
The Comparative Experimental Study of Sodium and Magnesium Dichloroacetate Effects on Pediatric PBT24 and SF8628 Cell Glioblastoma Tumors Using a Chicken Embryo Chorioallantoic Membrane Model and on Cells In Vitro


 , , and
, , and
Abstract
:1. Introduction
2. Results
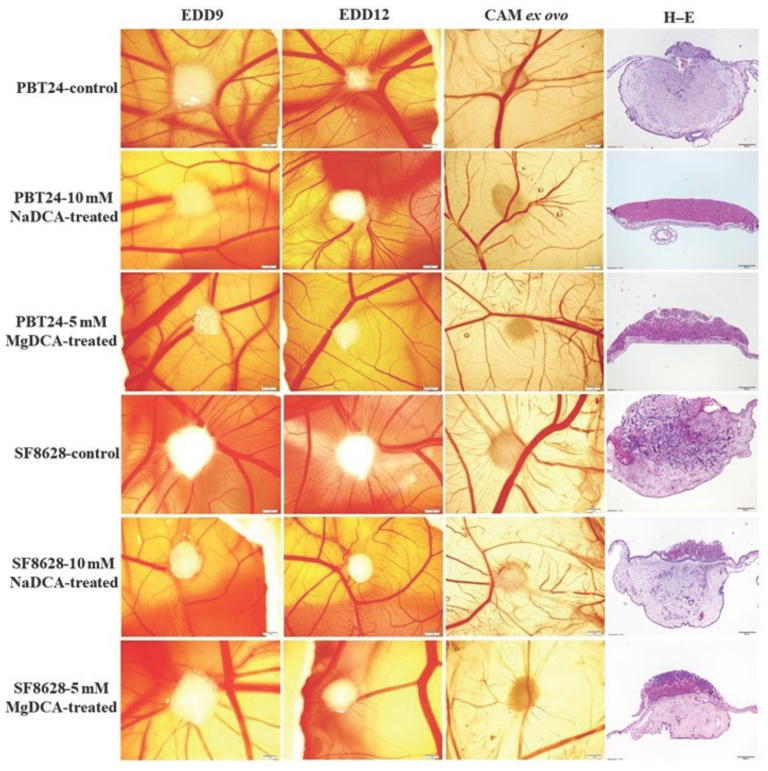
2.1. Stereomicroscopic Findings of Transplanted PBT24 and SF8628 Cell Tumors on CAM
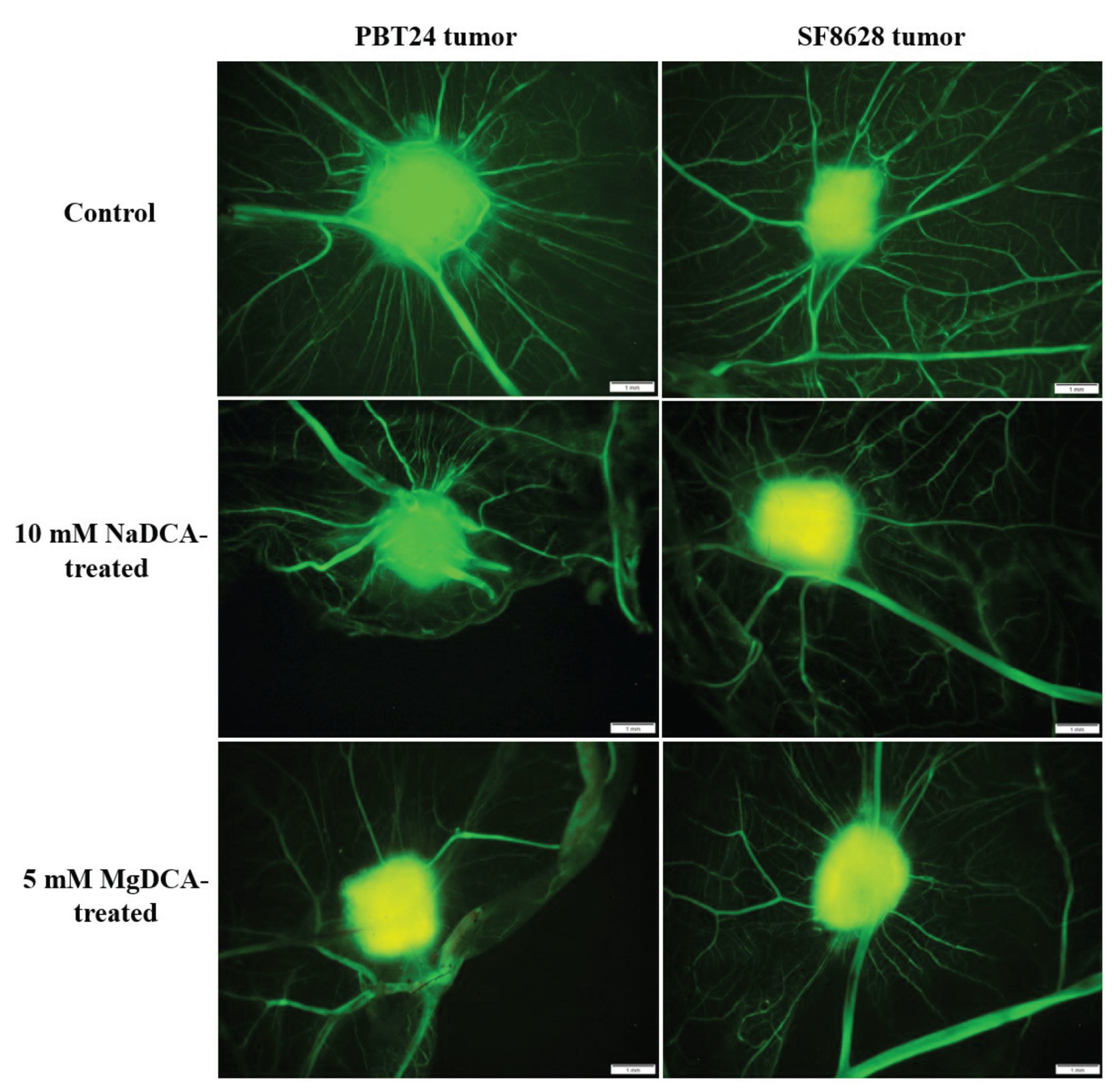
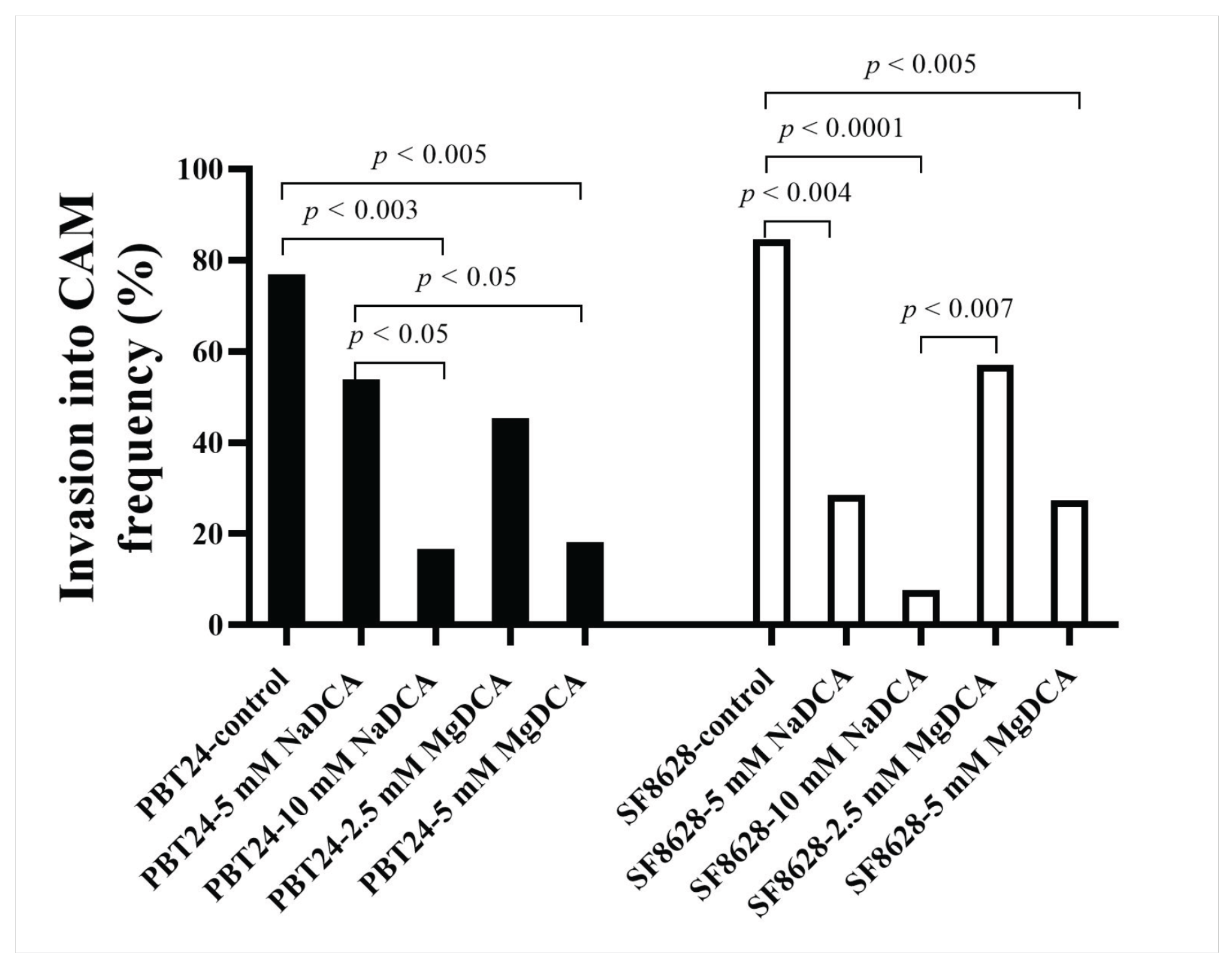
2.2. PBT24 and SF8628 Tumor Growth, Tumor Invasion into CAM Rate, the Number of Blood Vessels, and CAM Thickness
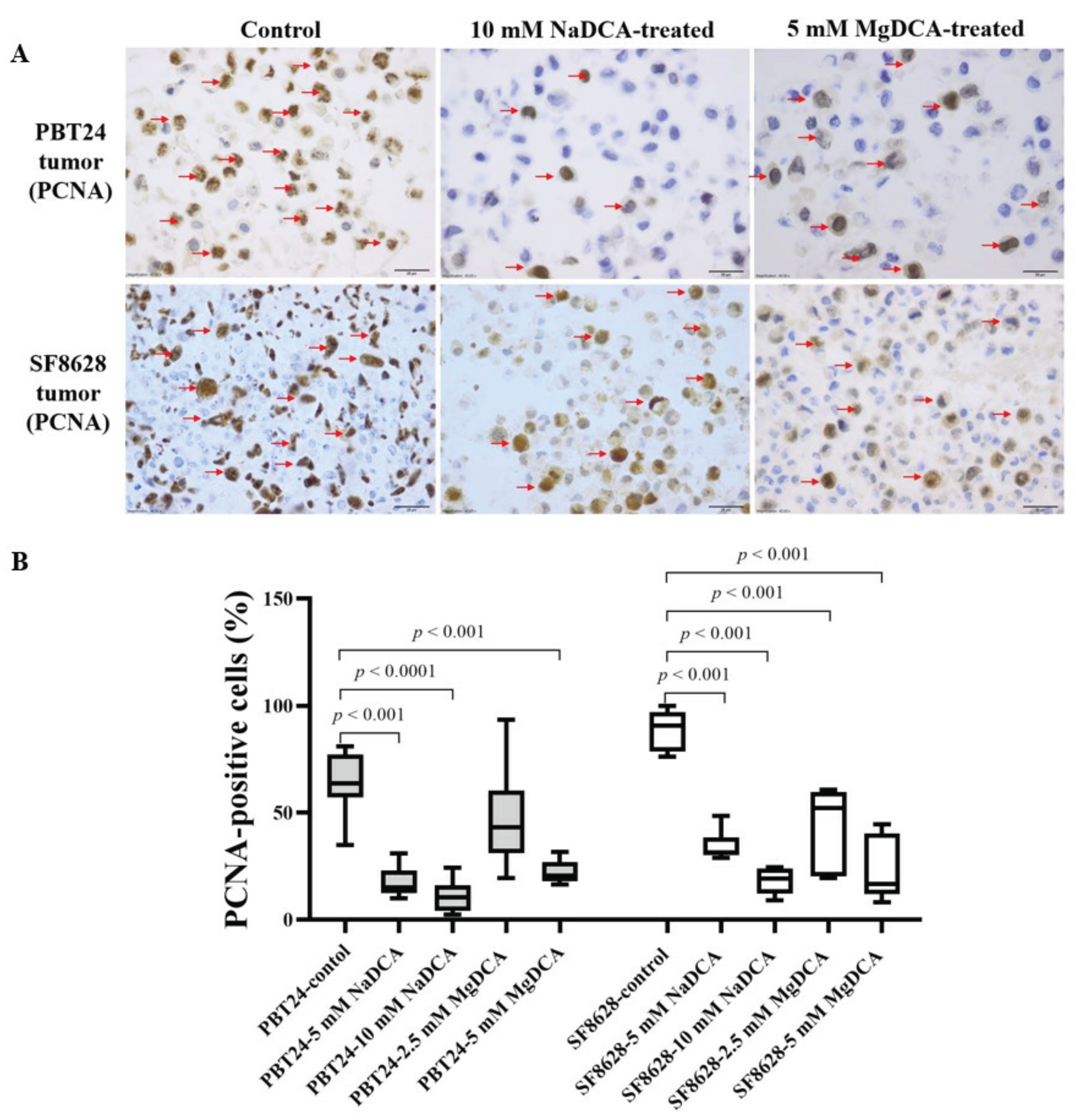
2.3. The PCNA Expression of PBT24 and SF8628 in Control and DCA-Treated Tumors
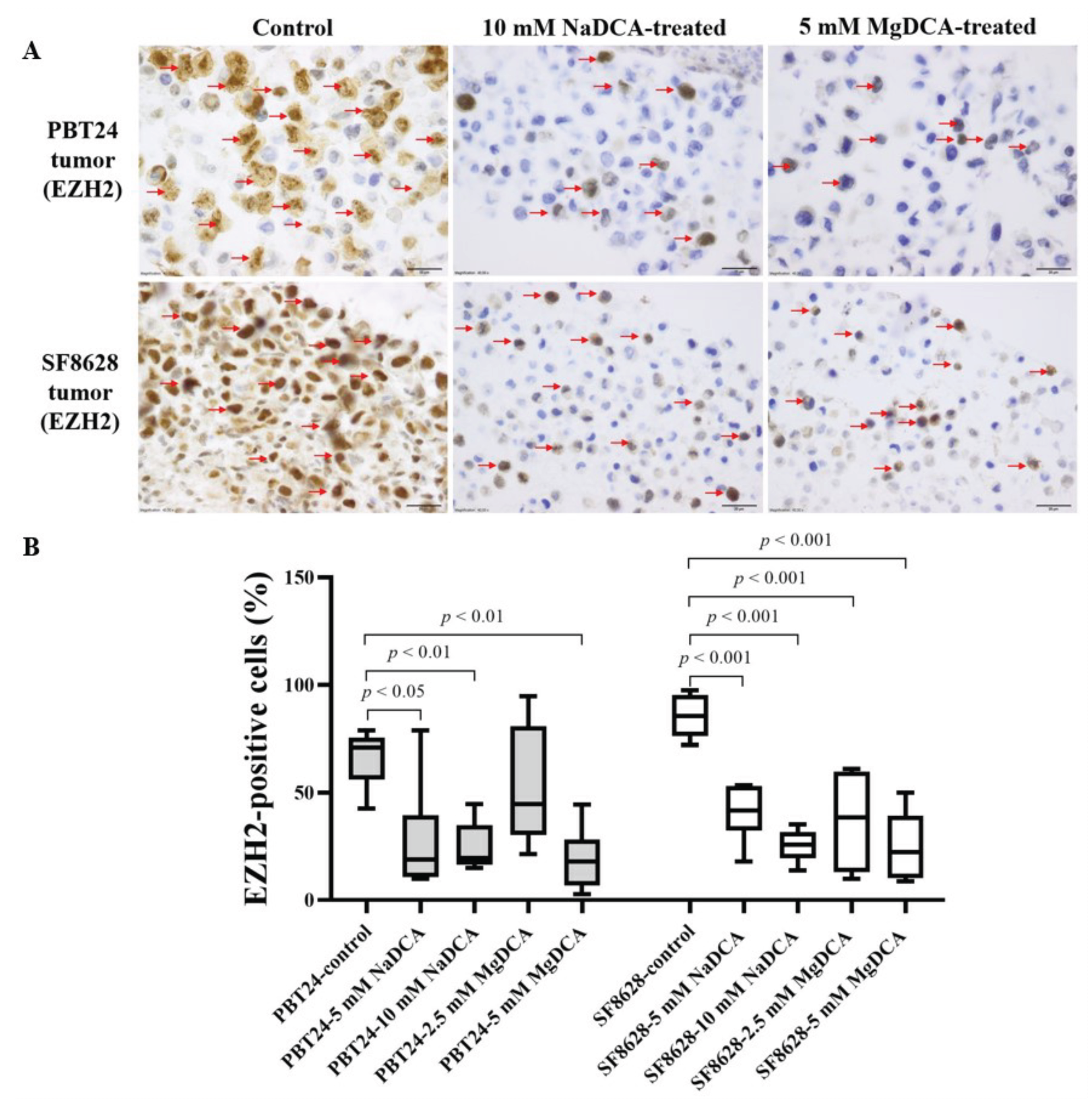
2.4. EZH2 Expression of PBT24 and SF8628 in Control and DCA-Treated Tumors
2.5. The p53 Expression in Control and DCA-Treated PBT24 and SF8628 Tumors
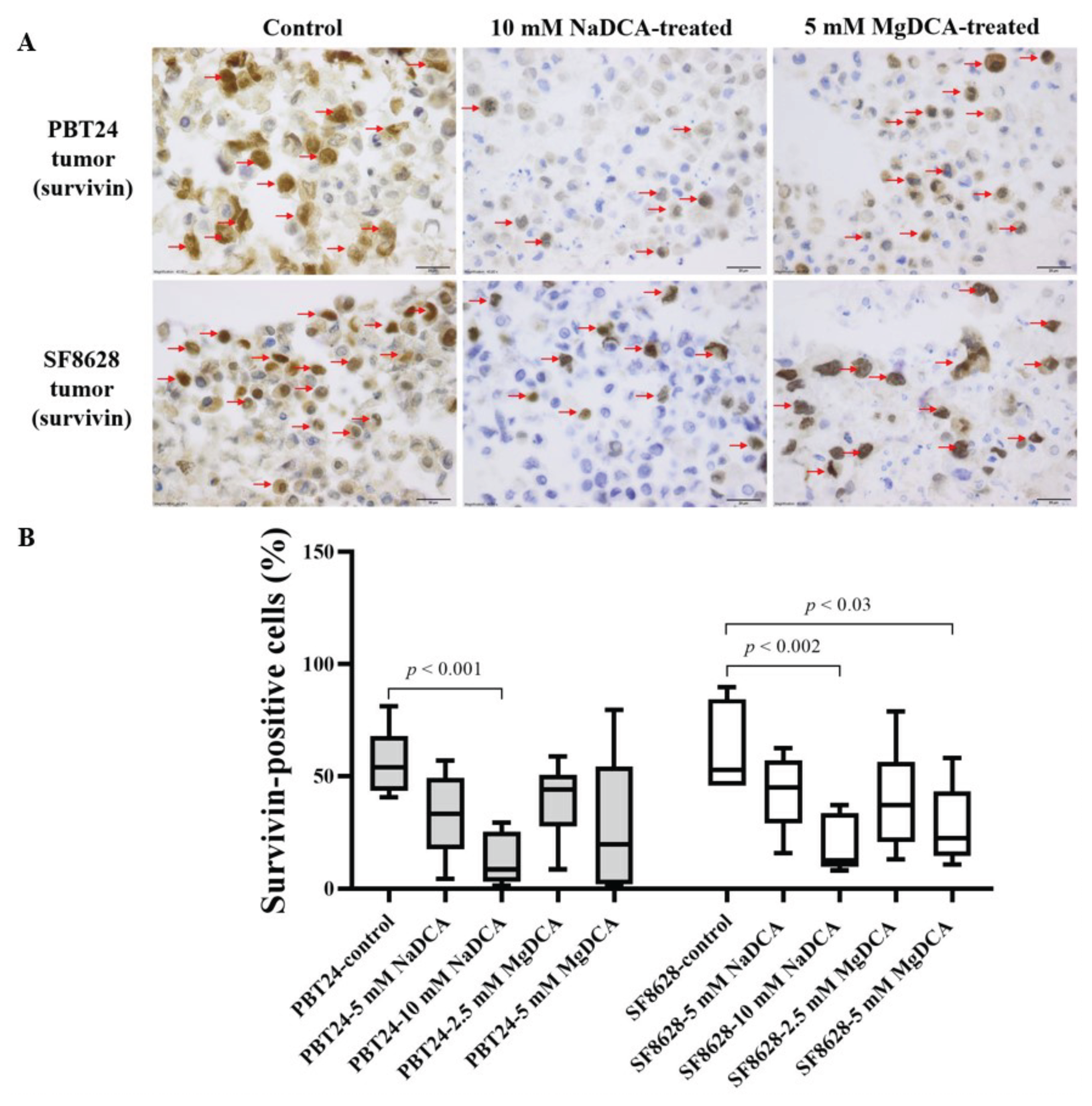
2.6. The Survivin Expression in the Control and DCA-Treated PBT24 and SF8628 Tumors
2.7. The Expression of NKCC1, KCC2, SLC5A8, E- and N-Cadherins Genes in the Studied PBT24 and SF8628 Cells Groups
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Cell Culture
4.2. Groups Studied for PBT24 and SF8628 Tumors on CAM
4.3. Application of the CAM Model to the Study of PBT24 and SF8626 Tumors on CAM
4.4. Immunohistochemical Study
4.5. Extraction of RNA from Study Cells and Determination of the Gene Expression in PBT24 and SF8628 Cells
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Antonucci, L.; Canciani, G.; Mastronuzzi, A.; Carai, A.; Del Baldo, G.; Del Bufalo, F. CAR-T Therapy for Pediatric High-Grade Gliomas: Peculiarities, Current Investigations and Future Strategies. Front. Immunol. 2022, 13, 867154. [Google Scholar] [CrossRef] [PubMed]
- Garofano, L.; Migliozzi, S.; Oh, Y.T.; D’Angelo, F.; Najac, R.D.; Ko, A.; Frangaj, B.; Caruso, F.P.; Yu, K.; Yuan, J.; et al. Pathway-Based Classification of Glioblastoma Uncovers a Mitochondrial Subtype with Therapeutic Vulnerabilities. Nat. Cancer 2021, 2, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.; Sweha, S.R.; Pratt, D.; Tamrazi, B.; Panwalkar, P.; Banda, A.; Bayliss, J.; Hawes, D.; Yang, F.; Lee, H.-J.; et al. Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas. Cancer Cell 2020, 38, 334–349.e9. [Google Scholar] [CrossRef] [PubMed]
- Parker, N.R.; Hudson, A.L.; Khong, P.; Parkinson, J.F.; Dwight, T.; Ikin, R.J.; Zhu, Y.; Cheng, Z.J.; Vafaee, F.; Chen, J.; et al. Intratumoral Heterogeneity Identified at the Epigenetic, Genetic and Transcriptional Level in Glioblastoma. Sci. Rep. 2016, 6, 22477. [Google Scholar] [CrossRef] [PubMed]
- Duraj, T.; García-Romero, N.; Carrión-Navarro, J.; Madurga, R.; Mendivil, A.O.; de Prat-Acin, R.; Garcia-Cañamaque, L.; Ayuso-Sacido, A. Beyond the Warburg Effect: Oxidative and Glycolytic Phenotypes Coexist within the Metabolic Heterogeneity of Glioblastoma. Cells 2021, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, A.H.; Kant, S.; Kesarwani, P.; Ahmed, K.; Forsyth, P.; Nakano, I.; Chinnaiyan, P. Integrative Cross-Platform Analyses Identify Enhanced Heterotrophy as a Metabolic Hallmark in Glioblastoma. Neuro-Oncol. 2019, 21, 337–347. [Google Scholar] [CrossRef]
- Kant, S.; Kesarwani, P.; Prabhu, A.; Graham, S.F.; Buelow, K.L.; Nakano, I.; Chinnaiyan, P. Enhanced Fatty Acid Oxidation Provides Glioblastoma Cells Metabolic Plasticity to Accommodate to Its Dynamic Nutrient Microenvironment. Cell Death Dis. 2020, 11, 253. [Google Scholar] [CrossRef]
- Shen, H.; Yu, M.; Tsoli, M.; Chang, C.; Joshi, S.; Liu, J.; Ryall, S.; Chornenkyy, Y.; Siddaway, R.; Hawkins, C.; et al. Targeting Reduced Mitochondrial DNA Quantity as a Therapeutic Approach in Pediatric High-Grade Gliomas. Neuro-Oncol. 2020, 22, 139–151. [Google Scholar] [CrossRef]
- Jones, C.; Karajannis, M.A.; Jones, D.T.W.; Kieran, M.W.; Monje, M.; Baker, S.J.; Becher, O.J.; Cho, Y.-J.; Gupta, N.; Hawkins, C.; et al. Pediatric High-Grade Glioma: Biologically and Clinically in Need of New Thinking. Neuro-Oncol. 2017, 19, 153–161. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Bi, J.; Chowdhry, S.; Wu, S.; Zhang, W.; Masui, K.; Mischel, P.S. Altered Cellular Metabolism in Gliomas—An Emerging Landscape of Actionable Co-Dependency Targets. Nat. Rev. Cancer 2020, 20, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.-L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of Tumor Metabolism Reveals Mitochondrial Glucose Oxidation in Genetically Diverse Human Glioblastomas in the Mouse Brain In Vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Griguer, C.E.; Oliva, C.R. Bioenergetics Pathways and Therapeutic Resistance in Gliomas: Emerging Role of Mitochondria. Curr. Pharm. Des. 2011, 17, 2421–2427. [Google Scholar] [CrossRef]
- Cook, K.M.; Shen, H.; McKelvey, K.J.; Gee, H.E.; Hau, E. Targeting Glucose Metabolism of Cancer Cells with Dichloroacetate to Radiosensitize High-Grade Gliomas. Int. J. Mol. Sci. 2021, 22, 7265. [Google Scholar] [CrossRef]
- Nile, D.L.; Rae, C.; Walker, D.J.; Waddington, J.C.; Vincent, I.; Burgess, K.; Gaze, M.N.; Mairs, R.J.; Chalmers, A.J. Inhibition of Glycolysis and Mitochondrial Respiration Promotes Radiosensitisation of Neuroblastoma and Glioma Cells. Cancer Metab. 2021, 9, 24. [Google Scholar] [CrossRef]
- McCrea, H.J.; Bander, E.D.; Venn, R.A.; Reiner, A.S.; Iorgulescu, J.B.; Puchi, L.A.; Schaefer, P.M.; Cederquist, G.; Greenfield, J.P. Sex, Age, Anatomic Location, and Extent of Resection Influence Outcomes in Children with High-Grade Glioma. Neurosurgery 2015, 77, 443–452; discussion 452–453. [Google Scholar] [CrossRef]
- Ippolito, J.E.; Yim, A.K.-Y.; Luo, J.; Chinnaiyan, P.; Rubin, J.B. Sexual Dimorphism in Glioma Glycolysis Underlies Sex Differences in Survival. JCI Insight 2017, 2, 92142. [Google Scholar] [CrossRef]
- Stakišaitis, D.; Juknevičienė, M.; Damanskienė, E.; Valančiūtė, A.; Balnytė, I.; Alonso, M.M. The Importance of Gender-Related Anticancer Research on Mitochondrial Regulator Sodium Dichloroacetate in Preclinical Studies In Vivo. Cancers 2019, 11, 1210. [Google Scholar] [CrossRef]
- Stakišaitis, D.; Damanskienė, E.; Curkūnavičiūtė, R.; Juknevičienė, M.; Alonso, M.M.; Valančiūtė, A.; Ročka, S.; Balnytė, I. The Effectiveness of Dichloroacetate on Human Glioblastoma Xenograft Growth Depends on Na+ and Mg2+ Cations. Dose-Response 2021, 19, 1559325821990166. [Google Scholar] [CrossRef] [PubMed]
- Abdelmalak, M.; Lew, A.; Ramezani, R.; Shroads, A.L.; Coats, B.S.; Langaee, T.; Shankar, M.N.; Neiberger, R.E.; Subramony, S.H.; Stacpoole, P.W. Long-Term Safety of Dichloroacetate in Congenital Lactic Acidosis. Mol. Genet. Metab. 2013, 109, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Belkahla, S.; Brualla, J.M.; Fayd’herbe de Maudave, A.; Falvo, P.; Allende-Vega, N.; Constantinides, M.; Khan, A.U.H.; Coenon, L.; Alexia, C.; Mitola, G.; et al. The Metabolism of Cells Regulates Their Sensitivity to NK Cells Depending on p53 Status. Sci. Rep. 2022, 12, 3234. [Google Scholar] [CrossRef] [PubMed]
- Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- McKelvey, K.J.; Wilson, E.B.; Short, S.; Melcher, A.A.; Biggs, M.; Diakos, C.I.; Howell, V.M. Glycolysis and Fatty Acid Oxidation Inhibition Improves Survival in Glioblastoma. Front. Oncol. 2021, 11, 633210. [Google Scholar] [CrossRef] [PubMed]
- James, M.O.; Jahn, S.C.; Zhong, G.; Smeltz, M.G.; Hu, Z.; Stacpoole, P.W. Therapeutic Applications of Dichloroacetate and the Role of Glutathione Transferase Zeta-1. Pharmacol. Ther. 2017, 170, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Colen, C.B.; Shen, Y.; Ghoddoussi, F.; Yu, P.; Francis, T.B.; Koch, B.J.; Monterey, M.D.; Galloway, M.P.; Sloan, A.E.; Mathupala, S.P. Metabolic Targeting of Lactate Efflux by Malignant Glioma Inhibits Invasiveness and Induces Necrosis: An In Vivo Study. Neoplasia 2011, 13, 620–632. [Google Scholar] [CrossRef]
- Crane, C.A.; Austgen, K.; Haberthur, K.; Hofmann, C.; Moyes, K.W.; Avanesyan, L.; Fong, L.; Campbell, M.J.; Cooper, S.; Oakes, S.A.; et al. Immune Evasion Mediated by Tumor-Derived Lactate Dehydrogenase Induction of NKG2D Ligands on Myeloid Cells in Glioblastoma Patients. Proc. Natl. Acad. Sci. USA 2014, 111, 12823–12828. [Google Scholar] [CrossRef]
- Stacpoole, P.W.; Nagaraja, N.V.; Hutson, A.D. Efficacy of Dichloroacetate as a Lactate-Lowering Drug. J. Clin. Pharmacol. 2003, 43, 683–691. [Google Scholar] [CrossRef]
- Khan, A.U.H.; Allende-Vega, N.; Gitenay, D.; Garaude, J.; Vo, D.-N.; Belkhala, S.; Gerbal-Chaloin, S.; Gondeau, C.; Daujat-Chavanieu, M.; Delettre, C.; et al. Mitochondrial Complex I Activity Signals Antioxidant Response through ERK5. Sci. Rep. 2018, 8, 7420. [Google Scholar] [CrossRef]
- Ward, N.P.; Poff, A.M.; Koutnik, A.P.; D’Agostino, D.P. Complex I Inhibition Augments Dichloroacetate Cytotoxicity through Enhancing Oxidative Stress in VM-M3 Glioblastoma Cells. PLoS ONE 2017, 12, e0180061. [Google Scholar] [CrossRef] [Green Version]
- Fangusaro, J. Pediatric High Grade Glioma: A Review and Update on Tumor Clinical Characteristics and Biology. Front. Oncol. 2012, 2, 105. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Alentorn, A.; Duran-Peña, A.; Pingle, S.C.; Piccioni, D.E.; Idbaih, A.; Kesari, S. Molecular Profiling of Gliomas: Potential Therapeutic Implications. Expert Rev. Anticancer. Ther. 2015, 15, 955–962. [Google Scholar] [CrossRef]
- Li, S.; Gai, X.; Myint, S.S.; Arroyo, K.; Morimoto, L.; Metayer, C.; de Smith, A.J.; Walsh, K.M.; Wiemels, J.L. Mitochondrial 1555 G>A Variant as a Potential Risk Factor for Childhood Glioblastoma. Neuro-Oncol. Adv. 2022, 4, vdac045. [Google Scholar] [CrossRef]
- Jane, E.P.; Premkumar, D.R.; Rajasundaram, D.; Thambireddy, S.; Reslink, M.C.; Agnihotri, S.; Pollack, I.F. Reversing Tozasertib Resistance in Glioma through Inhibition of Pyruvate Dehydrogenase Kinases. Mol. Oncol. 2022, 16, 219–249. [Google Scholar] [CrossRef]
- Klose, K.; Packeiser, E.-M.; Müller, P.; Granados-Soler, J.L.; Schille, J.T.; Goericke-Pesch, S.; Kietzmann, M.; Murua Escobar, H.; Nolte, I. Metformin and Sodium Dichloroacetate Effects on Proliferation, Apoptosis, and Metabolic Activity Tested Alone and in Combination in a Canine Prostate and a Bladder Cancer Cell Line. PLoS ONE 2021, 16, e0257403. [Google Scholar] [CrossRef] [PubMed]
- Damanskienė, E.; Balnytė, I.; Valančiūtė, A.; Alonso, M.M.; Preikšaitis, A.; Stakišaitis, D. The Different Temozolomide Effects on Tumorigenesis Mechanisms of Pediatric Glioblastoma PBT24 and SF8628 Cell Tumor in CAM Model and on Cells In Vitro. Int. J. Mol. Sci. 2022, 23, 2001. [Google Scholar] [CrossRef] [PubMed]
- Damanskienė, E.; Balnytė, I.; Valančiūtė, A.; Alonso, M.M.; Stakišaitis, D. Different Effects of Valproic Acid on SLC12A2, SLC12A5 and SLC5A8 Gene Expression in Pediatric Glioblastoma Cells as an Approach to Personalised Therapy. Biomedicines 2022, 10, 968. [Google Scholar] [CrossRef] [PubMed]
- Chandra, S.; Parker, D.J.; Barth, R.F.; Pannullo, S.C. Quantitative Imaging of Magnesium Distribution at Single-Cell Resolution in Brain Tumors and Infiltrating Tumor Cells with Secondary Ion Mass Spectrometry (SIMS). J. Neuro-Oncol. 2016, 127, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Tabata, S.; Shindo, Y.; Hotta, K.; Suzuki, K.; Soga, T.; Oka, K. Mitochondrial Mg(2+) Homeostasis Decides Cellular Energy Metabolism and Vulnerability to Stress. Sci. Rep. 2016, 6, 30027. [Google Scholar] [CrossRef]
- Maguire, D.; Neytchev, O.; Talwar, D.; McMillan, D.; Shiels, P.G. Telomere Homeostasis: Interplay with Magnesium. Int. J. Mol Sci. 2018, 19, E157. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.-H.; Zhang, S.; Wang, H.; Xue, J.-L.; Zhang, Z.-G. Emerging Experimental Models for Assessing Perineural Invasion in Human Cancers. Cancer Lett. 2022, 535, 215610. [Google Scholar] [CrossRef]
- Ribatti, D. The Chick Embryo Chorioallantoic Membrane (CAM). A Multifaceted Experimental Model. Mech. Dev. 2016, 141, 70–77. [Google Scholar] [CrossRef]
- Garcia, P.; Wang, Y.; Viallet, J.; Macek Jilkova, Z. The Chicken Embryo Model: A Novel and Relevant Model for Immune-Based Studies. Front. Immunol. 2021, 12, 791081. [Google Scholar] [CrossRef]
- Schneider-Stock, R.; Ribatti, D. The CAM Assay as an Alternative In Vivo Model for Drug Testing. Handb. Exp. Pharmacol. 2021, 265, 303–323. [Google Scholar] [CrossRef]
- Yang, L.; Moses, H.L. Transforming Growth Factor Beta: Tumor Suppressor or Promoter? Are Host Immune Cells the Answer? Cancer Res. 2008, 68, 9107–9111. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Nico, B.; Vacca, A.; Roncali, L.; Burri, P.H.; Djonov, V. Chorioallantoic Membrane Capillary Bed: A Useful Target for Studying Angiogenesis and Anti-Angiogenesis In Vivo. Anat. Rec. 2001, 264, 317–324. [Google Scholar] [CrossRef]
- Lee, M.S.; Moon, E.J.; Lee, S.W.; Kim, M.S.; Kim, K.W.; Kim, Y.J. Angiogenic Activity of Pyruvic Acid in In Vivo and In Vitro Angiogenesis Models. Cancer Res. 2001, 61, 3290–3293. [Google Scholar]
- Ribatti, D.; Crivellato, E.; Candussio, L.; Nico, B.; Vacca, A.; Roncali, L.; Dammacco, F. Mast Cells and Their Secretory Granules Are Angiogenic in the Chick Embryo Chorioallantoic Membrane. Clin. Exp. Allergy 2001, 31, 602–608. [Google Scholar] [CrossRef]
- Rema, R.B.; Rajendran, K.; Ragunathan, M. Angiogenic Efficacy of Heparin on Chick Chorioallantoic Membrane. Vasc. Cell 2012, 4, 8. [Google Scholar] [CrossRef]
- Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.-L.; Mackey, J.R.; Fulton, D.; et al. Metabolic Modulation of Glioblastoma with Dichloroacetate. Sci. Transl. Med. 2010, 2, 31ra34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lv, Q.; Zhang, J.; Yi, Y.; Huang, Y.; Wang, Y.; Wang, Y.; Zhang, W. Proliferating Cell Nuclear Antigen Has an Association with Prognosis and Risks Factors of Cancer Patients: A Systematic Review. Mol. Neurobiol. 2016, 53, 6209–6217. [Google Scholar] [CrossRef] [PubMed]
- de Vries, N.A.; Hulsman, D.; Akhtar, W.; de Jong, J.; Miles, D.C.; Blom, M.; van Tellingen, O.; Jonkers, J.; van Lohuizen, M. Prolonged Ezh2 Depletion in Glioblastoma Causes a Robust Switch in Cell Fate Resulting in Tumor Progression. Cell Rep. 2015, 10, 383–397. [Google Scholar] [CrossRef]
- Cordero, F.J.; Huang, Z.; Grenier, C.; He, X.; Hu, G.; McLendon, R.E.; Murphy, S.K.; Hashizume, R.; Becher, O.J. Histone H3.3K27M Represses p16 to Accelerate Gliomagenesis in a Murine Model of DIPG. Mol. Cancer Res. 2017, 15, 1243–1254. [Google Scholar] [CrossRef]
- Mohammad, F.; Weissmann, S.; Leblanc, B.; Pandey, D.P.; Højfeldt, J.W.; Comet, I.; Zheng, C.; Johansen, J.V.; Rapin, N.; Porse, B.T.; et al. EZH2 Is a Potential Therapeutic Target for H3K27M-Mutant Pediatric Gliomas. Nat. Med. 2017, 23, 483–492. [Google Scholar] [CrossRef]
- Piunti, A.; Hashizume, R.; Morgan, M.A.; Bartom, E.T.; Horbinski, C.M.; Marshall, S.A.; Rendleman, E.J.; Ma, Q.; Takahashi, Y.-H.; Woodfin, A.R.; et al. Therapeutic Targeting of Polycomb and BET Bromodomain Proteins in Diffuse Intrinsic Pontine Gliomas. Nat. Med. 2017, 23, 493–500. [Google Scholar] [CrossRef]
- von Knebel Doeberitz, N.; Paech, D.; Sturm, D.; Pusch, S.; Turcan, S.; Saunthararajah, Y. Changing Paradigms in Oncology: Toward Noncytotoxic Treatments for Advanced Gliomas. Int. J. Cancer 2022, 1–16. [Google Scholar] [CrossRef]
- Dhar, S.; Gadd, S.; Patel, P.; Vaynshteyn, J.; Raju, G.P.; Hashizume, R.; Brat, D.J.; Becher, O.J. A Tumor Suppressor Role for EZH2 in Diffuse Midline Glioma Pathogenesis. Acta Neuropathol. Commun. 2022, 10, 47. [Google Scholar] [CrossRef]
- Karlowee, V.; Amatya, V.J.; Takayasu, T.; Takano, M.; Yonezawa, U.; Takeshima, Y.; Sugiyama, K.; Kurisu, K.; Yamasaki, F. Immunostaining of Increased Expression of Enhancer of Zeste Homolog 2 (EZH2) in Diffuse Midline Glioma H3K27M-Mutant Patients with Poor Survival. Pathobiology 2019, 86, 152–161. [Google Scholar] [CrossRef]
- Litak, J.; Grajkowska, W.; Szumiło, J.; Krukow, P.; Maciejewski, R.; Roliński, J.; Grochowski, C. PD-L1 Expression Correlated with p53 Expression in Pediatric Glioblastoma Multiforme. Brain Sci. 2021, 11, 262. [Google Scholar] [CrossRef]
- Zavras, P.D.; Shastri, A.; Goldfinger, M.; Verma, A.K.; Saunthararajah, Y. Clinical Trials Assessing Hypomethylating Agents Combined with Other Therapies: Causes for Failure and Potential Solutions. Clin. Cancer Res. 2021, 27, 6653–6661. [Google Scholar] [CrossRef] [PubMed]
- Rosa, J.; Canovas, P.; Islam, A.; Altieri, D.C.; Doxsey, S.J. Survivin Modulates Microtubule Dynamics and Nucleation throughout the Cell Cycle. Mol. Biol. Cell 2006, 17, 1483–1493. [Google Scholar] [CrossRef] [PubMed]
- Fenstermaker, R.A.; Figel, S.A.; Qiu, J.; Barone, T.A.; Dharma, S.S.; Winograd, E.K.; Galbo, P.M.; Wiltsie, L.M.; Ciesielski, M.J. Survivin Monoclonal Antibodies Detect Survivin Cell Surface Expression and Inhibit Tumor Growth In Vivo. Clin. Cancer Res. 2018, 24, 2642–2652. [Google Scholar] [CrossRef]
- Adida, C.; Crotty, P.L.; McGrath, J.; Berrebi, D.; Diebold, J.; Altieri, D.C. Developmentally Regulated Expression of the Novel Cancer Anti-Apoptosis Gene Survivin in Human and Mouse Differentiation. Am. J. Pathol. 1998, 152, 43–49. [Google Scholar]
- Chakravarti, A.; Noll, E.; Black, P.M.; Finkelstein, D.F.; Finkelstein, D.M.; Dyson, N.J.; Loeffler, J.S. Quantitatively Determined Survivin Expression Levels Are of Prognostic Value in Human Gliomas. J. Clin. Oncol. 2002, 20, 1063–1068. [Google Scholar] [CrossRef]
- Kajiwara, Y.; Yamasaki, F.; Hama, S.; Yahara, K.; Yoshioka, H.; Sugiyama, K.; Arita, K.; Kurisu, K. Expression of Survivin in Astrocytic Tumors: Correlation with Malignant Grade and Prognosis. Cancer 2003, 97, 1077–1083. [Google Scholar] [CrossRef]
- Uematsu, M.; Ohsawa, I.; Aokage, T.; Nishimaki, K.; Matsumoto, K.; Takahashi, H.; Asoh, S.; Teramoto, A.; Ohta, S. Prognostic Significance of the Immunohistochemical Index of Survivin in Glioma: A Comparative Study with the MIB-1 Index. J. Neuro-Oncol. 2005, 72, 231–238. [Google Scholar] [CrossRef]
- Virrey, J.J.; Guan, S.; Li, W.; Schönthal, A.H.; Chen, T.C.; Hofman, F.M. Increased Survivin Expression Confers Chemoresistance to Tumor-Associated Endothelial Cells. Am. J. Pathol. 2008, 173, 575–585. [Google Scholar] [CrossRef]
- Thangaraju, M.; Carswell, K.N.; Prasad, P.D.; Ganapathy, V. Colon Cancer Cells Maintain Low Levels of Pyruvate to Avoid Cell Death Caused by Inhibition of HDAC1/HDAC3. Biochem. J. 2009, 417, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Garzon-Muvdi, T.; Schiapparelli, P.; ap Rhys, C.; Guerrero-Cazares, H.; Smith, C.; Kim, D.-H.; Kone, L.; Farber, H.; Lee, D.Y.; An, S.S.; et al. Regulation of Brain Tumor Dispersal by NKCC1 through a Novel Role in Focal Adhesion Regulation. PLoS Biol. 2012, 10, e1001320. [Google Scholar] [CrossRef]
- Pallud, J.; Le Van Quyen, M.; Bielle, F.; Pellegrino, C.; Varlet, P.; Cresto, N.; Baulac, M.; Duyckaerts, C.; Kourdougli, N.; Chazal, G.; et al. Cortical GABAergic Excitation Contributes to Epileptic Activities around Human Glioma. Sci. Transl. Med. 2014, 6, 244ra89. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Boer, K.; Redeker, S.; Spliet, W.G.M.; van Rijen, P.C.; Troost, D.; Gorter, J.A. Differential Expression Patterns of Chloride Transporters, Na+-K+-2Cl−-Cotransporter and K+-Cl−-Cotransporter, in Epilepsy-Associated Malformations of Cortical Development. Neuroscience 2007, 145, 185–196. [Google Scholar] [CrossRef]
- Okada, Y.; Maeno, E. Apoptosis, Cell Volume Regulation and Volume-Regulatory Chloride Channels. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2001, 130, 377–383. [Google Scholar] [CrossRef]
- Haas, B.R.; Cuddapah, V.A.; Watkins, S.; Rohn, K.J.; Dy, T.E.; Sontheimer, H. With-No-Lysine Kinase 3 (WNK3) Stimulates Glioma Invasion by Regulating Cell Volume. Am. J. Physiol. Cell Physiol. 2011, 301, C1150–C1160. [Google Scholar] [CrossRef]
- Haas, B.R.; Sontheimer, H. Inhibition of the Sodium-Potassium-Chloride Cotransporter Isoform-1 Reduces Glioma Invasion. Cancer Res. 2010, 70, 5597–5606. [Google Scholar] [CrossRef]
- Maeno, E.; Ishizaki, Y.; Kanaseki, T.; Hazama, A.; Okada, Y. Normotonic Cell Shrinkage Because of Disordered Volume Regulation Is an Early Prerequisite to Apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 9487–9492. [Google Scholar] [CrossRef]
- Bortner, C.D.; Cidlowski, J.A. Cell Shrinkage and Monovalent Cation Fluxes: Role in Apoptosis. Arch. Biochem. Biophys. 2007, 462, 176–188. [Google Scholar] [CrossRef]
- Cavallaro, U.; Christofori, G. Cell Adhesion and Signalling by Cadherins and Ig-CAMs in Cancer. Nat. Rev. Cancer 2004, 4, 118–132. [Google Scholar] [CrossRef]
- Johnson, K.S.; Hussein, S.; Chakraborty, P.; Muruganantham, A.; Mikhail, S.; Gonzalez, G.; Song, S.; Jolly, M.K.; Toneff, M.J.; Benton, M.L.; et al. CTCF Expression and Dynamic Motif Accessibility Modulates Epithelial-Mesenchymal Gene Expression. Cancers 2022, 14, 209. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Anastasiadis, P.Z. p120 Catenin Is Essential for Mesenchymal Cadherin-Mediated Regulation of Cell Motility and Invasiveness. J. Cell Biol. 2006, 174, 1087–1096. [Google Scholar] [CrossRef]
- Noh, M.-G.; Oh, S.-J.; Ahn, E.-J.; Kim, Y.-J.; Jung, T.-Y.; Jung, S.; Kim, K.-K.; Lee, J.-H.; Lee, K.-H.; Moon, K.-S. Prognostic Significance of E-Cadherin and N-Cadherin Expression in Gliomas. BMC Cancer 2017, 17, 583. [Google Scholar] [CrossRef] [PubMed]
- Bar, J.K.; Zub, L.; Lis-Nawara, A.; Noga, L.; Jeleń, M.; Paradowski, B. Expression and Interactions between Cell Adhesion Molecules CD44v6 and E-Cadherin in Human Gliomas. Adv. Clin. Exp. Med. 2014, 23, 827–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Urso, P.I.; D’Urso, O.F.; Storelli, C.; Catapano, G.; Gianfreda, C.D.; Montinaro, A.; Muscella, A.; Marsigliante, S. Retrospective Protein Expression and Epigenetic Inactivation Studies of CDH1 in Patients Affected by Low-Grade Glioma. J. Neuro-Oncol. 2011, 104, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Tuffin, L.J.; Rodriguez, F.; Giannini, C.; Scheithauer, B.; Necela, B.M.; Sarkaria, J.N.; Anastasiadis, P.Z. Misregulated E-Cadherin Expression Associated with an Aggressive Brain Tumor Phenotype. PLoS ONE 2010, 5, e13665. [Google Scholar] [CrossRef]
- Yang, W.; Warrington, N.M.; Taylor, S.J.; Whitmire, P.; Carrasco, E.; Singleton, K.W.; Wu, N.; Lathia, J.D.; Berens, M.E.; Kim, A.H.; et al. Sex Differences in GBM Revealed by Analysis of Patient Imaging, Transcriptome, and Survival Data. Sci. Transl. Med. 2019, 11, eaao5253. [Google Scholar] [CrossRef]
- Martinez-Velez, N.; Marigil, M.; García-Moure, M.; Gonzalez-Huarriz, M.; Aristu, J.J.; Ramos-García, L.-I.; Tejada, S.; Díez-Valle, R.; Patiño-García, A.; Becher, O.J.; et al. Delta-24-RGD Combined with Radiotherapy Exerts a Potent Antitumor Effect in Diffuse Intrinsic Pontine Glioma and Pediatric High Grade Glioma Models. Acta Neuropathol. Commun. 2019, 7, 64. [Google Scholar] [CrossRef]
- SF8628 Human DIPG H3.3-K27M Cell Line SF8628 Pediatric Diffuse Intrinsic Pontine Glioma (DIPG) Cell Line Harbors the Histone H3.3 Lys 27-to-Methionine (K27M) Mutation and Can Support Research and Drug Development Efforts Targeting DIPG. Sigma-Aldrich. Available online: http://www.sigmaaldrich.com/ (accessed on 8 December 2021).
- Mueller, S.; Hashizume, R.; Yang, X.; Kolkowitz, I.; Olow, A.K.; Phillips, J.; Smirnov, I.; Tom, M.W.; Prados, M.D.; James, C.D.; et al. Targeting Wee1 for the Treatment of Pediatric High-Grade Gliomas. Neuro-Oncol. 2014, 16, 352–360. [Google Scholar] [CrossRef]
- Kankotia, S.; Stacpoole, P.W. Dichloroacetate and Cancer: New Home for an Orphan Drug? Biochim. Biophys. Acta 2014, 1846, 617–629. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study Group | n | Invasion Frequency (%) | Number of Blood Vessels | CAM Thickness (µm) |
|---|---|---|---|---|
| Median (Range) | ||||
| PBT24-control | 13 | 76.9 | 15 (6–28) | 300.9 (65.2–700.9) |
| PBT24-5 mM NaDCA | 13 | 53.9 a,b | 12 (5–29) | 236.02 (31.3–484.4) |
| PBT24-10 mM NaDCA | 12 | 16.7 c | 11.5 (3–25) | 199.2 (54.4–627.6) |
| PBT24-2.5 mM MgDCA | 11 | 45.5 | 9 (2–23) | 138.6 (21.3–383.0) n |
| PBT24-5 mM MgDCA | 11 | 18.2 d | 7 (2–27) i | 96.5 (36.3–591.9) o |
| SF8628-control | 13 | 84.6 | 15 (5–21) | 282.5 (47.85–539.7) |
| SF8628-5 mM NaDCA | 14 | 28.6 e | 6 (1–22) j | 247.5 (86.76–879.3) |
| SF8628-10 mM NaDCA | 13 | 7.7 f | 5 (2–16) k | 199.4 (52.09–776.2) |
| SF8628-2.5 mM MgDCA | 14 | 57.1 g | 10.5 (5–23) l | 244.2 (55.02–409.1) |
| SF8628-5 mM MgDCA | 11 | 27.3 h | 8 (2–31) m | 140.5 (50.56–636.4) |
| Study Group | PBT24 Tumor PCNA-Positive Cells (%) | SF8628 Tumor PCNA-Positive Cells (%) | ||
|---|---|---|---|---|
| n | Median (Range) | n | Median (Range) | |
| Control | 9 | 63.8 (34.9–81.0) | 8 | 90.8 (76.3–100) |
| 5 mM NaDCA | 6 | 15.0 (10.0–31.1) a,b | 7 | 31.5 (29.0–48.5) g,h |
| 10 mM NaDCA | 8 | 10.5 (2.4–24.3) c,d | 7 | 19.1 (9.1–24.6) i |
| 2.5 mM MgDCA | 7 | 43.1 (19.5–93.6) | 6 | 52.2 (19.3–60.7) j,k,l |
| 5 mM MgDCA | 6 | 20.7 (16.4–31.7) e,f | 7 | 16.7 (8.2–44.6) m |
| Study Group | PBT24 Tumor EZH2-Positive Cells (%) | SF8628 Tumor EZH2-Positive Cells (%) | ||
|---|---|---|---|---|
| n | Median (Range) | n | Median (Range) | |
| Control | 6 | 71.0 (42.6–78.7) | 8 | 85.4 (72.0–97.5) |
| 5 mM NaDCA | 6 | 18.8 (9.8–78.7) a,b | 6 | 41.6 (18.0–53.5) g,h |
| 10 mM NaDCA | 7 | 19.6 (14.9–44.6) c,d | 7 | 25.9 (13.8–35.3) i |
| 2.5 mM MgDCA | 7 | 44.7 (21.5–94.8) | 6 | 38.4 (9.9–61.1) j |
| 5 mM MgDCA | 7 | 17.9 (2.8–44.4) e,f | 6 | 22.4 (8.9–50.0) k |
| Study Group | PBT24 Tumor p53-Positive Cells (%) | SF8628 Tumor p53-Positive Cells (%) | ||
|---|---|---|---|---|
| n | Median (Range) | n | Median (Range) | |
| Control | 7 | 75.1 (61.2–96.9) | 8 | 85.3 (56.7–92.9) |
| 5 mM NaDCA | 8 | 53.7 (33.9–75.5) a | 6 | 40.7 (27.1–48.9) h |
| 10 mM NaDCA | 8 | 29.6 (11.5–52.3) b,c,d | 6 | 19.5 (11.3–52.6) i |
| 2.5 mM MgDCA | 7 | 51.9 (32.0–91.0) e | 6 | 56.3 (27.8–87.1) j |
| 5 mM MgDCA | 6 | 24.2 (19.3–61.6) f,g | 7 | 44.9 (5.4–52.8) k |
| Study Group | PBT24 Tumor Survivin- Positive Cells (%) | SF8628 Tumor Survivin-Positive Cells (%) | ||
|---|---|---|---|---|
| n | Median (Range) | n | Median (Range) | |
| Control | 6 | 53.8 (40.6–81.1) | 6 | 52.8 (45.7–89.6) |
| 5 mM NaDCA | 6 | 33.1 (4.3–56.8) | 7 | 45.0 (15.6–62.3) |
| 10 mM NaDCA | 8 | 8.5 (1.3–29.3) a,b,c | 7 | 12.4 (8.1–37.0) d,e |
| 2.5 mM MgDCA | 7 | 43.9 (8.4–58.7) | 6 | 37.0 (13.0–8.6) |
| 5 mM MgDCA | 6 | 19.6 (0.9–79.4) | 6 | 22.4 (10.6–58.1) f |
| Study Group | n | CT Mean | ΔCT Mean ± SD | ΔΔCT | |
|---|---|---|---|---|---|
| SLC12A2 | GAPDH | ||||
| PBT24-control | 6 | 22.95 | 19.37 | 3.58 ± 0.73 | |
| PBT24-3 mM NaDCA | 6 | 22.72 | 18.69 | 4.03 ± 0.48 | 0.45 |
| PBT24-1.5 mM MgDCA | 6 | 23.00 | 19.31 | 3.69 ± 1.33 | 0.11 |
| SF8628-control | 6 | 22.89 | 19.02 | 3.88 ± 0.21 | |
| SF8628-3 mM NaDCA | 6 | 23.61 | 19.77 | 3.84 ± 0.29 | −0.04 |
| SF8628-1.5 mM MgDCA | 6 | 23.66 | 19.63 | 4.04 ± 0.27 | 0.16 |
| SLC12A5 | GAPDH | ΔCT Mean ± SD | ΔΔCT | ||
| PBT24-control | 6 | 32.56 | 19.37 | 13.19 ± 0.83 | |
| PBT24-3 mM NaDCA | 6 | 31.94 | 18.69 | 13.25 ± 0.72 | 0.06 |
| PBT24-1.5 mM MgDCA | 6 | 32.39 | 19.31 | 13.08 ± 1.19 | −0.11 |
| SF8628-control | 6 | 36.83 | 19.02 | 17.81 ± 0.43 a | |
| SF8628-3 mM NaDCA | 6 | 37.84 | 19.77 | 18.07 ± 0.81 b | 0.260 |
| SF8628-1.5 mM MgDCA | 6 | 37.66 | 19.63 | 18.04 ± 0.29 c | 0.223 |
| SLC5A8 | GAPDH | ΔCT Mean ± SD | ΔΔCT | ||
| PBT24-control | 6 | 39.42 | 19.37 | 20.04 ± 1.82 | |
| PBT24-3 mM NaDCA | 6 | 38.91 | 18.69 | 20.22 ± 1.81 | 0.18 |
| PBT24-1.5 mM MgDCA | 6 | 39.47 | 19.31 | 20.17 ± 2.24 | 0.12 |
| SF8628-control | 6 | 38.97 | 19.02 | 19.96 ± 0.59 | |
| SF8628-3 mM NaDCA | 6 | 40.06 | 19.77 | 20.29 ± 0.68 | 0.34 |
| SF8628-1.5 mM MgDCA | 6 | 40.03 | 19.63 | 20.41 ± 0.54 | 0.45 |
| Study Group | n | CT Mean | ΔCT Mean ± SD | ΔΔCT | |
|---|---|---|---|---|---|
| CDH1 | GAPDH | ||||
| PBT24-control | 6 | 33.48 | 19.37 | 14.104 ± 1.1 | |
| PBT24-3 mM NaDCA | 6 | 33.61 | 18.69 | 14.92 ± 1.24 | 0.82 |
| PBT24-1.5 mM MgDCA | 6 | 33.95 | 19.31 | 14.65 ± 1.56 | 0.54 |
| SF8628-control | 6 | 38.69 | 19.02 | 19.67 ± 0.51 a | |
| SF8628-3 mM NaDCA | 6 | 38.59 | 19.77 | 18.83 ± 0.79 b | −0.84 |
| SF8628-1.5 mM MgDCA | 6 | 37.36 | 19.63 | 17.73 ± 0.32 c,d,e | −1.94 |
| CDH2 | GAPDH | ΔCT Mean ± SD | ΔΔCT | ||
| PBT24-control | 6 | 22.92 | 19.37 | 3.55 ± 0.98 | |
| PBT24-3 mM NaDCA | 6 | 22.55 | 18.69 | 3.86 ± 0.85 | 0.31 |
| PBT24-1.5 mM MgDCA | 6 | 22.28 | 19.31 | 2.98 ± 1.09 | −0.57 |
| SF8628-control | 6 | 23.45 | 19.02 | 4.43 ± 0.23 | |
| SF8628-3 mM NaDCA | 6 | 23.95 | 19.77 | 4.18 ± 0.39 | −0.25 |
| SF8628-1.5 mM MgDCA | 6 | 24.14 | 19.63 | 4.52 ± 0.19 f | 0.08 |
| Control and Treated Group | Invasion, No. of Vessels, CAM Thickness | PCNA | EZH2 | p53 | Survivin | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| PBT24 n | SF8628 n | PBT24 n | SF8628 n | PBT24 n | SF8628 n | PBT24 n | SF8628 n | PBT24 n | SF8628 n | |
| Control | 13 | 13 | 9 | 8 | 6 | 8 | 7 | 8 | 6 | 6 |
| 5 mM NaDCA | 13 | 14 | 6 | 7 | 6 | 6 | 8 | 6 | 6 | 7 |
| 10 mM NaDCA | 12 | 13 | 8 | 7 | 7 | 7 | 8 | 6 | 8 | 7 |
| 2.5 mM MgDCA | 11 | 14 | 7 | 6 | 7 | 6 | 7 | 6 | 7 | 6 |
| 5 mM MgDCA | 11 | 11 | 6 | 7 | 7 | 6 | 6 | 7 | 6 | 6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damanskienė, E.; Balnytė, I.; Valančiūtė, A.; Lesauskaitė, V.; Alonso, M.M.; Stakišaitis, D. The Comparative Experimental Study of Sodium and Magnesium Dichloroacetate Effects on Pediatric PBT24 and SF8628 Cell Glioblastoma Tumors Using a Chicken Embryo Chorioallantoic Membrane Model and on Cells In Vitro. Int. J. Mol. Sci. 2022, 23, 10455. https://doi.org/10.3390/ijms231810455
Damanskienė E, Balnytė I, Valančiūtė A, Lesauskaitė V, Alonso MM, Stakišaitis D. The Comparative Experimental Study of Sodium and Magnesium Dichloroacetate Effects on Pediatric PBT24 and SF8628 Cell Glioblastoma Tumors Using a Chicken Embryo Chorioallantoic Membrane Model and on Cells In Vitro. International Journal of Molecular Sciences. 2022; 23(18):10455. https://doi.org/10.3390/ijms231810455
Chicago/Turabian StyleDamanskienė, Eligija, Ingrida Balnytė, Angelija Valančiūtė, Vaiva Lesauskaitė, Marta Marija Alonso, and Donatas Stakišaitis. 2022. "The Comparative Experimental Study of Sodium and Magnesium Dichloroacetate Effects on Pediatric PBT24 and SF8628 Cell Glioblastoma Tumors Using a Chicken Embryo Chorioallantoic Membrane Model and on Cells In Vitro" International Journal of Molecular Sciences 23, no. 18: 10455. https://doi.org/10.3390/ijms231810455
APA StyleDamanskienė, E., Balnytė, I., Valančiūtė, A., Lesauskaitė, V., Alonso, M. M., & Stakišaitis, D. (2022). The Comparative Experimental Study of Sodium and Magnesium Dichloroacetate Effects on Pediatric PBT24 and SF8628 Cell Glioblastoma Tumors Using a Chicken Embryo Chorioallantoic Membrane Model and on Cells In Vitro. International Journal of Molecular Sciences, 23(18), 10455. https://doi.org/10.3390/ijms231810455