factor 2 (RUNX2) and Osterix [47,52]. RUNX2 is a master regulator in BMSC-induced osteoblast differentiation induced [53]. RUNX2 was highly expressed in p53-deficient mouse embryonic fibroblasts, which had higher alkaline phosphatase (ALP) activity induced by bone morphogenetic protein 4 (BMP4) than normal fibroblasts [54]. Deletion of Runx2 in mouse primary osteoblasts impaired osteoblastogenesis and the expression of major bone matrix proteins while inhibiting osteoclastogenesis due to the decreased RANKL secretion in osteoblasts [55]. RUNX2 promoted osteoclastogenesis induced by RANKL-stimulated RAW264.7 cells, whereas its mutation led to the loss of osteoclast differentiation and function [56]. Osterix is a vital transcription factor involved in early-phase osteoblastogenesis derived from osteoblast precursors [47,57]. The Osterix zinc finger region interacts with the p53 DNA-binding domain under Osterix and p53 overexpression. However, this interaction was downregulated by p53-mediated Osterix transcriptional activity during osteoblast differentiation [58].
As mentioned above, there are few reports on how p53 directly regulates osteoclastogenesis. p53 deficiency in BMSCs greatly promoted serum OPG production, resulting in increased bone density and trabecular (and not cortical) bone thickness. This was manifested as damaged osteoclast differentiation induced by p53-deficient monocytes, suggesting that p53 negatively regulates osteoclastogenesis [57]. OPG acts as a decoy receptor of RANKL to repress osteoclastogenesis [59]. Previously, we demonstrated that p53 has a positive role in OPG-inhibited osteoclastogenesis in vitro and the inhibition of p53 activity by pifithrin-α (PFT-α) reversed this process [48].
Bone tumor diseases caused by different undefined reasons have an unusual relationship with p53 and osteoclast differentiation. Growth factor independent 1 (GFI1) expression was upregulated in multiple myeloma cells, while silencing GFI1 in p53 wild-type, p53 mutant, and p53-deficient multiple myeloma cells led to apoptosis. Conversely, Gfi1 overexpression in mouse multiple myeloma cells enhanced bone destruction by increasing osteoclast number and size, thereby promoting tumor growth [60]. There was a reciprocal inhibitory effect between translationally controlled tumor protein (TCTP) and p53 [61,62]. TCTP expression was elevated in osteoclastogenesis, while silencing TCTP led to suppressed osteoclast differentiation induced by human bone marrow cells (hBMCs) [63].
2.1. Lysosome-Associated Regulators
Osteoclasts transport different physical substances through membrane-bound intermediaries to resorb the bone matrix. Membrane trafficking involves the transportation of solutes and proteins and is performed by other eukaryotic cell macromolecules (the endocytic, secretory, and transcytotic pathways) [64]. In this process, the secretory lysosome is a specialized lysosome-related organelle in osteoclasts that represents the primary activation and warehouse sites for acid hydrolases, such as cathepsin K (CTSK) [65]. Osteoclastic lysosome-associated proteins participate in the degradation of bone matrix from the ruffled border (RB) in osteoclastic resorptive lacunae, including lysosome-associated membrane protein 1/2 (LAMP1/2) and the small Ras-related GTPase Rab7. In this review, we focus on how osteoclastic functional factors regulate osteoclastogenesis.
A cathepsin family member, CTSK is a lysosomal predominant protease and the only subtype expressed in osteoclasts [66,67]. CTSK is indispensable for bone resorption, resides in the lysosomes and cytoplasmic vesicles of osteoclasts, and is released to the bone matrix surface regulated by an integrin αvβ3-induced sealing zone with bone [67,68]. In mice, Ctsk knockout led to osteopetrosis and development of pycnodysostosis characteristics by decreasing osteoclastic resorptive activity and impairing CTSK secretion, which impeded bone organic matrix degradation and osteoclasts [69]. Deletion of the Ctsk gene in mouse hematopoietic cells or monocyte lineage cells reinforced bone volume, the bone formation rate, and osteoclast and osteoblast numbers [70]. In contrast, Ctsk overexpression recovered osteoclastic bone resorption in UTU17+/+ background mice, which also enhanced osteoblast formation by increasing ALP activity and soluble RANKL (sRANKL) secretion [71]. Trabecular bone volume was reduced in Ctsk-overexpressing transgenic mice, yet it accelerated osteoblast numbers, the bone turnover rate, and the amount of mineralizing surface [72].
CTSK is expressed abundantly in osteoclasts, replacing the other cathepsins to resorb the bone matrix [73,74]. CTSK is secreted from the lysosomes of mature osteoclasts and interacts with the E3 ubiquitin ligase Cb1 (Tyr737) adaptor protein and the PI3K p85 regulatory subunit [75]. A22, a specific potent inhibitor of CTSK, blocked osteoclast activation in vitro and enhanced the spine bone density in zebrafish [76]. In clinical tests, a series of pharmaceutical products were applied to inhibit CTSK activity, such as balicatib (AAE581), ONO-5534, and odanacatib, which were mostly efficacious for inhibiting osteoclast bone resorption and preventing bone loss. Nevertheless, these inhibitors were discontinued due to their adverse effects outside bone in clinical trials [66]. Angiogenesis-coupled osteogenesis was enhanced by treatment with the angiogenic factor platelet-derived growth factor (PDGF)-BB secreted from periosteal OCPs, especially for repairing fractures [77]. These studies suggested CTSK as a potential target for treating impaired osteoclastogenesis associated with bone-related diseases in vivo or in vitro.
LAMP is a crucial lysosomal membrane protein for phagosome fusion with lysosomes, has two isoforms (LAMP1 and LAMP2), and represents approximately 50% of the total amount of lysosomal membrane proteins [78]. LAMP2 is a key regulator of RANKL-induced osteoclastogenesis [78]. In osteoclasts, LAMP2 was enriched at the RB within actin rings while CTSK was transported into the resorptive lacuna with actin rings [79]. LAMP2 colocalized with Rab7 in mouse osteoclasts, but knockdown of Rab7 disrupted osteoclast polarization and the RB vesicles, which impaired bone resorption in vivo and in vitro [80,81]. Rab7 is a secreted lysosomal protein related to late endosomes/lysosomes and phagosomes that is localized to the osteoclast RB [64,82]. Rab7 has two GTP-dependent effectors [Rac1 and pleckstrin homology domain-containing protein family member 1 (PLEKHM1)], which are involved in the adaption of endosomal-lysosomal systems [83]. A Rab7-binding partner, Rac1 belongs to the small GTPase Rho family. Rab7 overexpression upregulated Rac1 activity, but silencing Rab7 inactivated Rac1 [84]. PLEKHM1 is present in osteoclast vesicles, but PLEKHM1 mutation or deletion was associated with the loss of function of osteoclast bone resorption caused by lysosomes and microtubules [79,83,85].
2.2. NFATc1 and c-Fos
NFATc1 is a master regulator responsible for osteoclastogenesis by regulating a series of downstream specific genes, such as that for CTSK, tartrate-resistant acid phosphatase (TRAP), c-Fos, and calcitonin receptor (CT) [86]. In the presence of RANKL, NFATc1-deficient embryonic cells failed to differentiate into osteoclasts, which suggested that NFATc1 is indispensable in osteoclastogenesis [87,88]. Conversely, NFATc1 overexpression reversed osteoclastogenesis in Fos-deficient mice [89]. In BMMs, NFATc1 overexpression induced osteoclastogenesis by enhancing the levels of the vacuolar ATPase V0 domain d2 isoform (Atp6v0d2) and dendritic cell (DC)-specific transmembrane protein (DC-STAMP) in the absence of RANKL and increased TRAP, CTSK, c-Src, and integrin αvβ3 expression [90]. Cyclosporin A (which blocks NFAT transcription) inhibited osteoclastogenesis in the presence of M-CSF and RANKL by downregulating the expression of osteoclastic-specific factors (Atp6v0d2 and DC-STAMP) in vitro [90]. These data suggested that NFATc1 is critical in osteoclastogenesis.
A Fos gene family member, c-Fos is essential in osteoclastogenesis [91]. RANKL binds to its receptor RANK in osteoclast precursors or the osteoclast surface and recruits the tumor necrosis factor receptor-associated factors (TRAFs) to activate downstream signaling [89,92]. TRAF6 binds to RANK to mediate c-Src, Akt, and PI3K activation [93]. Traf6-deficient mice exhibited osteopetrosis caused by reduced osteoclast numbers [94]. Likewise, osteoclast formation in Fos (c-Fos)-deficient mice failed due to the reduced RANK expression in macrophages, which resulted in osteopetrosis [91]. Interestingly, Fos overexpression upregulated RANK expression but RANK overexpression failed to rescue RANKL-induced osteoclastogenesis in Fos-deficient mice [91]. In BMMs, c-Fos overexpression rescued RANKL-stimulated osteoclastogenesis by increasing NFATc1 expression [95].
MicroRNAs (miRNAs) represent post-transcriptional regulators of protein-encoding genes and are important during osteoclastogenesis [96]. RANKL stimulated c-Fos activation by upregulating miR-21 expression during osteoclastogenesis, which resulted in the downregulation of programmed cell death 4 (PDCD4) levels [97]. miR-145 mediated the cooperation between the c-Fos complex and Smad3 in osteoclastic-specific precursors, which reduced phosphorylated Smad2/3 complex formation and suppressed c-Fos and NFATc1 transcription [98]. miR-20a attenuated hypoxia-induced osteoclastogenesis by reducing autophagy-related 16-like 1 (ATG16L1) and microtubule-associated light chain protein 3II (LC3II) expression and the levels of osteoclastic-specific markers (NFATc1, TRAP, TRAF6) [99]. These studies demonstrated that c-Fos regulates transcription factor and signaling pathway activity in RANKL-induced osteoclastogenesis.
2.3. OPG
A soluble receptor for RANKL, OPG inhibits osteoclast differentiation and bone resorption by decreasing osteoclast numbers and RANK signaling in vivo and in vitro [100]. OPG is a transmembrane protein that promotes bone remodeling [11,101]. Opg transgenic mice exhibited severe but non-lethal osteopetrosis via decreased osteoclast number and bone resorption [12]. OPG-deficient mice exhibited alveolar bone loss via reduced sclerostin levels and accelerated bone resorption [102]. Conversely, the high bone density phenotype caused by Opg overexpression in transgenic mice resulted in suppressed osteoclastogenesis [12]. OPG inhibited osteoclast adherent structure differentiation and formation and correspondingly attenuated the expression of RANK, TRAP, integrin β3, matrix metalloproteinase-9 (MMP-9), and carbonic anhydrase I (CAII), which are involved in autophagy [103]. Previously, we demonstrated that OPG inhibited the expression of c-Fos (an AP-1 family member), CTSK, and NFATc1 in a dose-dependent manner in osteoclastogenesis [8]. These data indicated that OPG is a potential target for inhibiting osteoclastogenesis in various regulatory mechanisms (Figure 1).
3. Autophagy Regulates Osteoclastogenesis
Eukaryotic cells feature three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). Macroautophagy (usually termed autophagy) is a conservative survival pathway that can use abandoned intracellular materials for cellular energy production [104]. Recent studies demonstrated that autophagy is an intercellular conservative mechanism for removing misfolded proteins, damaged organelles, and macromolecules [105,106]. During autophagy in eukaryotic cells, a total of 32 ATGs were tightly related to LC3 and the adaptor molecule sequestome 1 (SQSTM1)/p62 [107]. Autophagy is a vital intracellular regulatory mechanism associated with osteoclastogenesis [108,109,110]. Clearly, osteoclast differentiation and osteoclast-mediated bone resorption are highly complex processes in bone remodeling and are involved in regulating the cell signaling pathways, transcription factors, and external environment. The role of AMPK on autophagy in osteoclastogenesis has been described previously [32]. Therefore, in this review, we discuss how partial regulators mediate osteoclastogenesis and are involved in autophagy.
Many studies demonstrated that autophagy regulation of osteoclastogenesis involves multiple cellular signaling pathways, such as that of PI3K–Akt–mTOR, AMPK–mTOR–ribosomal protein S6 kinase beta-1 (S6K1), and p53 [8,47,110,111,112]. In the initial stage of autophagy, autophagy activation and stimulation are caused by signaling factors at amino acid sites, such as the phosphorylation of class III PI3K and Unc-51-like autophagy-activating kinase 1/2 (ULK1/2) [113,114]. Activated PI3K causes Akt phosphorylation at different amino acid residue sites. For example, Akt Ser473 is involved in regulating the mTOR complex and Akt Thr308 is activated partially for full Akt phosphorylation [115]. PI3K is part of the PI3K complex that includes Beclin1, Vps34, UVRAG, and Ambra1 [22].
M-CSF binds to its receptor c-Fms to promote OCP survival and proliferation and phosphorylate four tyrosine residues (Y559, Y697, Y721, Y921), causing its autophosphorylation and transphosphorylation [116,117,118]. c-Fms phosphorylation at Y559 and Y721 interacted with c-Src to form a complex to activate the PI3K–Akt signaling pathway [119]. In primary osteoclasts, excessive dexamethasone induced bone loss caused by PI3K–Akt–mTOR signaling pathway-mediated elevation of autophagy [120]. We previously reported that AMPK–mTOR–S6K1 signaling pathway-mediated autophagy was involved in OPG-inhibited osteoclastogenesis in primary osteoclasts and osteoclast-like cells (OLCs) [8]. These data suggested that the PI3K–Akt signaling pathway has a vital effect on regulating autophagy during osteoclastogenesis.
mTOR contains two distinct complexes: mTORC1 and mTORC2. mTORC1 includes mTOR, regulatory-associated protein of mTOR (Raptor), mammalian lethal with SEC13 protein 8 (mLST8 or G protein beta subunit-like [GβL]), and the non-core components proline-rich Akt substrate of 40 kDa (PRAS40) and DEP domain-containing mTOR-interacting protein (DEPTOR). mTORC2 includes mTOR partner, rapamycin-insensitive companion of mTOR (Rictor), GβL, and mammalian stress-activated protein kinase (SAPK)-interacting protein (mSin1) [121,122]. mTORC1 is a switch for osteoclastogenesis proliferation-to-differentiation, which is a critical role that causes bone loss by increasing osteoclast activity at low-dose rapamycin treatment or inactivating mTORC1 during RANKL-induced osteoclastogenesis via the calcineurin–mTORC1–NFATc1 signaling pathway [123]. Likewise, rapamycin or mTOR small interfering RNA treatment inhibited osteoclast differentiation and bone resorption [16,124]. However, deleting mTOR or Raptor significantly inhibited osteoclastogenesis in BMMs derived from mTOR flox/– and Raptor flox/flox mice in vitro, and in vivo research yielded analogous results [125,126]. Conversely, late Raptor deletion, but not Rictor deletion, in osteoclast precursors enhanced osteoclastogenesis in mice [123,127].
mTOR activity is regulated negatively by upstream tuberous sclerosis complex 1 (TSC1). Deletion of the Tsc1 gene in mouse bone marrow macrophages prevented osteoclast bone resorption and bone loss in vitro and in vivo [127]. TSC2 is associated with TSC1, forming the TSC complex, which negatively regulates mTOR mediated by the small G-protein Ras homologue enriched in brain (Rheb) [128,129]. Subsequently, the mTOR complex is activated by upstream Akt signaling, which causes p70S6K phosphorylation and the suppression of 4E-binding protein 1 (4E-BP1), leading to cell survival, proliferation, and metabolism [130,131]. p70S6K is an mTORC1 activity indicator that correspondingly decreased during osteoclastogenesis [132,133]. These studies demonstrated that mTOR-regulated autophagy is a major regulator in osteoclastogenesis.
Beclin1 is involved in initiating autophagy, which promoted the formation of autolysosomes from double-membrane autophagosomes fusing with lysosomes [134]. Beclin1 is indispensable for initiating autophagy in osteoclastogenesis [108], is pivotal in autophagy, and is enhanced in RANKL-stimulated osteoclastogenesis in vitro. 3-MA inhibition of autophagy suppressed osteoclast formation and resorption, accompanied by decreased Beclin1 and ATG5 expression [8,135]. Mice lacking Beclin1 exhibited increased cortical bone thickness caused by impeded osteoclast function and ATG-mediated autophagy in vivo [136]. Mice with Beclin1 knockout had increased cortical femoral bone mass but reduced trabecular bone quality and mass [108]. Conversely, lentiviral overexpression of Beclin1 stimulated autophagy, which increased osteoclastic bone resorption [137]. Beclin1 overexpression reversed osteoclastogenesis and autophagy deficiency in OCPs incubated with SP600125 (a JNK inhibitor), which resulted in suppressed autophagy and augmented apoptosis [138]. Beclin1 also plays a non-autophagic role and its expression was increased in a p38- and NF-κB-dependent manner during RANKL-induced osteoclastogenesis in vitro [139].
Osteoclastogenesis also requires the involvement of ATGs and autophagy-related proteins in bone resorption, then completes the degradation of old bone matrix with the specific zone. Beclin1, LC3, ATG5, ATG7, and ATG12 expression is upregulated in RANKL-induced osteoclastogenesis [140]. In autophagy, ATGs are essential for the degradation of intracellular useless cargos, and ATG7 aggregates the soluble forms of LC3I into LC3II, complementing normal cellular function [137]. The ATG5–ATG12 complex acts as a part of autophagosomes and prolongs phagocytic ring formation in the cytoplasm. Subsequently, the unnecessary molecules are transported to autolysosomes through ubiquitin p62 binding to LC3 [141]. The deletion of Atg5 or Atg7 suppressed osteoclast differentiation and impaired CTSK secretion respectively, leading to reduced osteoclast bone resorption [142,143]. Knockdown of p62 attenuated the formation of TRAP-positive osteoclasts and the F-actin ring, osteoclast-related gene expression, and LC3 accumulation [140]. p62 mutant mice had increased osteoclast numbers, which led to bone mass loss [144]. Based on these findings, ATGs have a more important regulatory role in osteoclastogenesis involving autophagy and the latter inversely regulates the former.
A negative regulator of bone metabolism, p53 can enhance osteoblast-dependent osteoclastogenesis and osteoclastic bone resorption by increasing M-CSF levels [47]. p53 negatively regulates osteoclastogenesis and is involved in the cell cycle and programed cell death, such as autophagy [48]. p53 activates autophagy, which is part of the p53 protective function, causing cell survival [145]. Activated p53 increased AMPKα phosphorylation by inhibiting mTOR-induced autophagy [146]. Autophagy has positive effects on tumor progression when p53 is activated, whereas autophagy is a part of tumor suppression when p53 function is lost [48,147]. Conversely, autophagy suppresses p53 and is important for tumor promotion. Previously, we demonstrated that p53 is critical during OPG-inhibited osteoclastogenesis by regulating TSC2-induced autophagy in vitro [48].
The lysosomal specific inhibitors chloroquine (CQ) and bafilomycin A1 (BAF1) reduce lysosome acidification by increasing the pH, which leads to mTOR activation and augments p62 levels to inhibit osteoclastogenesis while attenuating fusion and multinucleation [148,149]. mTORC1 activation requires a complex formed by the lysosomal membrane structure vacuolar-type H+-ATPase proton pump (V-ATPase). Correspondingly, BAF1 inhibited lysosomal activity in epiphyseal chondrocytes but potently caused mTORC1 signaling activation and p70S6K phosphorylation [150]. We demonstrated that CQ and BAF1 impaired osteoclast differentiation and function in the presence or absence of OPG [10]. In this process, inhibiting lysosomal function resulted in failure to bind to autophagosomes, but CQ increased autophagosome numbers and size, which promoted AMPK phosphorylation [10]. These data suggested that lysosomal regulators are critical in osteoclastogenesis by regulating autophagy.
4. Inflammation and Immunity Mediate Osteoclastogenesis
Recently, there has been evidence regarding bone cell inflammation and immunity, which includes bone resorption and bone remodeling, especially the interaction between bone marrow immune cells and osteoclasts [151,152,153]. Osteoimmunity is an interdisciplinary concept originated by Arron and Choi on bone-related disease in 2000 [153]. Importantly, the RANKL–RANK–OPG system is a core element in osteoclastogenesis, providing a solid foundation in osteoimmunity [151]. Various immune regulatory cells (T and B lymphocytes, macrophages, DCs) were implicated in osteoclast-induced bone resorption [154]. Immune cells release proinflammatory cytokines, such as interleukin-1β (IL-1β), IL-6, tumor necrosis factor-α (TNF-α), and IL-11 and/or also secrete anti-inflammatory cytokines, such as IL-4, IL-10, and interferon-β (IFN-β), which are associated with the skeletal system. For example, IL-1 derived from the immune cells induced osteoclastogenesis [155] and TNF-α promoted osteoclastogenesis and bone resorption by activating autophagy in patients with rheumatoid arthritis in vivo and in vitro [156]. Osteoclastogenesis is related to the regulation of autophagy, inflammation, and immunity and is extremely complex (Figure 2).
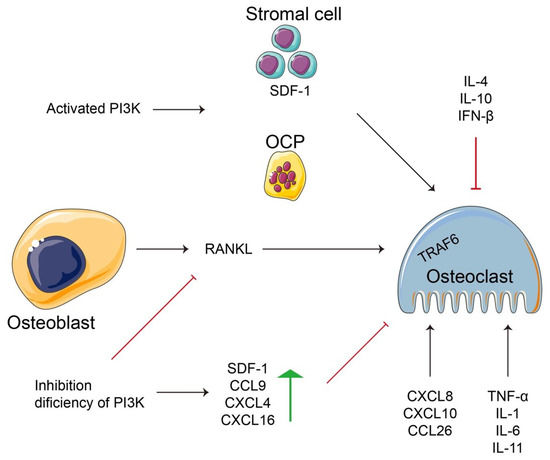
Figure 2.
A schematic diagram of the relationship between inflammation and immunity for regulating osteoclastogenesis or osteoblast-dependent osteoclastogenesis. PI3K regulates the cell types (stromal cells, OCPs, osteoblasts, osteoclasts) to control cellular events, such as the release of anti- and/or pro-inflammation factors. Additionally, a few inflammatory factors not only implement their basic function according to their own definition, but are also involved in regulating various cells, such as osteoclastogenesis, or fused from OCPs.
The PI3K–Akt signaling pathway is a critical promotor of cellular proliferation, differentiation, and metabolism [157,158]. The pathway regulates osteoclastogenesis through various regulatory mechanisms as described in Section 2.1 and Section 2.2. Inhibition of the PI3K–Akt signaling pathway attenuated RANKL expression during osteoclastogenesis [159]. However, the activation of PI3K enhanced stromal cell-derived factor 1 (SDF-1 or CXC ligand 12 [CXCL12]) production and osteoclast precursor numbers in mouse bone marrow cells [160]. Osteoclast-mediated bone resorption is a vital process against excessive bone formation to maintain appropriate calcium levels in the blood [101]. Under pathological conditions, such as inflammatory conditions or estrogen deficiency, accelerated bone resorption results in osteolysis [161]. However, PI3Kγ-deficient mice had decreased osteoclast numbers following impaired osteoclast formation in vivo and in vitro, and increased levels of inflammatory chemokines (SDF-1, chemokine ligand 9 [CCL9], CXCL4, CXCL16) [20]. Chemokines are secreted dynamic molecules that are important regulators in osteoclastogenesis [162]. Recent studies have reported that CXCL8, CXCL10, and CCL20 contributed to osteoclastogenesis in response to changes in inflammatory molecules [163]. Moreover, CCL4 regulates osteoclast motility by promoting PI3K activity [162,164]. Therefore, the PI3K–Akt signaling pathway positively regulates immunity and inflammation in osteoclastogenesis.
The proinflammatory factors TNF-α, IL-1, and IL-6 have positive regulatory roles in response to autophagy in osteoclastogenesis [32,165,166]. In mice, TNF-α knockout inhibited osteoclast differentiation and function accompanied by suppressed autophagy [137]. In osteoclastogenesis, TNF-α induced transcriptional repressor B lymphocyte-induced maturation protein-1 (Blimp1) expression. Silencing Blimp1 in OCPs markedly attenuated TNF-α production in osteoclast differentiation [167]. Furthermore, TNF-α cooperated with IL-1 to activate functional osteoclast differentiation [168]. IL-1 induces osteoclast differentiation in the presence of RANKL through JNK activation, which is downstream of TRAF6 and has additive effects [169,170]. IL-1β promotes RANKL production and IL-6 synthesis from osteoblasts or osteocytes to further stimulate osteoclastogenesis [171,172]. Conversely, IL-4 stimulated OPG production by regulating the RANKL–RANK–OPG axis and inhibited RANKL-induced osteoclastogenesis in BMMs and RAW264.7 cells [173]. IL-4 plays a negative role in osteoclastogenesis and functions by inhibiting NF-κB signaling and NFATc1 expression [174,175]. In bone metastasis of colorectal cancer, IL-4 promoted the proliferation of early OCPs by binding its receptor IL-4α, while IL-4 deficiency impaired bone resorption [176]. In TNF-α-activated stromal cells, IL-4 suppressed TNF-α-mediated osteoclast formation by inhibiting RANKL expression and directly inhibited TNF-α-activated osteoclast precursors in vivo via a T cell-independent mechanism [177]. IL-4 also acted with IL-10 to negatively regulate osteoclastogenesis by reducing NFATc1 expression, and IL-10 deficiency promoted bone loss in mice [178]. Therefore, inflammation may be required for osteoclast formation and function and can be modulated by different factors, such as chemokines, cytokines, and hormones.
The immune cell and the bone cell are tightly linked with osteoclast differentiation and function. Among them, the interaction between immune cells and osteoclasts in the bone marrow or joint cavity is the basis of osteoimmunity [151]. For example, activated T lymphocytes promotes osteoclastogenesis via producing RANKL [179]. However, overexpression of RANKL in T lymphocytes of RANKL-deficient mice can restore osteoclastsogenesis [151,180]. In addition to RANKL, the type 17 helper T (Th17) cell-derived IL-17 has long been recognized as a crucial pre-inflammatory cytokine that facilitate osteoclastogenesis [155,181,182]. IL-17 is a complex of inflammatory cytokines indirectly involved in osteoblast-induced osteoclastogenesis, and binding of the IL-17 receptor complex augmented bone mass through conditional deletion of IL-17a receptor (IL-17ra) in osteoclast precursors [183]. Similarly, low IL-17 concentrations increased RANKL-induced osteoclastogenesis via Beclin1–TAK1-binding protein 3 (TAB3)–extracellular signal-regulated kinase (ERK) pathway-mediated autophagy, but high IL-17 levels had the opposite effect [184]. In addition, monocytes or tissue-specific macrophages (macrophages resident in tissues) are a major bone cells, which is origin of osteoclasts in inflammatory and immune environment [151]. Macrophages represent a type of myeloid monocyte with phagocytic properties, and they are mainly involved in the inflammatory response and immune response to keep dynamic balance [185]. Osteoclastogenesis fused with osteoclast precursors, which are derived from monocyte-macrophages [186]. Several identified genes are directly involved in macrophages membrane fusion, including CD9, CD81, CD44, and CD47 [187,188,189]. CD9 and CD81 negatively regulate macrophages fusion into osteoclast differentiation, while CD44 promotes the osteoclastogenesis by the of NF-kB/NFATc1 signaling pathway, and CD47 also stimulates the fusion of osteoclast precursors [189,190]. In summary, immune cells and bone cells cooperate to execute the effect of osteo-related immunity, which is shared by a common microenvironment and regulatory molecules.
5. Conclusions and Perspectives
Osteoclastogenesis is an extremely complicated regulatory process that includes fusion between OCPs and cytokine-induced differentiation and is controlled by other regulatory factors. These actions involve two essential cytokines (M-CSF and RANKL) to control osteoclastic bone resorption. More evidence has demonstrated that osteoclastogenesis originated from the diversification of cell species, such as monocytes/macrophages and DCs. In vitro studies have the advantage of being able to focus on the regulatory mechanisms of osteoclast formation and function, but they are also subject to more restrictions. First, multiple regulators and cascade signaling pathways control osteoclastogenesis, revealing the intricacy of differentiation and function in vivo. Second, osteoclasts are responsible for bone resorption and are involved in different intracellular metabolisms, including autophagy, apoptosis, inflammation, and immunity. Finally, a certain regulator of osteoclastogenesis may participate in ≥1 regulatory modes or courses to perform functions other than its own. This may make it easier to control osteoclastogenesis equilibrium by either internal regulation or external stimulation.
Inflammation and immunity are intricately linked to bone-related diseases and interact and influence each other. The deepening and richness of bone mechanism research demonstrates not only the involvement of blood immunity mediated by inflammatory cytokines, but also presents the considerable challenge of the different bone cell regulatory contents. For example, recent studies concentrated on the positive role of inflammatory cytokines on osteocytes, including osteoblasts, osteoclasts, OCPs, and DCs, but might have neglected the effects of negative regulators. Nevertheless, we have only described the role of a few cytokines in osteoclastogenesis stimulated by different cells, such as OLPs or other cells with which they interact.
Lastly, endogenous hormones exert a critical effect on osteoclastogenesis by controlling inflammation and immunity and other factors, such as PGE2, parathyroid hormone, progesterone, estrogen, and vitamin D. These regulators feature large and convoluted mechanisms of action in the animal body, including bone. In particular, the bone marrow contains abundant cells and interacts with the blood, which acts on the tissues and organs and is susceptible to environmental or drug influences.
Author Contributions
Conceptualization: X.T. and G.Y.; Writing—original draft preparation: X.T., G.Y. and X.F.; Writing—review and editing: R.S., J.G. and Z.L.; Supervision: J.G. and Z.L.; Funding acquisition: X.T., J.G. and Z.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China grant numbers 32102732, 31872533 and 31872534, Natural Science Foundation of Jiangsu Province grant number BK20210806, the Open Project Program of the Joint International Research Laboratory of Agriculture and Agri-Product Safety, the Ministry of Education of China, Yangzhou University (JILAR-KF202110), and was funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jilka, R.L.; O’Brien, C.A. The Role of Osteocytes in Age-Related Bone Loss. Curr. Osteoporos. Rep. 2016, 14, 16–25. [Google Scholar] [CrossRef]
- Kim, J.-M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.-H. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells 2020, 9, 2073. [Google Scholar] [CrossRef] [PubMed]
- Llorente, I.; García-Castañeda, N.; Valero, C.; González-Álvaro, I.; Castañeda, S. Osteoporosis in Rheumatoid Arthritis: Dangerous Liaisons. Front. Med. 2020, 7, 601618. [Google Scholar] [CrossRef] [PubMed]
- Adami, G.; Saag, K.G. Osteoporosis Pathophysiology, Epidemiology, and Screening in Rheumatoid Arthritis. Curr. Rheumatol. Rep. 2019, 21, 34. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Chen, X.; Gao, S.; Yu, X.; Xiao, J.; Zhang, B.; Liu, X.; Dai, M. Key Triggers of Osteoclast-Related Diseases and Available Strategies for Targeted Therapies: A Review. Front. Med. 2017, 4, 234. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Takayanagi, H. Osteoclasts, rheumatoid arthritis, and osteoimmunology. Curr. Opin. Rheumatol. 2006, 18, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Hayashi, S.I.; Kunisada, T.; Ogawa, M.; Nishikawa, S.; Okamura, H.; Sudo, T.; Shultz, L.D.; Nishikawa, S.I. The murine mutation osteopetrosis is in the coding region of the macrophage colony stimulating factor gene. Nature 1990, 345, 442–444. [Google Scholar] [CrossRef]
- Tong, X.; Zhang, C.; Wang, D.; Song, R.; Ma, Y.; Cao, Y.; Zhao, H.; Bian, J.; Gu, J.; Liu, Z. Suppression of AMP-activated protein kinase reverses osteoprotegerin-induced inhibition of osteoclast differentiation by reducing autophagy. Cell Prolif. 2020, 53, e12714. [Google Scholar] [CrossRef]
- Boyce, B.F.; Xing, L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch. Biochem. Biophys. 2008, 473, 139–146. [Google Scholar] [CrossRef]
- Tong, X.S.; Gu, J.H.; Song, R.L.; Wang, D.; Sun, Z.Q.; Sui, C.; Zhang, C.; Liu, X.Z.; Bian, J.C.; Liu, Z.P. Osteoprotegerin inhibit osteoclast differentiation and bone resorption by enhancing autophagy via AMPK/mTOR/p70S6K signaling pathway in vitro. J. Cell. Biochem. 2018, 120, 1630–1642. [Google Scholar] [CrossRef]
- Udagawa, N.; Takahashi, N.; Yasuda, H.; Mizuno, A.; Itoh, K.; Ueno, Y.; Shinki, T.; Gillespie, M.T.; Martin, T.J.; Higashio, K.; et al. Osteoprotegerin produced by osteoblasts is an important regulator in osteoclast development and function. Endocrinology 2000, 141, 3478–3484. [Google Scholar] [CrossRef]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Lüthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Liu, R.; Jin, C.; Wang, Z.; Wang, Z.; Wang, J.; Wang, L. Effects of manganese deficiency on the microstructure of proximal tibia and OPG/RANKL gene expression in chicks. Vet. Res. Commun. 2015, 39, 31–37. [Google Scholar] [CrossRef]
- Cappariello, A.; Maurizi, A.; Veeriah, V.; Teti, A. The Great Beauty of the osteoclast. Arch. Biochem. Biophys. 2014, 558, 70–78. [Google Scholar] [CrossRef]
- Ono, T.; Nakashima, T. Recent advances in osteoclast biology. Histochem. Cell. Biol. 2018, 149, 325–341. [Google Scholar] [CrossRef]
- Glantschnig, H.; Fisher, J.E.; Wesolowski, G.; Rodan, G.A.; Reszka, A.A. M-CSF, TNFalpha and RANK ligand promote osteoclast survival by signaling through mTOR/S6 kinase. Cell. Death. Differ. 2003, 10, 1165–1177. [Google Scholar] [CrossRef]
- Chen, J.; He, J.Q.; Zhen, S.Y.; Huang, L.Q. OPG inhibits gene expression of RANK and CAII in mouse osteoclast-like cell. Rheumatol. Int. 2012, 32, 3993–3998. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Shinohara, M.; Nakamura, M.; Masuda, H.; Hirose, J.; Kadono, Y.; Iwasawa, M.; Nagase, Y.; Ueki, K.; Kadowaki, T.; Sasaki, T.; et al. Class IA phosphatidylinositol 3-kinase regulates osteoclastic bone resorption through protein kinase B-mediated vesicle transport. J. Bone Miner. Res. 2012, 27, 2464–2475. [Google Scholar] [CrossRef]
- Kang, H.; Chang, W.; Hurley, M.; Vignery, A.; Wu, D. Important roles of PI3Kgamma in osteoclastogenesis and bone homeostasis. Proc. Natl. Acad. Sci. USA 2010, 107, 12901–12906. [Google Scholar] [CrossRef]
- Ball, J.; Archer, S.; Ward, S. PI3K inhibitors as potential therapeutics for autoimmune disease. Drug Discov. Today 2014, 19, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Baehrecke, E.H. Eaten alive: Novel insights into autophagy from multicellular model systems. Trends Cell Biol. 2015, 25, 376–387. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol. 2010, 11, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.B.; Kim, J.H.; Kim, K.; Youn, B.U.; Ko, A.; Lee, S.Y.; Kim, N. Akt Induces Osteoclast Differentiation through Regulating the GSK3β/NFATc1 Signaling Cascade. J. Immunol. 2012, 188, 163. [Google Scholar] [CrossRef]
- Chen, X.; Chen, W.; Aung, Z.M.; Han, W.; Zhang, Y.; Chai, G. LY3023414 inhibits both osteogenesis and osteoclastogenesis through the PI3K/Akt/GSK3 signalling pathway. Bone Jt. Res. 2021, 10, 237–249. [Google Scholar] [CrossRef]
- Cohen, M.M., Jr. The AKT genes and their roles in various disorders. Am. J. Med. Genet. Part A 2013, 161, 2931–2937. [Google Scholar] [CrossRef]
- Kawamura, N.; Kugimiya, F.; Oshima, Y.; Ohba, S.; Ikeda, T.; Saito, T.; Shinoda, Y.; Kawasaki, Y.; Ogata, N.; Hoshi, K.; et al. Akt1 in osteoblasts and osteoclasts controls bone remodeling. PLoS ONE 2007, 2, e1058. [Google Scholar] [CrossRef]
- Hanada, M.; Feng, J.; Hemmings, B.A. Structure, regulation and function of PKB/AKT-a major therapeutic target. BBA-Proteins Proteom. 2004, 1697, 3–16. [Google Scholar] [CrossRef]
- Peng, X.D.; Xu, P.Z.; Chen, M.L.; Hahn-Windgassen, A.; Skeen, J.; Jacobs, J.; Sundararajan, D.; Chen, W.S.; Crawford, S.E.; Coleman, K.G.; et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003, 17, 1352–1365. [Google Scholar] [CrossRef]
- Phung, T.L.; Du, W.; Xue, Q.; Ayyaswamy, S.; Gerald, D.; Antonello, Z.; Nhek, S.; Perruzzi, C.A.; Acevedo, I.; Ramanna-Valmiki, R.; et al. Akt1 and akt3 exert opposing roles in the regulation of vascular tumor growth. Cancer Res. 2015, 75, 40–50. [Google Scholar] [CrossRef]
- Ding, L.; Biswas, S.; Morton, R.E.; Smith, J.D.; Hay, N.; Byzova, T.V.; Febbraio, M.; Podrez, E.A. Akt3 deficiency in macrophages promotes foam cell formation and atherosclerosis in mice. Cell Metab. 2012, 15, 861–872. [Google Scholar] [CrossRef]
- Tong, X.; Ganta, R.R.; Liu, Z. AMP-activated protein kinase (AMPK) regulates autophagy, inflammation and immunity and contributes to osteoclast differentiation and functionabs. Biol. Cell 2020, 112, 251–264. [Google Scholar] [CrossRef]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrin. Met. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Kim, M.; Shen, M.; Ngoy, S.; Karamanlidis, G.; Liao, R.; Tian, R. AMPK isoform expression in the normal and failing hearts. J. Mol. Cell. Cardiol. 2012, 52, 1066–1073. [Google Scholar] [CrossRef]
- Qi, D.; Young, L.H. AMPK: Energy sensor and survival mechanism in the ischemic heart. Trends Endocrinol. Metab. 2015, 26, 422–429. [Google Scholar] [CrossRef]
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem. J. 2016, 473, 189–199. [Google Scholar] [CrossRef]
- Kang, N.; Kim, K.W.; Shin, D.M. Humanin suppresses receptor activator of nuclear factor-κB ligand-induced osteoclast differentiation via AMP-activated protein kinase activation. Korean J. Physiol. Pharmacol. 2019, 23, 411–417. [Google Scholar] [CrossRef]
- Oh, S.J.; Gu, D.R.; Jin, S.H.; Park, K.H.; Lee, S.H. Cytosolic malate dehydrogenase regulates RANKL-mediated osteoclastogenesis via AMPK/c-Fos/NFATc1 signaling. Biochem. Biophys. Res. Commun. 2016, 475, 125–132. [Google Scholar] [CrossRef]
- Wang, Y.G.; Han, X.G.; Yang, Y.; Qiao, H.; Dai, K.R.; Fan, Q.M.; Tang, T.T. Functional differences between AMPK α1 and α2 subunits in osteogenesis, osteoblast-associated induction of osteoclastogenesis, and adipogenesis. Sci. Rep. 2016, 6, 32771. [Google Scholar] [CrossRef]
- Mai, Q.G.; Zhang, Z.M.; Xu, S.; Lu, M.; Zhou, R.P.; Zhao, L.; Jia, C.H.; Wen, Z.H.; Jin, D.D.; Bai, X.C. Metformin stimulates osteoprotegerin and reduces RANKL expression in osteoblasts and ovariectomized rats. J. Cell. Biochem. 2011, 112, 2902–2909. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, Y.S.; Lee, S.Y.; Kim, G.H.; Kim, B.J.; Lee, S.H.; Lee, K.U.; Kim, G.S.; Kim, S.W.; Koh, J.M. AMP kinase acts as a negative regulator of RANKL in the differentiation of osteoclasts. Bone 2010, 47, 926–937. [Google Scholar] [CrossRef]
- Kang, H.; Viollet, B.; Wu, D. Genetic deletion of catalytic subunits of AMP-activated protein kinase increases osteoclasts and reduces bone mass in young adult mice. J. Biol. Chem. 2013, 288, 12187–12196. [Google Scholar] [CrossRef] [PubMed]
- Kainuma, S.; Otsuka, T.; Kuroyanagi, G.; Yamamoto, N.; Matsushima-Nishiwaki, R.; Kozawa, O.; Tokuda, H. Regulation by AMP-activated protein kinase of PGE2-induced osteoprotegerin synthesis in osteoblasts. Mol. Med. Rep. 2016, 13, 3363–3369. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Quinn, J.M.; Tam, S.; Sims, N.A.; Saleh, H.; McGregor, N.E.; Poulton, I.J.; Scott, J.W.; Gillespie, M.T.; Kemp, B.E.; van Denderen, B.J. Germline deletion of AMP-activated protein kinase β subunits reduces bone mass without altering osteoclast differentiation or function. FASEB J. 2010, 24, 275–285. [Google Scholar] [CrossRef]
- Fong, J.E.; Le Nihouannen, D.; Tiedemann, K.; Sadvakassova, G.; Barralet, J.E.; Komarova, S.V. Moderate excess of pyruvate augments osteoclastogenesis. Biol. Open 2013, 2, 387–395. [Google Scholar] [CrossRef]
- Shah, M.; Kola, B.; Bataveljic, A.; Arnett, T.R.; Viollet, B.; Saxon, L.; Korbonits, M.; Chenu, C. AMP-activated protein kinase (AMPK) activation regulates in vitro bone formation and bone mass. Bone 2010, 47, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Kua, H.Y.; Hu, Y.; Guo, K.; Zeng, Q.; Wu, Q.; Ng, H.H.; Karsenty, G.; de Crombrugghe, B.; Yeh, J.; et al. P53 functions as a negative regulator of osteoblastogenesis, osteoblast-dependent osteoclastogenesis, and bone remodeling. J. Cell Biol. 2006, 172, 115–125. [Google Scholar] [CrossRef]
- Tong, X.; Gu, J.; Chen, M.; Wang, T.; Zou, H.; Song, R.; Zhao, H.; Bian, J.; Liu, Z. p53 positively regulates osteoprotegerin-mediated inhibition of osteoclastogenesis by downregulating TSC2-induced autophagy in vitro. Differentiation 2020, 114, 58–66. [Google Scholar] [CrossRef]
- Minami, A.; Ogino, M.; Nakano, N.; Ichimura, M.; Nakanishi, A.; Murai, T.; Kitagishi, Y.; Matsuda, S. Roles of oncogenes and tumor-suppressor genes in osteoclastogenesis (Review). Int. J. Mol. Med. 2017, 39, 261–267. [Google Scholar] [CrossRef]
- Rauch, D.A.; Hurchla, M.A.; Harding, J.C.; Deng, H.; Shea, L.K.; Eagleton, M.C.; Niewiesk, S.; Lairmore, M.D.; Piwnica-Worms, D.; Rosol, T.J.; et al. The ARF tumor suppressor regulates bone remodeling and osteosarcoma development in mice. PLoS ONE 2010, 5, e15755. [Google Scholar]
- Winkeler, C. Role of the ARF Tumor Suppressor in Osteoclasts; Washington University in St. Louis: St. Louis, MO, USA, 2011; p. 667. [Google Scholar]
- Jacques, C.; Tesfaye, R.; Lavaud, M.; Georges, S.; Baud’huin, M.; Lamoureux, F.; Ory, B. Implication of the p53-Related miR-34c, -125b, and -203 in the Osteoblastic Differentiation and the Malignant Transformation of Bone Sarcomas. Cells 2020, 9, 810. [Google Scholar] [CrossRef]
- Xu, J.; Li, Z.; Hou, Y.; Fang, W. Potential mechanisms underlying the Runx2 induced osteogenesis of bone marrow mesenchymal stem cells. Am. J. Transl. Res. 2015, 7, 2527–2535. [Google Scholar]
- Molchadsky, A.; Shats, I.; Goldfinger, N.; Pevsner-Fischer, M.; Olson, M.; Rinon, A.; Tzahor, E.; Lozano, G.; Zipori, D.; Sarig, R.; et al. P53 plays a role in mesenchymal differentiation programs, in a cell fate dependent manner. PLoS ONE 2008, 3, e3707. [Google Scholar] [CrossRef]
- Qin, X.; Jiang, Q.; Komori, H.; Sakane, C.; Fukuyama, R.; Matsuo, Y.; Ito, K.; Miyazaki, T.; Komori, T. Runt-related transcription factor-2 (Runx2) is required for bone matrix protein gene expression in committed osteoblasts in mice. J. Bone Miner. Res. 2021, 36, 2081–2095. [Google Scholar] [CrossRef]
- Xin, Y.; Liu, Y.; Liu, D.; Li, J.; Zhang, C.; Wang, Y.; Zheng, S. New Function of RUNX2 in Regulating Osteoclast Differentiation via the AKT/NFATc1/CTSK Axis. Calcif. Tissue Int. 2020, 106, 553–566. [Google Scholar] [CrossRef]
- Velletri, T.; Huang, Y.; Wang, Y.; Li, Q.; Hu, M.; Xie, N.; Yang, Q.; Chen, X.; Chen, Q.; Shou, P.; et al. Loss of p53 in mesenchymal stem cells promotes alteration of bone remodeling through negative regulation of osteoprotegerin. Cell Death Differ. 2021, 28, 156–169. [Google Scholar] [CrossRef]
- Artigas, N.; Gámez, B.; Cubillos-Rojas, M.; Sánchez-de Diego, C.; Valer, J.A.; Pons, G.; Rosa, J.L.; Ventura, F. P53 inhibits SP7/Osterix activity in the transcriptional program of osteoblast differentiation. Cell Death Differ. 2017, 24, 2022–2031. [Google Scholar] [CrossRef]
- Kearns, A.E.; Khosla, S.; Kostenuik, P.J. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr. Rev. 2008, 29, 155–192. [Google Scholar] [CrossRef]
- Petrusca, D.N.; Toscani, D.; Wang, F.-M.; Park, C.; Crean, C.D.; Anderson, J.L.; Marino, S.; Mohammad, K.S.; Zhou, D.; Silbermann, R.; et al. Growth factor independence 1 expression in myeloma cells enhances their growth, survival, and osteoclastogenesis. J. Hematol. Oncol. 2018, 11, 123. [Google Scholar] [CrossRef]
- Kim, E.M.; Jung, C.-H.; Kim, J.; Hwang, S.-G.; Park, J.K.; Um, H.-D. The p53/p21 Complex Regulates Cancer Cell Invasion and Apoptosis by Targeting Bcl-2 Family Proteins. Cancer Res. 2017, 77, 3092. [Google Scholar] [CrossRef]
- Amson, R.; Pece, S.; Lespagnol, A.; Vyas, R.; Mazzarol, G.; Tosoni, D.; Colaluca, I.; Viale, G.; Rodrigues-Ferreira, S.; Wynendaele, J.; et al. Reciprocal repression between P53 and TCTP. Nat. Med. 2011, 18, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-W.; Shin, H.K.; Yang-Yen, H.-F.; Lee, M.S.; Lee, C.H.; Park, S.-J.; Kim, K.-J.; Lee, K.; Kim, S.H. Osteoclastogenic activity of translationally-controlled tumor protein (TCTP) with reciprocal repression of p21. FEBS Lett. 2014, 588, 4026–4031. [Google Scholar] [CrossRef]
- Ng, P.Y.; Brigitte Patricia Ribet, A.; Pavlos, N.J. Membrane trafficking in osteoclasts and implications for osteoporosis. Biochem. Soc. Trans. 2019, 47, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Hackmann, Y.; Dieckmann, N.M.; Griffiths, G.M. The biogenesis of lysosomes and lysosome-related organelles. Cold Spring Harb. Perspect. Biol. 2014, 6, a016840. [Google Scholar] [CrossRef] [PubMed]
- Dai, R.; Wu, Z.; Chu, H.Y.; Lu, J.; Lyu, A.; Liu, J.; Zhang, G. Cathepsin K: The Action in and Beyond Bone. Front. Cell Dev. Biol. 2020, 8, 433. [Google Scholar] [CrossRef]
- Drake, M.T.; Clarke, B.L.; Oursler, M.J.; Khosla, S. Cathepsin K Inhibitors for Osteoporosis: Biology, Potential Clinical Utility, and Lessons Learned. Endocr. Rev. 2017, 38, 325–350. [Google Scholar] [CrossRef]
- Gu, J.H.; Tong, X.S.; Chen, G.H.; Wang, D.; Chen, Y.; Yuan, Y.; Liu, X.Z.; Bian, J.C.; Liu, Z.P. Effects of 1alpha, 25-(OH)2D3 on the formation and activity of osteoclasts in RAW264.7 cells. J. Steroid. Biochem. 2015, 152, 25–33. [Google Scholar] [CrossRef]
- Troen, B.R. The role of cathepsin K in normal bone resorption. Drug News Perspect. 2004, 17, 19–28. [Google Scholar] [CrossRef]
- Lotinun, S.; Kiviranta, R.; Matsubara, T.; Alzate, J.A.; Neff, L.; Luth, A.; Koskivirta, I.; Kleuser, B.; Vacher, J.; Vuorio, E.; et al. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J. Clin. Investig. 2013, 123, 666–681. [Google Scholar] [CrossRef]
- Morko, J.; Kiviranta, R.; Mulari, M.T.; Ivaska, K.K.; Vaananen, H.K.; Vuorio, E.; Laitala-Leinonen, T. Overexpression of cathepsin K accelerates the resorption cycle and osteoblast differentiation in vitro. Bone 2009, 44, 717–728. [Google Scholar] [CrossRef]
- Kiviranta, R.; Morko, J.; Uusitalo, H.; Aro, H.T.; Vuorio, E.; Rantakokko, J. Accelerated turnover of metaphyseal trabecular bone in mice overexpressing cathepsin K. J. Bone Miner. Res. 2001, 16, 1444–1452. [Google Scholar] [CrossRef]
- Costa, A.G.; Cusano, N.E.; Silva, B.C.; Cremers, S.; Bilezikian, J.P. Cathepsin K: Its skeletal actions and role as a therapeutic target in osteoporosis. Nat. Rev. Rheumatol. 2011, 7, 447–456. [Google Scholar] [CrossRef]
- Drake, F.H.; Dodds, R.A.; James, I.E.; Connor, J.R.; Debouck, C.; Richardson, S.; Lee-Rykaczewski, E.; Coleman, L.; Rieman, D.; Barthlow, R.; et al. Cathepsin K, but not cathepsins B, L, or S, is abundantly expressed in human osteoclasts. J. Biol. Chem. 1996, 271, 12511–12516. [Google Scholar] [CrossRef]
- Yu, J.; Adapala, N.S.; Doherty, L.; Sanjay, A. Cbl-PI3K interaction regulates Cathepsin K secretion in osteoclasts. Bone 2019, 127, 376–385. [Google Scholar]
- Xue, S.-T.; Wang, Y.-l.; Han, X.-W.; Yi, H.; Jiang, W.; Si, S.-Y.; Guo, H.-F.; Li, Z.-R. Novel cathepsin K inhibitors block osteoclasts in vitro and increase spinal bone density in zebrafish. RSC Adv. 2019, 9, 8600–8607. [Google Scholar]
- Walia, B.; Lingenheld, E.; Duong, L.; Sanjay, A.; Drissi, H. A novel role for cathepsin K in periosteal osteoclast precursors during fracture repair. Ann. N. Y. Acad. Sci. 2018, 1415, 57–68. [Google Scholar] [CrossRef]
- Jansen, I.D.C.; Tigchelaar-Gutter, W.; Hogervorst, J.M.A.; de Vries, T.J.; Saftig, P.; Everts, V. LAMP-2 Is Involved in Surface Expression of RANKL of Osteoblasts In Vitro. Int. J. Mol. Sci. 2020, 21, 6110. [Google Scholar] [CrossRef]
- Fujiwara, T.; Ye, S.; Castro-Gomes, T.; Winchell, C.G.; Andrews, N.W.; Voth, D.E.; Varughese, K.I.; Mackintosh, S.G.; Feng, Y.; Pavlos, N.; et al. PLEKHM1/DEF8/RAB7 complex regulates lysosome positioning and bone homeostasis. JCI Insight 2016, 1, e86330. [Google Scholar] [CrossRef]
- Shimada-Sugawara, M.; Sakai, E.; Okamoto, K.; Fukuda, M.; Izumi, T.; Yoshida, N.; Tsukuba, T. Rab27A Regulates Transport of Cell Surface Receptors Modulating Multinucleation and Lysosome-Related Organelles in Osteoclasts. Sci. Rep. 2015, 5, 9620. [Google Scholar] [CrossRef]
- Zhao, H.; Laitala-Leinonen, T.; Parikka, V.; Vaananen, H.K. Downregulation of small GTPase Rab7 impairs osteoclast polarization and bone resorption. J. Biol. Chem. 2001, 276, 39295–39302. [Google Scholar] [CrossRef]
- Guerra, F.; Bucci, C. Multiple Roles of the Small GTPase Rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Roux, S. Rab GTPases in Osteoclastic Bone Resorption and Autophagy. Int. J. Mol. Sci. 2020, 21, 7655. [Google Scholar] [CrossRef] [PubMed]
- Pan, Z.-N.; Pan, M.-H.; Sun, M.-H.; Li, X.-H.; Zhang, Y.; Sun, S.-C. RAB7 GTPase regulates actin dynamics for DRP1-mediated mitochondria function and spindle migration in mouse oocyte meiosis. FASEB J. 2020, 34, 9615–9627. [Google Scholar] [CrossRef] [PubMed]
- Van Wesenbeeck, L.; Odgren, P.R.; Coxon, F.P.; Frattini, A.; Moens, P.; Perdu, B.; MacKay, C.A.; Van Hul, E.; Timmermans, J.-P.; Vanhoenacker, F.; et al. Involvement of PLEKHM1 in osteoclastic vesicular transport and osteopetrosis in incisors absent rats and humans. J. Clin. Investig. 2007, 117, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kim, N. Regulation of NFATc1 in Osteoclast Differentiation. J. Bone Metab. 2014, 21, 233–241. [Google Scholar] [CrossRef]
- Asagiri, M.; Takayanagi, H. The molecular understanding of osteoclast differentiation. Bone 2007, 40, 251–264. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Boyce, B.F.; Yamashita, T.; Yao, Z.; Zhang, Q.; Li, F.; Xing, L. Roles for NF-kappaB and c-Fos in osteoclasts. J. Bone Miner. Metab. 2005, 23, 11–15. [Google Scholar] [CrossRef]
- Kim, K.; Lee, S.H.; Ha Kim, J.; Choi, Y.; Kim, N. NFATc1 induces osteoclast fusion via up-regulation of Atp6v0d2 and the dendritic cell-specific transmembrane protein (DC-STAMP). Mol. Endocrinol. 2008, 22, 176–185. [Google Scholar]
- Arai, A.; Mizoguchi, T.; Harada, S.; Kobayashi, Y.; Nakamichi, Y.; Yasuda, H.; Penninger, J.M.; Yamada, K.; Udagawa, N.; Takahashi, N. Fos plays an essential role in the upregulation of RANK expression in osteoclast precursors within the bone microenvironment. J. Cell Sci. 2012, 125 Pt 12, 2910–2917. [Google Scholar]
- Xu, S.; Li, S.; Liu, X.; Tan, K.; Zhang, J.; Li, K.; Bai, X.; Zhang, Y. Rictor Is a Novel Regulator of TRAF6/TRAF3 in Osteoclasts. J. Bone Miner. Res. 2021, 36, 2053–2064. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.R.; Besser, D.; Kim, N.; Arron, J.R.; Vologodskaia, M.; Hanafusa, H.; Choi, Y. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-Src. Mol. Cell 1999, 4, 1041–1049. [Google Scholar] [CrossRef]
- Armstrong, A.P.; Tometsko, M.E.; Glaccum, M.; Sutherland, C.L.; Cosman, D.; Dougall, W.C. A RANK/TRAF6-dependent signal transduction pathway is essential for osteoclast cytoskeletal organization and resorptive function. J. Biol. Chem. 2002, 277, 44347–44356. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chang, E.J.; Ryu, J.; Lee, Z.H.; Lee, Y.; Kim, H.H. Induction of c-Fos and NFATc1 during RANKL-stimulated osteoclast differentiation is mediated by the p38 signaling pathway. Biochem. Biophys. Res. Commun. 2006, 351, 99–105. [Google Scholar] [CrossRef]
- Lozano, C.; Duroux-Richard, I.; Firat, H.; Schordan, E.; Apparailly, F. MicroRNAs: Key Regulators to Understand Osteoclast Differentiation? Front. Immunol. 2019, 10, 375. [Google Scholar] [CrossRef]
- Sugatani, T.; Vacher, J.; Hruska, K.A. A microRNA expression signature of osteoclastogenesis. Blood J. Am. Soc. Hematol. 2011, 117, 3648–3657. [Google Scholar] [CrossRef]
- Yu, F.Y.; Xie, C.Q.; Sun, J.T.; Peng, W.; Huang, X.W. Overexpressed miR-145 inhibits osteoclastogenesis in RANKL-induced bone marrow-derived macrophages and ovariectomized mice by regulation of Smad3. Life Sci. 2018, 202, 11–20. [Google Scholar] [CrossRef]
- Sun, K.T.; Chen, M.Y.; Tu, M.G.; Wang, I.K.; Chang, S.S.; Li, C.Y. MicroRNA-20a regulates autophagy related protein-ATG16L1 in hypoxia-induced osteoclast differentiation. Bone 2015, 73, 145–153. [Google Scholar] [CrossRef]
- Kang, J.H.; Ko, H.M.; Moon, J.S.; Yoo, H.I.; Jung, J.Y.; Kim, M.S.; Koh, J.T.; Kim, W.J.; Kim, S.H. Osteoprotegerin expressed by osteoclasts: An autoregulator of osteoclastogenesis. J. Dent. Res. 2014, 93, 1116–1123. [Google Scholar] [CrossRef]
- Udagawa, N.; Koide, M.; Nakamura, M.; Nakamichi, Y.; Yamashita, T.; Uehara, S.; Kobayashi, Y.; Furuya, Y.; Yasuda, H.; Fukuda, C.; et al. Osteoclast differentiation by RANKL and OPG signaling pathways. J. Bone Miner. Metab. 2021, 39, 19–26. [Google Scholar] [CrossRef]
- Ozaki, Y.; Koide, M.; Furuya, Y.; Ninomiya, T.; Yasuda, H.; Nakamura, M.; Kobayashi, Y.; Takahashi, N.; Yoshinari, N.; Udagawa, N. Treatment of OPG-deficient mice with WP9QY, a RANKL-binding peptide, recovers alveolar bone loss by suppressing osteoclastogenesis and enhancing osteoblastogenesis. PLoS ONE 2017, 12, e0184904. [Google Scholar] [CrossRef]
- Zhao, H.; Gu, J.; Dai, N.; Gao, Q.; Wang, D.; Song, R.; Liu, W.; Yuan, Y.; Bian, J.; Liu, X.; et al. Osteoprotegerin exposure at different stages of osteoclastogenesis differentially affects osteoclast formation and function. Cytotechnology 2016, 68, 1325–1335. [Google Scholar] [CrossRef]
- Florencio-Silva, R.; Sasso, G.R.; Simoes, M.J.; Simoes, R.S.; Baracat, M.C.; Sasso-Cerri, E.; Cerri, P.S. Osteoporosis and autophagy: What is the relationship? Rev. Assoc. Med. Bras. 2017, 63, 173–179. [Google Scholar] [CrossRef]
- Lőrincz, P.; Juhász, G. Autophagosome-Lysosome Fusion. J. Mol. Biol. 2020, 432, 2462–2482. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Starling, S. A role for autophagy in bone biology. Nat. Rev. Endocrinol. 2019, 15, 438–439. [Google Scholar] [CrossRef]
- Valenti, M.T.; Carbonare, L.D.; Mottes, M. Role of autophagy in bone and muscle biology. World J. Stem. Cell. 2016, 8, 396–398. [Google Scholar] [CrossRef]
- Pierrefite-Carle, V.; Santucci-Darmanin, S.; Breuil, V.; Camuzard, O.; Carle, G.F. Autophagy in bone: Self-eating to stay in balance. Ageing Res. Rev. 2015, 24 Pt B, 206–217. [Google Scholar] [CrossRef]
- Shen, G.; Ren, H.; Shang, Q.; Qiu, T.; Yu, X.; Zhang, Z.; Huang, J.; Zhao, W.; Zhang, Y.; Liang, D.; et al. Autophagy as a target for glucocorticoid-induced osteoporosis therapy. Cell Mol. Life Sci. 2018, 75, 2683–2693. [Google Scholar] [CrossRef]
- Ma, Y.; Qi, M.; An, Y.; Zhang, L.; Yang, R.; Doro, D.H.; Liu, W.; Jin, Y. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging Cell 2018, 17, e12709. [Google Scholar]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [PubMed]
- Rodon, J.; Dienstmann, R.; Serra, V.; Tabernero, J. Development of PI3K inhibitors: Lessons learned from early clinical trials. Nat. Rev. Clin. Oncol. 2013, 10, 143–153. [Google Scholar]
- Yu, W.; Chen, J.; Xiong, Y.; Pixley, F.J.; Yeung, Y.G.; Stanley, E.R. Macrophage proliferation is regulated through CSF-1 receptor tyrosines 544, 559, and 807. J. Biol. Chem. 2012, 287, 13694–13704. [Google Scholar]
- Takeshita, S.; Faccio, R.; Chappel, J.; Zheng, L.; Feng, X.; Weber, J.D.; Teitelbaum, S.L.; Ross, F.P. c-Fms tyrosine 559 is a major mediator of M-CSF-induced proliferation of primary macrophages. J. Biol. Chem. 2007, 282, 18980–18990. [Google Scholar]
- Hamilton, J.A. CSF-1 signal transduction. J. Leukoc. Biol. 1997, 62, 145–155. [Google Scholar]
- Kim, J.H.; Kim, N. Signaling Pathways in Osteoclast Differentiation. Chonnam. Med. J. 2016, 52, 12–17. [Google Scholar]
- Fu, L.; Wu, W.; Sun, X.; Zhang, P. Glucocorticoids Enhanced Osteoclast Autophagy Through the PI3K/Akt/mTOR Signaling Pathway. Calcif. Tissue Int. 2020, 107, 60–71. [Google Scholar]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR Interacts with Raptor to Form a Nutrient-Sensitive Complex that Signals to the Cell Growth Machinery. Cell 2002, 110, 163–175. [Google Scholar]
- Huynh, H.; Wan, Y. mTORC1 impedes osteoclast differentiation via calcineurin and NFATc1. Commun. Biol. 2018, 1, 29. [Google Scholar]
- Sugatani, T.; Hruska, K.A. Akt1/Akt2 and mammalian target of rapamycin/Bim play critical roles in osteoclast differentiation and survival, respectively, whereas Akt is dispensable for cell survival in isolated osteoclast precursors. J. Biol. Chem. 2005, 280, 3583–3589. [Google Scholar]
- Indo, Y.; Takeshita, S.; Ishii, K.A.; Hoshii, T.; Aburatani, H.; Hirao, A.; Ikeda, K. Metabolic regulation of osteoclast differentiation and function. J. Bone Miner. Res. 2013, 28, 2392–2399. [Google Scholar]
- Hussein, O.; Tiedemann, K.; Murshed, M.; Komarova, S.V. Rapamycin inhibits osteolysis and improves survival in a model of experimental bone metastases. Cancer Lett. 2012, 314, 176–184. [Google Scholar]
- Hiraiwa, M.; Ozaki, K.; Yamada, T.; Iezaki, T.; Park, G.; Fukasawa, K.; Horie, T.; Kamada, H.; Tokumura, K.; Motono, M.; et al. mTORC1 Activation in Osteoclasts Prevents Bone Loss in a Mouse Model of Osteoporosis. Front. Pharmacol. 2019, 10, 684. [Google Scholar]
- Ren, S.; Luo, Y.; Chen, H.; Warburton, D.; Lam, H.C.; Wang, L.L.; Chen, P.; Henske, E.P.; Shi, W. Inactivation of Tsc2 in Mesoderm-Derived Cells Causes Polycystic Kidney Lesions and Impairs Lung Alveolarization. Am. J. Pathol. 2016, 186, 3261–3272. [Google Scholar] [PubMed]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar]
- She, Q.B.; Halilovic, E.; Ye, Q.; Zhen, W.; Shirasawa, S.; Sasazuki, T.; Solit, D.B.; Rosen, N. 4E-BP1 is a key effector of the oncogenic activation of the AKT and ERK signaling pathways that integrates their function in tumors. Cancer Cell 2010, 18, 39–51. [Google Scholar]
- Lu, C.; Willingham, M.C.; Furuya, F.; Cheng, S.Y. Activation of phosphatidylinositol 3-kinase signaling promotes aberrant pituitary growth in a mouse model of thyroid-stimulating hormone-secreting pituitary tumors. Endocrinology 2008, 149, 3339–3345. [Google Scholar]
- Tiedemann, K.; Le Nihouannen, D.; Fong, J.E.; Hussein, O.; Barralet, J.E.; Komarova, S.V. Regulation of Osteoclast Growth and Fusion by mTOR/raptor and mTOR/rictor/Akt. Front. Cell Dev. Biol. 2017, 5, 54. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. Regulation of mTORC1 and its impact on gene expression at a glance. J. Cell. Sci. 2013, 126 Pt 8, 1713–1719. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Luo, C.; Xiao, W. Induction of osteoclast formation by LOX mutant (LOXG473A) through regulation of autophagy. Ann. Transl. Med. 2021, 9, 1474. [Google Scholar] [CrossRef]
- Arai, A.; Kim, S.; Goldshteyn, V.; Kim, T.; Park, N.-H.; Wang, C.-Y.; Kim, R.H. Beclin1 Modulates Bone Homeostasis by Regulating Osteoclast and Chondrocyte Differentiation. J. Bone Miner. Res. 2019, 34, 1753–1766. [Google Scholar] [CrossRef]
- Lin, N.-Y.; Beyer, C.; Gießl, A.; Kireva, T.; Scholtysek, C.; Uderhardt, S.; Munoz, L.E.; Dees, C.; Distler, A.; Wirtz, S.; et al. Autophagy regulates TNFα-mediated joint destruction in experimental arthritis. Ann. Rheum. Dis. 2013, 72, 761–768. [Google Scholar] [CrossRef]
- Ke, D.; Ji, L.; Wang, Y.; Fu, X.; Chen, J.; Wang, F.; Zhao, D.; Xue, Y.; Lan, X.; Hou, J. JNK1 regulates RANKL-induced osteoclastogenesis via activation of a novel Bcl-2-Beclin1-autophagy pathway. FASEB J. 2019, 33, 11082–11095. [Google Scholar] [CrossRef]
- Chung, Y.H.; Jang, Y.; Choi, B.; Song, D.H.; Lee, E.J.; Kim, S.M.; Song, Y.; Kang, S.W.; Yoon, S.Y.; Chang, E.J. Beclin-1 is required for RANKL-induced osteoclast differentiation. J. Cell. Physiol. 2014, 229, 1963. [Google Scholar] [CrossRef]
- Li, R.F.; Chen, G.; Ren, J.G.; Zhang, W.; Wu, Z.X.; Liu, B.; Zhao, Y.; Zhao, Y.F. The adaptor protein p62 is involved in RANKL-induced autophagy and osteoclastogenesis. J. Histochem. Cytochem. 2014, 62, 879–888. [Google Scholar] [CrossRef]
- Nakatogawa, H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013, 55, 39–50. [Google Scholar]
- Deselm, C.J.; Miller, B.C.; Zou, W.; Beatty, W.L.; Van, M.E.; Takahata, Y.; Klumperman, J.; Tooze, S.A.; Teitelbaum, S.L.; Virgin, H.W. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev. Cell. 2011, 21, 966–974. [Google Scholar] [CrossRef]
- Chung, Y.H.; Choi, B.; Song, D.H.; Song, Y.; Kang, S.W.; Yoon, S.Y.; Kim, S.W.; Lee, H.K.; Chang, E.J. Interleukin-1beta promotes the LC3-mediated secretory function of osteoclast precursors by stimulating the Ca2+-dependent activation of ERK. Int. J. Biochem. Cell Biol. 2014, 54, 198–207. [Google Scholar] [CrossRef]
- Kurihara, N.; Hiruma, Y.; Zhou, H.; Subler, M.A.; Dempster, D.W.; Singer, F.R.; Reddy, S.V.; Gruber, H.E.; Windle, J.J.; Roodman, G.D. Mutation of the sequestosome 1 (p62) gene increases osteoclastogenesis but does not induce Paget disease. J. Clin. Investig. 2007, 117, 133–142. [Google Scholar] [CrossRef]
- White, E. Autophagy and p53. Cold Spring Harb. Perspect. Med. 2016, 6, a026120. [Google Scholar] [CrossRef]
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209. [Google Scholar] [CrossRef]
- Zhang, J.; Yang, Z.; Dong, J. P62: An emerging oncotarget for osteolytic metastasis. J. Bone Oncol. 2016, 5, 30–37. [Google Scholar] [CrossRef]
- Hu, Y.; Carraro-Lacroix, L.R.; Wang, A.; Owen, C.; Bajenova, E.; Corey, P.N.; Brumell, J.H.; Voronov, I. Lysosomal pH Plays a Key Role in Regulation of mTOR Activity in Osteoclasts. J. Cell. Biochem. 2016, 117, 413–425. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Rea, S.L.; Cheng, T.; Feng, H.T.; Walsh, J.P.; Ratajczak, T.; Tickner, J.; Pavlos, N.; Xu, H.Z.; Xu, J. Bafilomycin A1 Attenuates Osteoclast Acidification and Formation, Accompanied by Increased Levels of SQSTM1/p62 Protein. J. Cell. Biochem. 2016, 117, 1464–1470. [Google Scholar] [CrossRef]
- Newton, P.T.; Vuppalapati, K.K.; Bouderlique, T.; Chagin, A.S. Pharmacological inhibition of lysosomes activates the MTORC1 signaling pathway in chondrocytes in an autophagy-independent manner. Autophagy 2015, 11, 1594–1607. [Google Scholar] [CrossRef]
- Yao, Y.; Cai, X.; Ren, F.; Ye, Y.; Wang, F.; Zheng, C.; Qian, Y.; Zhang, M. The Macrophage-Osteoclast Axis in Osteoimmunity and Osteo-Related Diseases. Front. Immunol. 2021, 12, 664871. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Arron, J.R.; Choi, Y. Bone versus immune system. Nature 2000, 408, 535–536. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Xiao, Y. The Autophagy in Osteoimmonology: Self-Eating, Maintenance, and Beyond. Front. Endocrinol. 2019, 10, 490. [Google Scholar] [CrossRef] [PubMed]
- Dewhirst, F.E.; Stashenko, P.P.; Mole, J.E.; Tsurumachi, T. Purification and partial sequence of human osteoclast-activating factor: Identity with interleukin 1 beta. J. Immunol. 1985, 135, 2562–2568. [Google Scholar]
- Lin, N.Y.; Stefanica, A.; Distler, J.H. Autophagy: A key pathway of TNF-induced inflammatory bone loss. Autophagy 2013, 9, 1253–1255. [Google Scholar] [CrossRef]
- Cui, J.; Li, X.; Wang, S.; Su, Y.; Chen, X.; Cao, L.; Zhi, X.; Qiu, Z.; Wang, Y.; Jiang, H.; et al. Triptolide prevents bone loss via suppressing osteoclastogenesis through inhibiting PI3K-AKT-NFATc1 pathway. J. Cell. Mol. Med. 2020, 24, 6149–6161. [Google Scholar] [CrossRef]
- Chen, L.L.; Huang, M.; Tan, J.Y.; Chen, X.T.; Lei, L.H.; Wu, Y.M.; Zhang, D.Y. PI3K/AKT pathway involvement in the osteogenic effects of osteoclast culture supernatants on preosteoblast cells. Tissue Eng. Part. A 2013, 19, 2226–2232. [Google Scholar] [CrossRef]
- Han, J.; Li, L.; Zhang, C.; Huang, Q.; Wang, S.; Li, W.; Zong, J.; Li, L.; Zhao, Z.; Zhang, Z.; et al. Eucommia, Cuscuta, and Drynaria Extracts Ameliorate Glucocorticoid-Induced Osteoporosis by Inhibiting Osteoclastogenesis Through PI3K/Akt Pathway. Front. Pharmacol. 2021, 12, 772944. [Google Scholar] [CrossRef]
- Adapala, N.S.; Root, S.; Lorenzo, J.; Aguila, H.; Sanjay, A. PI3K activation increases SDF-1 production and number of osteoclast precursors, and enhances SDF-1-mediated osteoclast precursor migration. Bone Rep. 2019, 10, 100203. [Google Scholar] [CrossRef]
- Lee, J.-W.; Lee, I.-H.; Iimura, T.; Kong, S.W. Two macrophages, osteoclasts and microglia: From development to pleiotropy. Bone Res. 2021, 9, 11. [Google Scholar] [CrossRef]
- Sun, Y.; Li, J.; Xie, X.; Gu, F.; Sui, Z.; Zhang, K.; Yu, T. Macrophage-Osteoclast Associations: Origin, Polarization, and Subgroups. Front. Immunol. 2021, 12, 778078. [Google Scholar] [CrossRef]
- Pathak, J.L.; Bakker, A.D.; Verschueren, P.; Lems, W.F.; Luyten, F.P.; Klein-Nulend, J.; Bravenboer, N. CXCL8 and CCL20 Enhance Osteoclastogenesis via Modulation of Cytokine Production by Human Primary Osteoblasts. PLoS ONE 2015, 10, e0131041. [Google Scholar] [CrossRef]
- Kwak, H.B.; Ha, H.; Kim, H.N.; Lee, J.H.; Kim, H.S.; Lee, S.; Kim, H.M.; Kim, J.Y.; Kim, H.H.; Song, Y.W.; et al. Reciprocal cross-talk between RANKL and interferon-gamma-inducible protein 10 is responsible for bone-erosive experimental arthritis. Arthritis. Rheum. 2008, 58, 1332–1342. [Google Scholar] [CrossRef]
- Qian, M.; Fang, X.; Wang, X. Autophagy and inflammation. Clin. Transl. Med. 2017, 6, 24. [Google Scholar] [CrossRef]
- Jones, S.A.; Mills, K.H.; Harris, J. Autophagy and inflammatory diseases. Immunol. Cell Biol. 2013, 91, 250–258. [Google Scholar] [CrossRef]
- Wu, L.; Guo, Q.; Yang, J.; Ni, B. Tumor Necrosis Factor Alpha Promotes Osteoclast Formation Via PI3K/Akt Pathway-Mediated Blimp1 Expression Upregulation. J. Cell. Biochem. 2017, 118, 1308–1315. [Google Scholar] [CrossRef]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S.L. IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef]
- Lee, Y.M.; Fujikado, N.; Manaka, H.; Yasuda, H.; Iwakura, Y. IL-1 plays an important role in the bone metabolism under physiological conditions. Int. Immunol. 2010, 22, 805–816. [Google Scholar] [CrossRef]
- Lee, S.K.; Gardner, A.E.; Kalinowski, J.F.; Jastrzebski, S.L.; Lorenzo, J.A. RANKL-stimulated osteoclast-like cell formation in vitro is partially dependent on endogenous interleukin-1 production. Bone 2006, 38, 678–685. [Google Scholar] [CrossRef]
- Li, Y.; Ling, J.; Jiang, Q. Inflammasomes in Alveolar Bone Loss. Front. Immunol. 2021, 12, 691013. [Google Scholar] [CrossRef]
- Kato, K.; Tokuda, H.; Matsushima-Nishiwaki, R.; Natsume, H.; Kondo, A.; Ito, Y.; Kozawa, O.; Otsuka, T. AMPK limits IL-1-stimulated IL-6 synthesis in osteoblasts: Involvement of IκB/NF-κB pathway. Cell. Signal. 2012, 24, 1706–1712. [Google Scholar] [CrossRef] [PubMed]
- Bendixen, A.C.; Shevde, N.K.; Dienger, K.M.; Willson, T.M.; Funk, C.D.; Pike, J.W. IL-4 inhibits osteoclast formation through a direct action on osteoclast precursors via peroxisome proliferator-activated receptor gamma 1. Proc. Natl. Acad. Sci. USA 2001, 98, 2443–2448. [Google Scholar] [CrossRef] [PubMed]
- Mangashetti, L.S.; Khapli, S.M.; Wani, M.R. IL-4 inhibits bone-resorbing activity of mature osteoclasts by affecting NF-kappa B and Ca2+ signaling. J. Immunol. 2005, 175, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Abu-Amer, Y. IL-4 abrogates osteoclastogenesis through STAT6-dependent inhibition of NF-kappaB. J. Clin. Investig. 2001, 107, 1375–1385. [Google Scholar] [CrossRef]
- Jin, Q.; Yang, H.; Jing, Z.; Hong-Hua, W.; Ben-Jing, S.; Li-Ting, W.; Li-Juan, Y.; Wei, X.; Xia, K.; Juan, W.; et al. IL4/IL4R signaling promotes the osteolysis in metastatic bone of CRC through regulating the proliferation of osteoclast precursors. Mol. Med. 2021, 27, 152. [Google Scholar] [CrossRef]
- Fujii, T.; Kitaura, H.; Kimura, K.; Hakami, Z.W.; Takano-Yamamoto, T. IL-4 inhibits TNF-α-mediated osteoclast formation by inhibition of RANKL expression in TNF-α-activated stromal cells and direct inhibition of TNF-α-activated osteoclast precursors via a T-cell-independent mechanism in vivo. Bone 2012, 51, 771–780. [Google Scholar] [CrossRef]
- Evans, K.E.; Fox, S.W. Interleukin-10 inhibits osteoclastogenesis by reducing NFATc1 expression and preventing its translocation to the nucleus. BMC Cell Biol. 2007, 8, 4. [Google Scholar] [CrossRef]
- Wu, L.; Su, Y.; Lin, F.; Zhu, S.; Wang, J.; Hou, Y.; Du, J.; Liu, Y.; Guo, L. MicroRNA-21 promotes orthodontic tooth movement by modulating the RANKL/OPG balance in T cells. Oral Dis. 2020, 26, 370–380. [Google Scholar] [CrossRef]
- Kim, N.; Odgren, P.R.; Kim, D.K.; Marks, S.C., Jr.; Choi, Y. Diverse roles of the tumor necrosis factor family member TRANCE in skeletal physiology revealed by TRANCE deficiency and partial rescue by a lymphocyte-expressed TRANCE transgene. Proc. Natl. Acad. Sci. USA 2000, 97, 10905–10910. [Google Scholar] [CrossRef]
- Korn, T.; Mitsdoerffer, M.; Croxford, A.L.; Awasthi, A.; Dardalhon, V.A.; Galileos, G.; Vollmar, P.; Stritesky, G.L.; Kaplan, M.H.; Waisman, A.; et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 18460–18465. [Google Scholar] [CrossRef]
- Kimura, A.; Naka, T.; Kishimoto, T. IL-6-dependent and -independent pathways in the development of interleukin 17-producing T helper cells. Proc. Natl. Acad. Sci. USA 2007, 104, 12099–12104. [Google Scholar] [CrossRef]
- Roberts, J.L.; Mella-Velazquez, G.; Dar, H.Y.; Liu, G.; Drissi, H. Deletion of IL-17ra in osteoclast precursors increases bone mass by decreasing osteoclast precursor abundance. Bone 2022, 157, 116310. [Google Scholar] [CrossRef]
- Zhong, J.; Wang, Z.; Yuan, W.; Shen, Y.; Chen, L. Interleukin-17 promotes osteoclastogenesis and periodontal damage via autophagy in vitro and in vivo. Int. Immunopharmacol. 2022, 107, 108631. [Google Scholar] [CrossRef]
- Gordon, S. Elie Metchnikoff: Father of natural immunity. Eur. J. Immunol. 2008, 38, 3257–3264. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Takeda, Y.; Tachibana, I.; Miyado, K.; Kobayashi, M.; Miyazaki, T.; Funakoshi, T.; Kimura, H.; Yamane, H.; Saito, Y.; Goto, H.; et al. Tetraspanins CD9 and CD81 function to prevent the fusion of mononuclear phagocytes. J. Cell Biol. 2003, 161, 945–956. [Google Scholar] [CrossRef]
- Cui, W.; Ke, J.Z.; Zhang, Q.; Ke, H.Z.; Chalouni, C.; Vignery, A. The intracellular domain of CD44 promotes the fusion of macrophages. Blood 2006, 107, 796–805. [Google Scholar] [CrossRef]
- Møller, A.M.; Delaissé, J.M.; Søe, K. Osteoclast Fusion: Time-Lapse Reveals Involvement of CD47 and Syncytin-1 at Different Stages of Nuclearity. J. Cell. Physiol. 2017, 232, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Hobolt-Pedersen, A.S.; Delaissé, J.M.; Søe, K. Osteoclast fusion is based on heterogeneity between fusion partners. Calcif. Tissue Int. 2014, 95, 73–82. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).