
The Role of Tumor Microenvironment in the Pathogenesis of Sézary Syndrome

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Tumor Microenvironment
2.1. CD8+ T-Cells
2.2. Regulatory T-Cells
2.3. Regulatory B-Cells
2.4. NK Cells
2.5. Dendritic Cells
2.6. Myeloid-Derived Suppressor Cells
2.7. M2 Macrophages
2.8. Neutrophils
2.9. Mast Cells
2.10. Eosinophils
2.11. Keratinocytes
2.12. Fibroblasts
2.13. Malignant Cells
3. Angiogenesis and Lymphangiogenesis
4. Skin Barrier
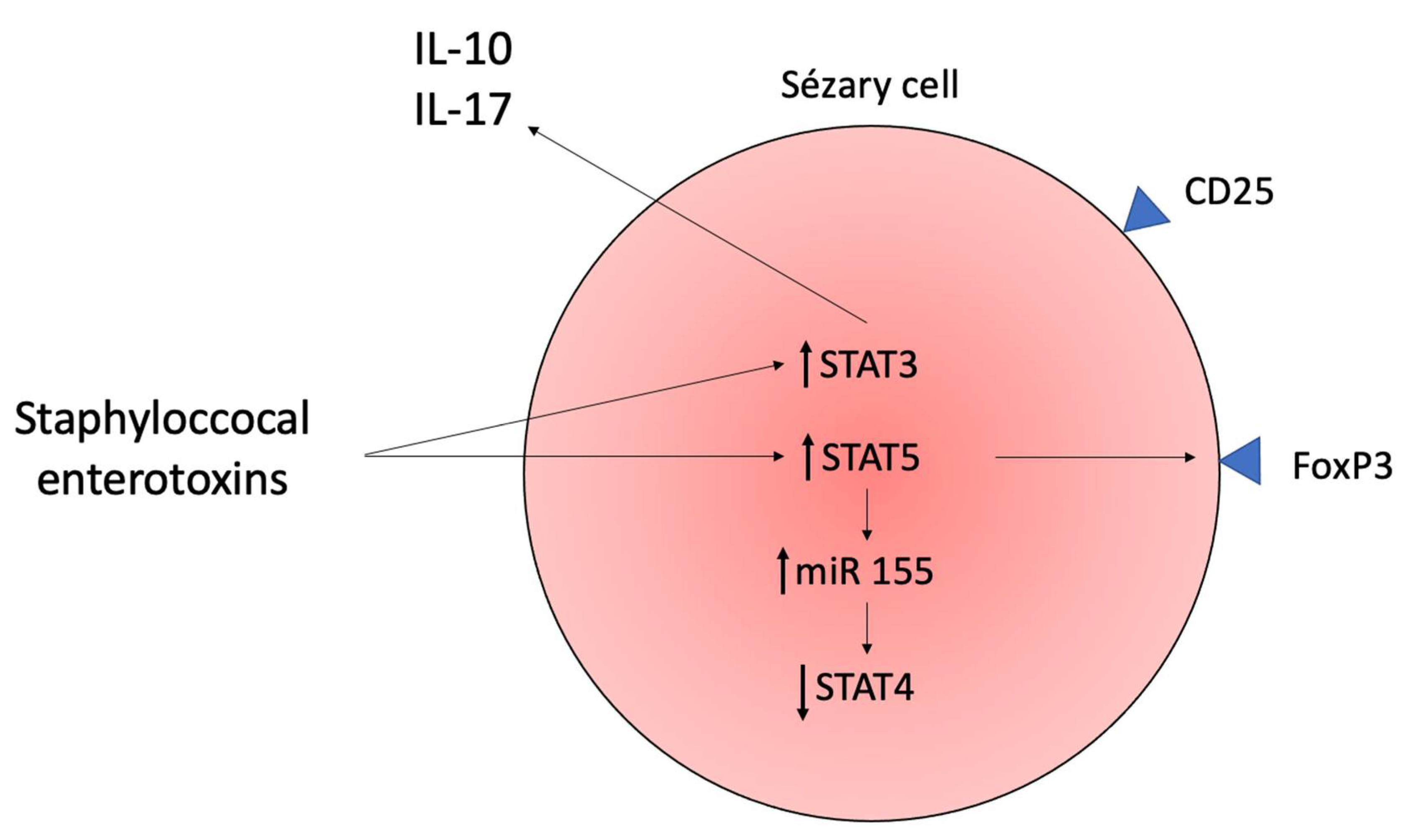
4.1. Staphylococcus aureus
4.2. Galectins
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sézary, A.; Bouvrain, Y. Érythrodermie avec présence de cellules monstrueuses dans le derme et le sang circulant. Bull. Société Fr. Dermatol. Syphiligr. Paris 1938, 45, 254–260. [Google Scholar]
- Kubica, A.W.; Davis, M.D.; Weaver, A.L.; Killian, J.M.; Pittelkow, M.R. Sézary syndrome: A study of 176 patients at Mayo Clinic. J. Am. Acad. Dermatol. 2012, 67, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Miyashiro, D.; Sanches, J.A. Erythroderma: A prospective study of 309 patients followed for 12 years in a tertiary center. Sci. Rep. 2020, 10, 9774. [Google Scholar] [CrossRef]
- Molloy, K.; Jonak, C.; Woei-A.-Jin, F.J.S.H.; Guenova, E.; Busschots, A.M.; Bervoets, A.; Hauben, E.; Knobler, R.; Porkert, S.; Fassnacht, C.; et al. Characteristics associated with significantly worse quality of life in mycosis fungoides/Sézary syndrome from the Prospective Cutaneous Lymphoma International Prognostic Index ( PROCLIPI ) study. Br. J. Dermatol. 2019, 182, 770–779. [Google Scholar] [CrossRef]
- Morris, L.; Tran, J.; Duvic, M. Non-Classic Signs of Sézary Syndrome: A Review. Am. J. Clin. Dermatol. 2020, 21, 383–391. [Google Scholar] [CrossRef]
- Willemze, R.; Jaffe, E.S.; Burg, G.; Cerroni, L.; Berti, E.; Swerdlow, S.H.; Ralfkiaer, E.; Chimenti, S.; Diaz-Perez, J.L.; Duncan, L.M.; et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005, 105, 3768–3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, E.; Vonderheid, E.; Pimpinelli, N.; Willemze, R.; Kim, Y.; Knobler, R.; Zackheim, H.; Duvic, M.; Estrach, T.; Lamberg, S.; et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: A proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 2007, 110, 1713–1722. [Google Scholar] [CrossRef] [Green Version]
- Litvinov, I.V.; Shtreis, A.; Kobayashi, K.; Glassman, S.; Tsang, M.; Woetmann, A.; Sasseville, D.; Ødum, N.; Duvic, M. Investigating potential exogenous tumor initiating and promoting factors for Cutaneous T-Cell Lymphomas (CTCL), a rare skin malignancy. Oncoimmunology 2016, 5, e1175799. [Google Scholar] [CrossRef] [Green Version]
- Nicolay, J.P.; Wobser, M. Cutaneous B-cell lymphomas—Pathogenesis, diagnostic workup, and therapy. J. Dtsch. Dermatol. Ges. 2016, 14, 1207–1224. [Google Scholar] [CrossRef]
- Campbell, J.J.; Clark, R.A.; Watanabe, R.; Kupper, T.S. Sézary syndrome and mycosis fungoides arise from distinct T-cell subsets: A biologic rationale for their distinct clinical behaviors. Blood 2010, 116, 767–771. [Google Scholar] [CrossRef]
- Scarisbrick, J.J.; Prince, H.M.; Vermeer, M.; Quaglino, P.; Horwitz, S.; Porcu, P.; Stadler, R.; Wood, G.S.; Beylot-Barry, M.; Pham-Ledard, A.; et al. Cutaneous Lymphoma International Consortium Study of Outcome in Advanced Stages of Mycosis Fungoides and Sézary Syndrome: Effect of Specific Prognostic Markers on Survival and Development of a Prognostic Model. J. Clin. Oncol. 2015, 33, 3766–3773. [Google Scholar] [CrossRef] [PubMed]
- Trautinger, F.; Eder, J.; Assaf, C.; Bagot, M.; Cozzio, A.; Dummer, R.; Gniadecki, R.; Klemke, C.-D.; Ortiz-Romero, P.L.; Papadavid, E.; et al. European Organisation for Research and Treatment of Cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome—Update 2017. Eur. J. Cancer 2017, 77, 57–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phyo, Z.; Shanbhag, S.; Rozati, S. Update on Biology of Cutaneous T-Cell Lymphoma. Front. Oncol. 2020, 10, 765. [Google Scholar] [CrossRef]
- Pileri, A.; Guglielmo, A.; Grandi, V.; Violetti, S.A.; Fanoni, D.; Fava, P.; Agostinelli, C.; Berti, E.; Quaglino, P.; Pimpinelli, N. The Microenvironment’s Role in Mycosis Fungoides and Sézary Syndrome: From Progression to Therapeutic Implications. Cells 2021, 10, 2780. [Google Scholar] [CrossRef]
- Krejsgaard, T.; Lindahl, L.M.; Mongan, N.P.; Wasik, M.A.; Litvinov, I.V.; Iversen, L.; Langhoff, E.; Woetmann, A.; Odum, N. Malignant inflammation in cutaneous T-cell lymphoma-a hostile takeover. Semin. Immunopathol. 2017, 39, 269–282. [Google Scholar] [CrossRef] [PubMed]
- DeSimone, J.A.; Sodha, P.; Ignatova, D.; Dummer, R.; Cozzio, A.; Guenova, E. Recent advances in primary cutaneous T-cell lymphoma. Curr. Opin. Oncol. 2015, 27, 128–133. [Google Scholar] [CrossRef]
- Miyagaki, T.; Sugaya, M. Immunological milieu in mycosis fungoides and Sézary syndrome. J. Dermatol. 2014, 41, 11–18. [Google Scholar] [CrossRef]
- Quaglino, P.; Fava, P.; Pileri, A.; Grandi, V.; Sanlorenzo, M.; Panasiti, V.; Guglielmo, A.; Alberti-Violetti, S.; Novelli, M.; Astrua, C.; et al. Phenotypical Markers, Molecular Mutations, and Immune Microenvironment as Targets for New Treatments in Patients with Mycosis Fungoides and/or Sézary Syndrome. J. Investig. Dermatol. 2021, 141, 484–495. [Google Scholar] [CrossRef]
- Torrealba, M.P.; Manfrere, K.C.; Miyashiro, D.; Lima, J.F.; Oliveira, L.D.M.; Pereira, N.Z.; Cury-Martins, J.; Pereira, J.; Duarte, A.J.; Sato, M.N.; et al. Chronic activation profile of circulating CD8+ T cells in Sézary syndrome. Oncotarget 2018, 9, 3497–3506. [Google Scholar] [CrossRef] [Green Version]
- Saed, G.; Fivenson, D.P.; Naidu, Y.; Nickoloff, B.J. Mycosis Fungoides Exhibits a Th1-Type Cell-Mediated Cytokine Profile Whereas Sezary Syndrome Express a Th2-Type Profile. J. Investig. Dermatol. 1994, 103, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Samimi, S.; Benoit, B.; Evans, K.; Wherry, E.J.; Showe, L.; Wysocka, M.; Rook, A.H. Increased programmed death-1 expression on CD4+ T cells in cutaneous T-cell lymphoma: Implications for immune suppression. Arch. Dermatol. 2010, 146, 1382–1388. [Google Scholar] [CrossRef] [Green Version]
- Guenova, E.; Watanabe, R.; Teague, J.E.; Desimone, J.A.; Jiang, Y.; Dowlatshahi, M.; Schlapbach, C.; Schaekel, K.; Rook, A.H.; Tawa, M.; et al. TH2 cytokines from malignant cells suppress TH1 responses and enforce a global TH2 bias in leukemic cutaneous T-cell lymphoma. Clin. Cancer Res. 2013, 19, 3755–3763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durgin, J.S.; Weiner, D.M.; Wysocka, M.; Rook, A.H. The immunopathogenesis and immunotherapy of cutaneous T cell lymphoma: Pathways and targets for immune restoration and tumor eradication. J. Am. Acad. Dermatol. 2021, 84, 587–595. [Google Scholar] [CrossRef]
- Woollard, W.J.; Pullabhatla, V.; Lorenc, A.; Patel, V.M.; Butler, R.M.; Bayega, A.; Begum, N.; Bakr, F.; Dedhia, K.; Fisher, J.; et al. Candidate driver genes involved in genome maintenance and DNA repair in Sézary syndrome. Blood 2016, 127, 3387–3397. [Google Scholar] [CrossRef] [Green Version]
- McGirt, L.Y.; Jia, P.; Baerenwald, D.A.; Duszynski, R.J.; Dahlman, K.B.; Zic, J.A.; Zwerner, J.P.; Hucks, D.; Dave, U.; Zhao, Z.; et al. Whole-genome sequencing reveals oncogenic mutations in mycosis fungoides. Blood 2015, 126, 508–519. [Google Scholar] [CrossRef] [Green Version]
- Willerslev-Olsen, A.; Krejsgaard, T.F.; Lindahl, L.M.; Litvinov, I.V.; Fredholm, S.; Petersen, D.L.; Nastasi, C.; Gniadecki, R.; Mongan, N.P.; Sasseville, D.; et al. Staphylococcal enterotoxin A (SEA) stimulates STAT3 activation and IL-17 expression in cutaneous T-cell lymphoma. Blood 2016, 127, 1287–1296. [Google Scholar] [CrossRef]
- Kopp, K.L.; Ralfkiaer, U.; Gjerdrum, L.M.R.; Helvad, R.; Pedersen, I.H.; Litman, T.; Jønson, L.; Hagedorn, P.H.; Krejsgaard, T.; Gniadecki, R.; et al. STAT5-mediated expression of oncogenic miR-155 in cutaneous T-cell lymphoma. Cell Cycle 2013, 12, 1939–1947. [Google Scholar] [CrossRef] [PubMed]
- Litvinov, I.V.; Cordeiro, B.; Fredholm, S.; Odum, N.; Zargham, H.; Huang, Y.; Zhou, Y.; Pehr, K.; Kupper, T.S.; Woetmann, A.; et al. Analysis of STAT4 expression in cutaneous T-cell lymphoma (CTCL) patients and patient-derived cell lines. Cell Cycle 2014, 13, 2975–2982. [Google Scholar] [CrossRef] [Green Version]
- Ho, I.-C.; Tai, T.-S.; Pai, S.-Y. GATA3 and the T-cell lineage: Essential functions before and after T-helper-2-cell differentiation. Nat. Rev. Immunol. 2009, 9, 125–135. [Google Scholar] [CrossRef] [Green Version]
- Stolearenco, V.; Namini, M.R.J.; Hasselager, S.S.; Gluud, M.; Buus, T.B.; Willerslev-Olsen, A.; Ødum, N.; Krejsgaard, T. Cellular Interactions and Inflammation in the Pathogenesis of Cutaneous T-Cell Lymphoma. Front. Cell Dev. Biol. 2020, 8, 851. [Google Scholar] [CrossRef]
- Vermeer, M.H.; Van Doorn, R.; Dukers, D.; Bekkenk, M.W.; Meijer, C.J.; Willemze, R. CD8+ T Cells in Cutaneous T-Cell Lymphoma: Expression of Cytotoxic Proteins, Fas Ligand, and Killing Inhibitory Receptors and Their Relationship With Clinical Behavior. J. Clin. Oncol. 2001, 19, 4322–4329. [Google Scholar] [CrossRef]
- Tang, R.; Rangachari, M.; Kuchroo, V.K. Tim-3: A co-receptor with diverse roles in T cell exhaustion and tolerance. Semin. Immunol. 2019, 42, 101302. [Google Scholar] [CrossRef]
- Canale, F.P.; Ramello, M.C.; Núñez, N.; Furlan, C.L.A.; Bossio, S.N.; Serrán, M.G.; Boari, J.T.; del Castillo, A.; Ledesma, M.; Sedlik, C.; et al. CD39 Expression Defines Cell Exhaustion in Tumor-Infiltrating CD8+ T Cells. Cancer Res. 2018, 78, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Jabri, B.; Abadie, V. IL-15 functions as a danger signal to regulate tissue-resident T cells and tissue destruction. Nat. Rev. Immunol. 2015, 15, 771–783. [Google Scholar] [CrossRef]
- Qin, J.-Z.; Kamarashev, J.; Zhang, C.-L.; Dummer, R.; Burg, G.; Döbbeling, U. Constitutive and Interleukin-7- and Interleukin-15-Stimulated DNA Binding of STAT and Novel Factors in Cutaneous T Cell Lymphoma Cells. J. Investig. Dermatol. 2001, 117, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Tripathi, P.; Kurtulus, S.; Wojciechowski, S.; Sholl, A.; Hoebe, K.; Morris, S.C.; Finkelman, F.D.; Grimes, H.L.; Hildeman, D.A. STAT5 Is Critical To Maintain Effector CD8+T Cell Responses. J. Immunol. 2010, 185, 2116–2124. [Google Scholar] [CrossRef] [Green Version]
- Kurtulus, S.; Tripathi, P.; Moreno-Fernandez, M.E.; Sholl, A.; Katz, J.D.; Grimes, H.L.; Hildeman, D.A. Bcl-2 Allows Effector and Memory CD8+ T Cells To Tolerate Higher Expression of Bim. J. Immunol. 2011, 186, 5729–5737. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, D.; Melão, A.; Van Boxtel, R.; Santos, C.I.; Silva, A.; Silva, M.C.; Cardoso, B.A.; Coffer, P.J.; Barata, J.T. STAT5 is essential for IL-7–mediated viability, growth, and proliferation of T-cell acute lymphoblastic leukemia cells. Blood Adv. 2018, 2, 2199–2213. [Google Scholar] [CrossRef] [PubMed]
- Berger, C.L.; Tigelaar, R.; Cohen, J.; Mariwalla, K.; Trinh, J.; Wang, N.; Edelson, R.L. Cutaneous T-cell lymphoma: Malignant proliferation of T-regulatory cells. Blood 2005, 105, 1640–1647. [Google Scholar] [CrossRef]
- Safinia, N.; Scottà, C.; Vaikunthanathan, T.; Lechler, R.I.; Lombardi, G. Regulatory T Cells: Serious Contenders in the Promise for Immunological Tolerance in Transplantation. Front. Immunol. 2015, 6, 438. [Google Scholar] [CrossRef] [Green Version]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Krejsgaard, T.; Odum, N.; Geisler, C.; Wasik, M.A.; Woetmann, A. Regulatory T cells and immunodeficiency in mycosis fungoides and Sézary syndrome. Leukemia 2012, 26, 424–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heid, J.B.; Schmidt, A.; Oberle, N.; Goerdt, S.; Krammer, P.H.; Suri-Payer, E.; Klemke, C.D. FOXP3+CD25- tumor cells with regulatory function in Sezary syndrome. J. Investig. Dermatol. 2009, 129, 2875–2885. [Google Scholar] [CrossRef] [Green Version]
- Whittaker, S. Global Patterns of Methylation in Sézary Syndrome Provide Insight into the Role of Epigenetics in Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2016, 136, 1753–1754. [Google Scholar] [CrossRef] [Green Version]
- van Doorn, R.; Slieker, R.C.; Boonk, S.E.; Zoutman, W.H.; Goeman, J.J.; Bagot, M.; Michel, L.; Tensen, C.P.; Willemze, R.; Heijmans, B.T.; et al. Epigenomic Analysis of Sezary Syndrome Defines Patterns of Aberrant DNA Methylation and Identifies Diagnostic Markers. J. Investig. Dermatol. 2016, 136, 1876–1884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiemessen, M.M.; Mitchell, T.J.; Hendry, L.; Whittaker, S.J.; Taams, L.S.; John, S. Lack of Suppressive CD4+CD25+FOXP3+ T Cells in Advanced Stages of Primary Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2006, 126, 2217–2223. [Google Scholar] [CrossRef] [Green Version]
- Krejsgaard, T.; Gjerdrum, L.M.; Ralfkiaer, E.; Lauenborg, B.; Eriksen, K.W.; Mathiesen, A.-M.; Bovin, L.F.; Gniadecki, R.; Geisler, C.; Ryder, L.P.; et al. Malignant Tregs express low molecular splice forms of FOXP3 in Sézary syndrome. Leukemia 2008, 22, 2230–2239. [Google Scholar] [CrossRef]
- Krejsgaard, T.F.; Willerslev-Olsen, A.; Lindahl, L.M.; Bonefeld, C.M.; Koralov, S.; Geisler, C.; Wasik, M.A.; Gniadecki, R.; Kilian, M.; Iversen, L.; et al. Staphylococcal enterotoxins stimulate lymphoma-associated immune dysregulation. Blood 2014, 124, 761–770. [Google Scholar] [CrossRef]
- Willerslev-Olsen, A.; Buus, T.B.; Nastasi, C.; Blümel, E.; Gluud, M.; Bonefeld, C.M.; Geisler, C.; Lindahl, L.M.; Vermeer, M.; Wasik, M.A.; et al. Staphylococcus aureus enterotoxins induce FOXP3 in neoplastic T cells in Sézary syndrome. Blood Cancer J. 2020, 10, 57. [Google Scholar] [CrossRef]
- Saulite, I.; Ignatova, D.; Chang, Y.-T.; Fassnacht, C.; Dimitriou, F.; Varypataki, E.; Anzengruber, F.; Nägeli, M.; Cozzio, A.; Dummer, R.; et al. Blockade of programmed cell death protein 1 (PD-1) in Sézary syndrome reduces Th2 phenotype of non-tumoral T lymphocytes but may enhance tumor proliferation. OncoImmunology 2020, 9, 1738797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geskin, L.J.; Akilov, O.E.; Kwon, S.; Schowalter, M.; Watkins, S.; Whiteside, T.L.; Butterfield, L.H.; Falo, L.D. Therapeutic reduction of cell-mediated immunosuppression in mycosis fungoides and Sézary syndrome. Cancer Immunol. Immunother. 2018, 67, 423–434. [Google Scholar] [CrossRef]
- Catalán, D.; Mansilla, M.A.; Ferrier, A.; Soto, L.; Oleinika, K.; Aguillón, J.C.; Aravena, O. Immunosuppressive Mechanisms of Regulatory B Cells. Front. Immunol. 2021, 12, 611795. [Google Scholar] [CrossRef] [PubMed]
- Akatsuka, T.; Miyagaki, T.; Nakajima, R.; Kamijo, H.; Oka, T.; Takahashi, N.; Suga, H.; Yoshizaki, A.; Asano, Y.; Sugaya, M.; et al. Decreased IL-10-producing regulatory B cells in patients with advanced mycosis fungoides. Eur. J. Dermatol. 2018, 28, 314–319. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Qian, H.; Liu, Y.; Duan, L.; Li, Y.; Shi, G. The Roles of Regulatory B Cells in Cancer. J. Immunol. Res. 2014, 2014, 215471. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.; Fehniger, T.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Maghazachi, A.A. Role of Chemokines in the Biology of Natural Killer Cells. Curr. Top. Microbiol. Immunol. 2010, 341, 37–58. [Google Scholar] [CrossRef] [PubMed]
- Groh, V.; Wu, J.; Yee, C.; Spies, T. Tumour-derived soluble MIC ligands impair expression of NKG2D and T-cell activation. Nature 2002, 419, 734–738. [Google Scholar] [CrossRef]
- Manfrere, K.C.G.; Torrealba, M.P.; Miyashiro, D.; Pereira, N.Z.; Yoshikawa, F.; Oliveira, L.D.M.; Cury-Martins, J.; Duarte, A.J.; Sanches, J.A.; Sato, M.N. Profile of differentially expressed Toll-like receptor signaling genes in the natural killer cells of patients with Sézary syndrome. Oncotarget 2017, 8, 92183–92194. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Nakayama, M.; Kawano, M.; Ishii, T.; Harigae, H.; Ogasawara, K. NK-cell fratricide: Dynamic crosstalk between NK and cancer cells. OncoImmunology 2013, 2, e26529. [Google Scholar] [CrossRef] [Green Version]
- Bouaziz, J.-D.; Ortonne, N.; Giustiniani, J.; Schiavon, V.; Huet, D.; Bagot, M.; Bensussan, A. Circulating Natural Killer Lymphocytes Are Potential Cytotoxic Effectors Against Autologous Malignant Cells in Sezary Syndrome Patients. J. Investig. Dermatol. 2005, 125, 1273–1278. [Google Scholar] [CrossRef]
- Eisele, G.; Wischhusen, J.; Mittelbronn, M.; Meyermann, R.; Waldhauer, I.; Steinle, A.; Weller, M.; Friese, M.A. TGF-beta and metalloproteinases differentially suppress NKG2D ligand surface expression on malignant glioma cells. Brain 2006, 129 Pt 9, 2416–2425. [Google Scholar] [CrossRef]
- Kared, H.; Martelli, S.; Ng, T.P.; Pender, S.L.; Larbi, A. CD57 in human natural killer cells and T-lymphocytes. Cancer Immunol. Immunother. 2016, 65, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Lerias, J.R.; de Sousa, E.; Paraschoudi, G.; Martins, J.; Condeco, C.; Figueiredo, N.; Carvalho, C.; Dodoo, E.; Maia, A.; Castillo-Martin, M.; et al. Trained Immunity for Personalized Cancer Immunotherapy: Current Knowledge and Future Opportunities. Front. Microbiol. 2019, 10, 2924. [Google Scholar] [CrossRef]
- Fujii, K. New Therapies and Immunological Findings in Cutaneous T-Cell Lymphoma. Front. Oncol. 2018, 8, 198. [Google Scholar] [CrossRef] [PubMed]
- Thumann, P.; Luftl, M.; Moc, I.; Bagot, M.; Bensussan, A.; Schuler, G.; Jenne, L. Interaction of cutaneous lymphoma cells with reactive T cells and dendritic cells: Implications for dendritic cell-based immunotherapy. Br. J. Dermatol. 2003, 149, 1128–1142. [Google Scholar] [CrossRef]
- Luftl, M.; Feng, A.; Licha, E.; Schuler, G. Dendritic cells and apoptosis in mycosis fungoides. Br. J. Dermatol. 2002, 147, 1171–1179. [Google Scholar] [CrossRef]
- Schlapbach, C.; Ochsenbein, A.; Kaelin, U.; Hassan, A.S.; Hunger, R.E.; Yawalkar, N. High numbers of DC-SIGN+ dendritic cells in lesional skin of cutaneous T-cell lymphoma. J. Am. Acad. Dermatol. 2010, 62, 995–1004. [Google Scholar] [CrossRef]
- He, T.; Tang, C.; Xu, S.; Moyana, T.; Xiang, J. Interferon gamma stimulates cellular maturation of dendritic cell line DC2.4 leading to induction of efficient cytotoxic T cell responses and antitumor immunity. Cell. Mol. Immunol. 2007, 4, 105–111. [Google Scholar]
- Tada, K.; Hamada, T.; Asagoe, K.; Umemura, H.; Mizuno-Ikeda, K.; Aoyama, Y.; Otsuka, M.; Yamasaki, O.; Iwatsuki, K. Increase of DC-LAMP+ mature dendritic cell subsets in dermatopathic lymphadenitis of mycosis fungoides. Eur. J. Dermatol. 2014, 24, 670–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysocka, M.; Benoit, B.M.; Newton, S.; Azzoni, L.; Montaner, L.J.; Rook, A.H. Enhancement of the host immune responses in cutaneous T-cell lymphoma by CpG oligodeoxynucleotides and IL-15. Blood 2004, 104, 4142–4149. [Google Scholar] [CrossRef]
- Wysocka, M.; Zaki, M.H.; French, L.E.; Chehimi, J.; Shapiro, M.; Everetts, S.E.; McGinnis, K.S.; Montaner, L.; Rook, A.H. Sezary syndrome patients demonstrate a defect in dendritic cell populations: Effects of CD40 ligand and treatment with GM-CSF on dendritic cell numbers and the production of cytokines. Blood 2002, 100, 3287–3294. [Google Scholar] [CrossRef] [Green Version]
- Swiecki, M.; Colonna, M. The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 2015, 15, 471–485. [Google Scholar] [CrossRef]
- Tadmor, T.; Attias, D.; Polliack, A. Myeloid-derived suppressor cells—their role in haemato-oncological malignancies and other cancers and possible implications for therapy. Br. J. Haematol. 2011, 153, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Papafragkos, I.; Markaki, E.; Kalpadakis, C.; Verginis, P. Decoding the Myeloid-Derived Suppressor Cells in Lymphoid Malignancies. J. Clin. Med. 2021, 10, 3462. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, P.C.; Quiceno, D.G.; Ochoa, A.C. L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 2007, 109, 1568–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzoni, A.; Bronte, V.; Visintin, A.; Spitzer, J.H.; Apolloni, E.; Serafini, P.; Zanovello, P.; Segal, D.M. Myeloid Suppressor Lines Inhibit T Cell Responses by an NO-Dependent Mechanism. J. Immunol. 2002, 168, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Wang, X.; Wang, L.; Ma, X.; Gong, Z.; Zhang, S.; Li, Y. Targeting the IDO1 pathway in cancer: From bench to bedside. J. Hematol. Oncol. 2018, 11, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ku, A.W.; Muhitch, J.; Powers, C.A.; Diehl, M.; Kim, M.; Fisher, D.T.; Sharda, A.P.; Clements, V.K.; O’Loughlin, K.; Minderman, H.; et al. Tumor-induced MDSC act via remote control to inhibit L-selectin-dependent adaptive immunity in lymph nodes. eLife 2016, 5, e17375. [Google Scholar] [CrossRef]
- Ohmatsu, H.; Humme, D.; Gonzalez, J.; Gulati, N.; Möbs, M.; Sterry, W.; Krueger, J.G. IL-32 induces indoleamine 2,3-dioxygenase+CD1c+ dendritic cells and indoleamine 2,3-dioxygenase+CD163+ macrophages: Relevance to mycosis fungoides progression. OncoImmunology 2017, 6, e1181237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Guindy, D.M.; Elgarhy, L.H.; Elkholy, R.A.; Ali, D.A.; Helal, D.S. Potential role of tumor-associated macrophages and CD163/CD68 ratio in mycosis fungoides and Sezary syndrome in correlation with serum sCD163 and CCL22. J. Cutan. Pathol. 2021. [Google Scholar] [CrossRef]
- Islam, S.A.; Ling, M.; Leung, J.; Shreffler, W.G.; Luster, A.D. Identification of human CCR8 as a CCL18 receptor. J. Exp. Med. 2013, 210, 1889–1898. [Google Scholar] [CrossRef]
- Günther, C.; Zimmermann, N.; Berndt, N.; Großer, M.; Stein, A.; Koch, A.; Meurer, M. Up-Regulation of the Chemokine CCL18 by Macrophages Is a Potential Immunomodulatory Pathway in Cutaneous T-Cell Lymphoma. Am. J. Pathol. 2011, 179, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, M.; Miyagaki, T.; Ohmatsu, H.; Suga, H.; Kai, H.; Kamata, M.; Fujita, H.; Asano, Y.; Tada, Y.; Kadono, T.; et al. Association of the numbers of CD163+ cells in lesional skin and serum levels of soluble CD163 with disease progression of cutaneous T cell lymphoma. J. Dermatol. Sci. 2012, 68, 45–51. [Google Scholar] [CrossRef]
- Wu, X.; Schulte, B.C.; Zhou, Y.; Haribhai, D.; Mackinnon, A.; Plaza, J.A.; Williams, C.B.; Hwang, S.T. Depletion of M2-Like Tumor-Associated Macrophages Delays Cutaneous T-Cell Lymphoma Development In Vivo. J. Investig. Dermatol. 2014, 134, 2814–2822. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.D.S.; Banerjee, S.; Kruglov, O.; Viller, N.N.; Horwitz, S.M.; Lesokhin, A.; Zain, J.; Querfeld, C.; Chen, R.; Okada, C.; et al. Targeting CD47 in Sezary syndrome with SIRPalphaFc. Blood Adv. 2019, 3, 1145–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russ, A.; Hua, A.B.; Montfort, W.R.; Rahman, B.; Riaz, I.B.; Khalid, M.U.; Carew, J.S.; Nawrocki, S.T.; Persky, D.; Anwer, F. Blocking “don’t eat me” signal of CD47-SIRPalpha in hematological malignancies, an in-depth review. Blood Rev. 2018, 32, 480–489. [Google Scholar] [CrossRef]
- Fierro, M.T.; Cuffini, A.M.; Novelli, M.; Banche, G.; Allizond, V.; Comessatti, A.; Brizio, M.; Scalas, D.; Merlino, C.; Quaglino, P.; et al. Functional and phenotypical alterations of polymorphonuclear cells in Sézary syndrome patients. Eur. J. Dermatol. 2011, 21, 921–929. [Google Scholar] [CrossRef]
- Masucci, M.T.; Minopoli, M.; del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11, 1749. [Google Scholar] [CrossRef] [PubMed]
- Goddard, D.S.; Yamanaka, K.-I.; Kupper, T.S.; Jones, D.A. Activation of Neutrophils in Cutaneous T-Cell Lymphoma. Clin. Cancer Res. 2005, 11, 8243–8249. [Google Scholar] [CrossRef] [Green Version]
- Cirée, A.; Michel, L.; Camilleri-Bröet, S.; Louis, F.J.; Oster, M.; Flageul, B.; Senet, P.; Fossiez, F.; Fridman, W.H.; Bachelez, H.; et al. Expression and activity of IL-17 in cutaneous T-cell lymphomas (mycosis fungoides and sezary syndrome). Int. J. Cancer 2004, 112, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, F.; Shimizu, K.; Hamasakia, Y.-I.; Tanaka, Y.; Katayama, I.; Yamada, Y.; Tomonaga, M. Spontaneous IL-8 production by CD4(+), CD7(+) leukemia cells in erythrodermic Sézary syndrome. Leuk. Lymphoma 2002, 43, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Vidal, S.; Puig, L.; Carrascosa-Carrillo, J.-M.; González-Cantero, Á.; Ruiz-Carrascosa, J.-C.; Velasco-Pastor, A.-M. From Messengers to Receptors in Psoriasis: The Role of IL-17RA in Disease and Treatment. Int. J. Mol. Sci. 2021, 22, 6740. [Google Scholar] [CrossRef]
- Scala, E.; Cacciapuoti, S.; Garzorz-Stark, N.; Megna, M.; Marasca, C.; Seiringer, P.; Volz, T.; Eyerich, K.; Fabbrocini, G. Hidradenitis Suppurativa: Where We Are and Where We Are Going. Cells 2021, 10, 2094. [Google Scholar] [CrossRef]
- Wu, L.; Saxena, S.; Singh, R.K. Neutrophils in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1224, 1–20. [Google Scholar]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil chemoattractant receptors in health and disease: Double-edged swords. Cell Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef]
- Miyagaki, T.; Sugaya, M.; Suga, H.; Kamata, M.; Ohmatsu, H.; Fujita, H.; Asano, Y.; Tada, Y.; Kadono, T.; Sato, S. IL-22, but Not IL-17, Dominant Environment in Cutaneous T-cell Lymphoma. Clin. Cancer Res. 2011, 17, 7529–7538. [Google Scholar] [CrossRef] [Green Version]
- Krejsgaard, T.; Ralfkiaer, U.; Clasen-Linde, E.; Eriksen, K.W.; Kopp, K.L.; Bonefeld, C.M.; Geisler, C.; Dabelsteen, S.; Wasik, M.A.; Ralfkiaer, E.; et al. Malignant cutaneous T-cell lymphoma cells express IL-17 utilizing the Jak3/Stat3 signaling pathway. J. Investig. Dermatol. 2011, 131, 1331–1338. [Google Scholar] [CrossRef] [Green Version]
- Shelburne, C.P.; Abraham, S.N. The Mast Cell in Innate and Adaptive Immunity. Adv. Exp. Med. Biol. 2011, 716, 162–185. [Google Scholar] [CrossRef]
- Tsai, M.; Grimbaldeston, M.; Galli, S.J. Mast Cells and Immunoregulation/Immunomodulation. Best Pract. Health Care 2011, 716, 186–211. [Google Scholar] [CrossRef]
- Rabenhorst, A.; Schlaak, M.; Heukamp, L.C.; Förster, A.; Theurich, S.; von Bergwelt-Baildon, M.; Büttner, R.; Kurschat, P.; Mauch, C.; Roers, A.; et al. Mast cells play a protumorigenic role in primary cutaneous lymphoma. Blood 2012, 120, 2042–2054. [Google Scholar] [CrossRef] [Green Version]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Marone, G.; Iannone, R.; Marone, G.; Granata, F. Are Mast Cells MASTers in Cancer? Front. Immunol. 2017, 8, 424. [Google Scholar] [CrossRef] [Green Version]
- Eder, J.; Rogojanu, R.; Jerney, W.; Erhart, F.; Dohnal, A.; Kitzwögerer, M.; Steiner, G.; Moser, J.; Trautinger, F. Mast Cells Are Abundant in Primary Cutaneous T-Cell Lymphomas: Results from a Computer-Aided Quantitative Immunohistological Study. PLoS ONE 2016, 11, e0163661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Katayama, I.; Nishioka, K. Role of mast cell and stem cell factor in hyperpigmented mycosis fungoides. Blood 1997, 90, 1338–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siiskonen, H.; Harvima, I. Mast Cells and Sensory Nerves Contribute to Neurogenic Inflammation and Pruritus in Chronic Skin Inflammation. Front. Cell Neurosci. 2019, 13, 422. [Google Scholar] [CrossRef] [PubMed]
- Flier, J.S.; Underhill, L.H.; Weller, P.F. The Immunobiology of Eosinophils. N. Engl. J. Med. 1991, 324, 1110–1118. [Google Scholar] [CrossRef]
- Marzano, A.V.; Genovese, G. Eosinophilic Dermatoses: Recognition and Management. Am. J. Clin. Dermatol. 2020, 21, 525–539. [Google Scholar] [CrossRef]
- Furuta, G.T.; Katzka, D.A. Eosinophilic Esophagitis. N. Engl. J. Med. 2015, 373, 1640–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barry, J.; Gadre, A.; Akuthota, P. Hypersensitivity pneumonitis, allergic bronchopulmonary aspergillosis and other eosinophilic lung diseases. Curr. Opin. Immunol. 2020, 66, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, M. Chemokines and cutaneous lymphoma. J. Dermatol. Sci. 2010, 59, 81–85. [Google Scholar] [CrossRef]
- Davoine, F.; Lacy, P. Eosinophil Cytokines, Chemokines, and Growth Factors: Emerging Roles in Immunity. Front. Immunol. 2014, 5, 570. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Poschke, I.; Kiessling, R. Tumour-induced immune suppression: Role of inflammatory mediators released by myelomonocytic cells. J. Intern. Med. 2014, 276, 154–170. [Google Scholar] [CrossRef]
- Fredholm, S.; Gjerdrum, L.M.R.; Willerslev-Olsen, A.; Petersen, D.L.; Nielsen, I.Ø.; Kauczok, C.-S.; Wobser, M.; Ralfkiaer, U.; Bonefeld, C.M.; Wasik, M.A.; et al. STAT3 activation and infiltration of eosinophil granulocytes in mycosis fungoides. Anticancer. Res. 2014, 34, 5277–5286. [Google Scholar]
- Ionescu, M.A.; Rivet, J.; Daneshpouy, M.; Briere, J.; Morel, P.; Janin, A. In situ eosinophil activation in 26 primary cutaneous T-cell lymphomas with blood eosinophilia. J. Am. Acad. Dermatol. 2005, 52, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Tancrède-Bohin, E.; Ionescu, M.A.; de la Salmonière, P.; Dupuy, A.; Rivet, J.; Rybojad, M.; Dubertret, L.; Bachelez, H.; Lebbé, C.; Morel, P. Prognostic Value of Blood Eosinophilia in Primary Cutaneous T-Cell Lymphomas. Arch. Dermatol. 2004, 140, 1057–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchin, K.R.; Cassin, M.; Gottleib, S.L.; Sood, S.; Cucchiara, A.J.; Vonderheid, E.C.; Rook, A.H. Increased interleukin 5 production in eosinophilic Sézary syndrome: Regulation by interferon alfa and interleukin. J. Am. Acad. Dermatol. 2001, 44, 28–32. [Google Scholar] [CrossRef]
- Fujita, Y.; Abe, R.; Sasaki, M.; Honda, A.; Furuichi, M.; Asano, Y.; Norisugi, O.; Shimizu, T.; Shimizu, H. Presence of Circulating CCR10+ T cells and Elevated Serum CTACK/CCL27 in the Early Stage of Mycosis Fungoides. Clin. Cancer Res. 2006, 12, 2670–2675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugaya, M. Is blocking IL-4 receptor alpha beneficial for patients with mycosis fungoides or Sézary syndrome? J. Dermatol. 2021, 48, e225–e226. [Google Scholar] [CrossRef]
- Miyagaki, T.; Sugaya, M.; Fujita, H.; Saeki, H.; Tamaki, K. Increased serum thymic stromal lymphopoietin levels in patients with cutaneous T cell lymphoma. Clin. Exp. Dermatol. 2009, 34, 539–540. [Google Scholar] [CrossRef] [PubMed]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell–mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef]
- Herrera, A.; Fredholm, S.; Cheng, A.; Mimitou, E.P.; Seffens, A.; Bar-Natan, M.; Sun, A.; Latkowski, J.-A.; Willerslew-Olsen, A.; Buus, T.B.; et al. Low SATB1 Expression Promotes IL-5 and IL-9 Expression in Sézary Syndrome. J. Investig. Dermatol. 2020, 140, 713–716. [Google Scholar] [CrossRef]
- Nakajima, R.; Miyagaki, T.; Hirakawa, M.; Oka, T.; Takahashi, N.; Suga, H.; Yoshizaki, A.; Fujita, H.; Asano, Y.; Sugaya, M.; et al. Interleukin-25 is involved in cutaneous T-cell lymphoma progression by establishing a T helper 2-dominant microenvironment. Br. J. Dermatol. 2018, 178, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Geskin, L.J.; Viragova, S.; Stolz, D.B.; Fuschiotti, P. Interleukin-13 is overexpressed in cutaneous T-cell lymphoma cells and regulates their proliferation. Blood 2015, 125, 2798–2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottevanger, R.; van Beugen, S.; Evers, A.; Willemze, R.; Vermeer, M.; Quint, K. Quality of life in patients with Mycosis Fungoides and Sézary Syndrome: A systematic review of the literature. J. Eur. Acad. Dermatol. Venereol. 2021, 35, 2377–2387. [Google Scholar] [CrossRef]
- Suga, H.; Sugaya, M.; Miyagaki, T.; Ohmatsu, H.; Fujita, H.; Kagami, S.; Asano, Y.; Tada, Y.; Kadono, T.; Sato, S. Association of Nerve Growth Factor, Chemokine (C-C motif) Ligands and Immunoglobulin E with Pruritus in Cutaneous T-cell Lymphoma. Acta Derm. Venereol. 2013, 93, 144–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagaki, T.; Sugaya, M.; Suga, H.; Akamata, K.; Ohmatsu, H.; Fujita, H.; Asano, Y.; Tada, Y.; Kadono, T.; Sato, S. Angiogenin levels are increased in lesional skin and sera in patients with erythrodermic cutaneous T cell lymphoma. Arch. Dermatol. Res. 2012, 304, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Kohnken, R.; Fabbro, S.; Hastings, J.; Porcu, P.; Mishra, A. Sézary Syndrome: Clinical and Biological Aspects. Curr. Hematol. Malign. Rep. 2016, 11, 468–479. [Google Scholar] [CrossRef]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, definitions, and functions in health and disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Mao, X.; Xu, J.; Wang, W.; Liang, C.; Hua, J.; Liu, J.; Zhang, B.; Meng, Q.; Yu, X.; Shi, S. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: New findings and future perspectives. Mol. Cancer 2021, 20, 131. [Google Scholar] [CrossRef] [PubMed]
- Miyagaki, T.; Sugaya, M.; Suga, H.; Morimura, S.; Ohmatsu, H.; Fujita, H.; Asano, Y.; Tada, Y.; Kadono, T.; Sato, S. Low Herpesvirus Entry Mediator (HVEM) Expression on Dermal Fibroblasts Contributes to a Th2-Dominant Microenvironment in Advanced Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2012, 132, 1280–1289. [Google Scholar] [CrossRef] [Green Version]
- Narducci, M.G.; Scala, E.; Bresin, A.; Caprini, E.; Picchio, M.C.; Remotti, D.; Ragone, G.; Nasorri, F.; Frontani, M.; Arcelli, D.; et al. Skin homing of Sezary cells involves SDF-1-CXCR4 signaling and down-regulation of CD26/dipeptidylpeptidase IV. Blood 2006, 107, 1108–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzec, M.; Halasa, K.; Kasprzycka, M.; Wysocka, M.; Liu, X.; Tobias, J.W.; Baldwin, N.; Zhang, Q.; Odum, N.; Rook, A.H.; et al. Differential Effects of Interleukin-2 and Interleukin-15 versus Interleukin-21 on CD4+ Cutaneous T-Cell Lymphoma Cells. Cancer Res. 2008, 68, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- Mishra, A.; la Perle, K.; Kwiatkowski, S.; Sullivan, L.A.; Sams, G.H.; Johns, J.; Curphey, D.P.; Wen, J.; McConnell, K.; Qi, J.; et al. Mechanism, Consequences, and Therapeutic Targeting of Abnormal IL15 Signaling in Cutaneous T-cell Lymphoma. Cancer Discov. 2016, 6, 986–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suga, H.; Sugaya, M.; Miyagaki, T.; Kawaguchi, M.; Fujita, H.; Asano, Y.; Tada, Y.; Kadono, T.; Sato, S. The Role of IL-32 in Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2014, 134, 1428–1435. [Google Scholar] [CrossRef] [Green Version]
- Kopp, K.L.M.; Kauczok, C.S.; Lauenborg, B.; Krejsgaard, T.; Eriksen, K.W.; Zhang, Q.; Wasik, M.A.; Geisler, C.; Ralfkiaer, E.; Becker, J.C.; et al. COX-2-dependent PGE2 acts as a growth factor in mycosis fungoides (MF). Leukemia 2010, 24, 1179–1185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, H.Y.; Wei, F.; Liu, X.; Paterson, J.C.; Roy, D.; Mihova, D.; Woetmann, A.; Ptasznik, A.; Odum, N.; et al. Cutaneous T Cell Lymphoma Expresses Immunosuppressive CD80 (B7-1) Cell Surface Protein in a STAT5-Dependent Manner. J. Immunol. 2014, 192, 2913–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contassot, E.; Kerl, K.; Roques, S.; Shane, R.; Gaide, O.; Dupuis, M.; Rook, A.H.; French, L.E. Resistance to FasL and tumor necrosis factor-related apoptosis-inducing ligand-mediated apoptosis in Sézary syndrome T-cells associated with impaired death receptor and FLICE-inhibitory protein expression. Blood 2008, 111, 4780–4787. [Google Scholar] [CrossRef] [Green Version]
- Rendón-Serna, N.; Correa-Londoño, L.A.; Velásquez-Lopera, M.M.; Bermudez-Muñoz, M. Cell signaling in cutaneous T-cell lymphoma microenvironment: Promising targets for molecular-specific treatment. Int. J. Dermatol. 2021, 60, 1462–1480. [Google Scholar] [CrossRef]
- Pileri, A.; Agostinelli, C.; Righi, S.; Fuligni, F.; Bacci, F.; Sabattini, E.; Patrizi, A.; Pileri, S.A.; Piccaluga, P.P. Vascular endothelial growth factor A (VEGFA) expression in mycosis fungoides. Histopathology 2014, 66, 173–181. [Google Scholar] [CrossRef]
- Karpova, M.B.; Fujii, K.; Jenni, D.; Dummer, R.; Urosevic-Maiwald, M. Evaluation of lymphangiogenic markers in Sézary syndrome. Leuk. Lymphoma 2010, 52, 491–501. [Google Scholar] [CrossRef]
- Sakamoto, M.; Miyagaki, T.; Kamijo, H.; Oka, T.; Takahashi, N.; Suga, H.; Yoshizaki, A.; Asano, Y.; Sugaya, M.; Sato, S. Serum vascular endothelial growth factor A levels reflect itch severity in mycosis fungoides and Sézary syndrome. J. Dermatol. 2018, 45, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Jankowska-Konsur, A.; Kobierzycki, C.; Grzegrzółka, J.; Piotrowska, A.; Gomulkiewicz, A.; Glatzel-Plucinska, N.; Reich, A.; Podhorska-Okołów, M.; Dzięgiel, P.; Szepietowski, J. Podoplanin Expression Correlates with Disease Progression in Mycosis Fungoides. Acta Derm. Venereol. 2017, 97, 235–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kähäri, V.-M.; Saarialho-Kere, U. Matrix metalloproteinases in skin. Exp. Dermatol. 1997, 6, 199–213. [Google Scholar] [CrossRef]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Lambert, E.; Dassé, E.; Haye, B.; Petitfrère, E. TIMPs as multifacial proteins. Crit. Rev. Oncol. 2004, 49, 187–198. [Google Scholar] [CrossRef]
- Rasheed, H.; Fawzi, M.M.T.; Abdel-Halim, M.R.E.; Eissa, A.M.; Salem, N.M.; Mahfouz, S. Immunohistochemical Study of the Expression of Matrix Metalloproteinase-9 in Skin Lesions of Mycosis Fungoides. Am. J. Dermatopathol. 2010, 32, 162–169. [Google Scholar] [CrossRef]
- Napoli, S.; Scuderi, C.; Gattuso, G.; di Bella, V.; Candido, S.; Basile, M.S.; Libra, M.; Falzone, L. Functional Roles of Matrix Metalloproteinases and Their Inhibitors in Melanoma. Cells 2020, 9, 1151. [Google Scholar] [CrossRef]
- Lee, M.; Kistler, C.; Hartmann, T.B.; Li, F.; Dummer, R.; Dippel, E.; Booken, N.; Klemke, C.D.; Schadendorf, D.; Eichmüller, S.B. Immunoscreening of a cutaneous T-cell lymphoma library for plasma membrane proteins. Cancer Immunol. Immunother. 2007, 56, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Talpur, R.; Bassett, R.; Duvic, M. Prevalence and treatment of Staphylococcus aureus colonization in patients with mycosis fungoides and Sézary syndrome. Br. J. Dermatol. 2008, 159, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Mitsui, H.; Witte, K.; Gellrich, S.; Gulati, N.; Humme, D.; Witte, E.; Gonsior, M.; Beyer, M.; Kadin, M.E.; et al. Deficient Cutaneous Antibacterial Competence in Cutaneous T-Cell Lymphomas: Role of Th2-Mediated Biased Th17 Function. Clin. Cancer Res. 2014, 20, 5507–5516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modenutti, C.; Capurro, J.I.B.; di Lella, S.; Martí, M.A. The Structural Biology of Galectin-Ligand Recognition: Current Advances in Modeling Tools, Protein Engineering, and Inhibitor Design. Front. Chem. 2019, 7, 823. [Google Scholar] [CrossRef]
- Giordano, M.; Croci, D.O.; Rabinovich, G.A. Galectins in hematological malignancies. Curr. Opin. Hematol. 2013, 20, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Cedeno-Laurent, F.; Watanabe, R.; Teague, J.E.; Kupper, T.S.; Clark, R.A.; Dimitroff, C.J. Galectin-1 inhibits the viability, proliferation, and Th1 cytokine production of nonmalignant T cells in patients with leukemic cutaneous T-cell lymphoma. Blood 2012, 119, 3534–3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thode, C.; Woetmann, A.; Wandall, H.H.; Carlsson, M.C.; Qvortrup, K.; Kauczok, C.S.; Wobser, M.; Printzlau, A.; Ødum, N.; Dabelsteen, S. Malignant T Cells Secrete Galectins and Induce Epidermal Hyperproliferation and Disorganized Stratification in a Skin Model of Cutaneous T-Cell Lymphoma. J. Investig. Dermatol. 2015, 135, 238–246. [Google Scholar] [CrossRef] [Green Version]
- Koh, H.S.; Lee, C.; Lee, K.S.; Park, E.J.; Seong, R.H.; Hong, S.; Jeon, S.H. Twist2 regulates CD7 expression and galectin-1-induced apoptosis in mature T-cells. Mol. Cells 2009, 28, 553–558. [Google Scholar] [CrossRef]
- Rappl, G.; Abken, H.; Muche, J.M.; Sterry, W.; Tilgen, W.; André, S.; Kaltner, H.; Ugurel, S.; Gabius, H.-J.; Reinhold, U. CD4+CD7− leukemic T cells from patients with Sézary syndrome are protected from galectin-1-triggered T cell death. Leukemia 2002, 16, 840–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.A.; Amano, M.; Felten, C.; Galvan, M.; Sulur, G.; Pinter-Brown, L.; Dobbeling, U.; Burg, G.; Said, J.; Baum, L.G. Galectin-1–Mediated Apoptosis in Mycosis Fungoides: The Roles of CD7 and Cell Surface Glycosylation. Mod. Pathol. 2003, 16, 543–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, R.; Miyagaki, T.; Kamijo, H.; Oka, T.; Shishido, N.; Suga, H.; Sugaya, M.; Sato, S. Possible therapeutic applicability of galectin-9 in cutaneous T-cell lymphoma. J. Dermatol. Sci. 2019, 96, 134–142. [Google Scholar] [CrossRef] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miyashiro, D.; Souza, B.d.C.e.; Torrealba, M.P.; Manfrere, K.C.G.; Sato, M.N.; Sanches, J.A. The Role of Tumor Microenvironment in the Pathogenesis of Sézary Syndrome. Int. J. Mol. Sci. 2022, 23, 936. https://doi.org/10.3390/ijms23020936
Miyashiro D, Souza BdCe, Torrealba MP, Manfrere KCG, Sato MN, Sanches JA. The Role of Tumor Microenvironment in the Pathogenesis of Sézary Syndrome. International Journal of Molecular Sciences. 2022; 23(2):936. https://doi.org/10.3390/ijms23020936
Chicago/Turabian StyleMiyashiro, Denis, Bruno de Castro e Souza, Marina Passos Torrealba, Kelly Cristina Gomes Manfrere, Maria Notomi Sato, and José Antonio Sanches. 2022. "The Role of Tumor Microenvironment in the Pathogenesis of Sézary Syndrome" International Journal of Molecular Sciences 23, no. 2: 936. https://doi.org/10.3390/ijms23020936