
Granulocytic MDSC with Deficient CCR5 Alleviates Lipogenesis and Inflammation in Nonalcoholic Fatty Liver Disease

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
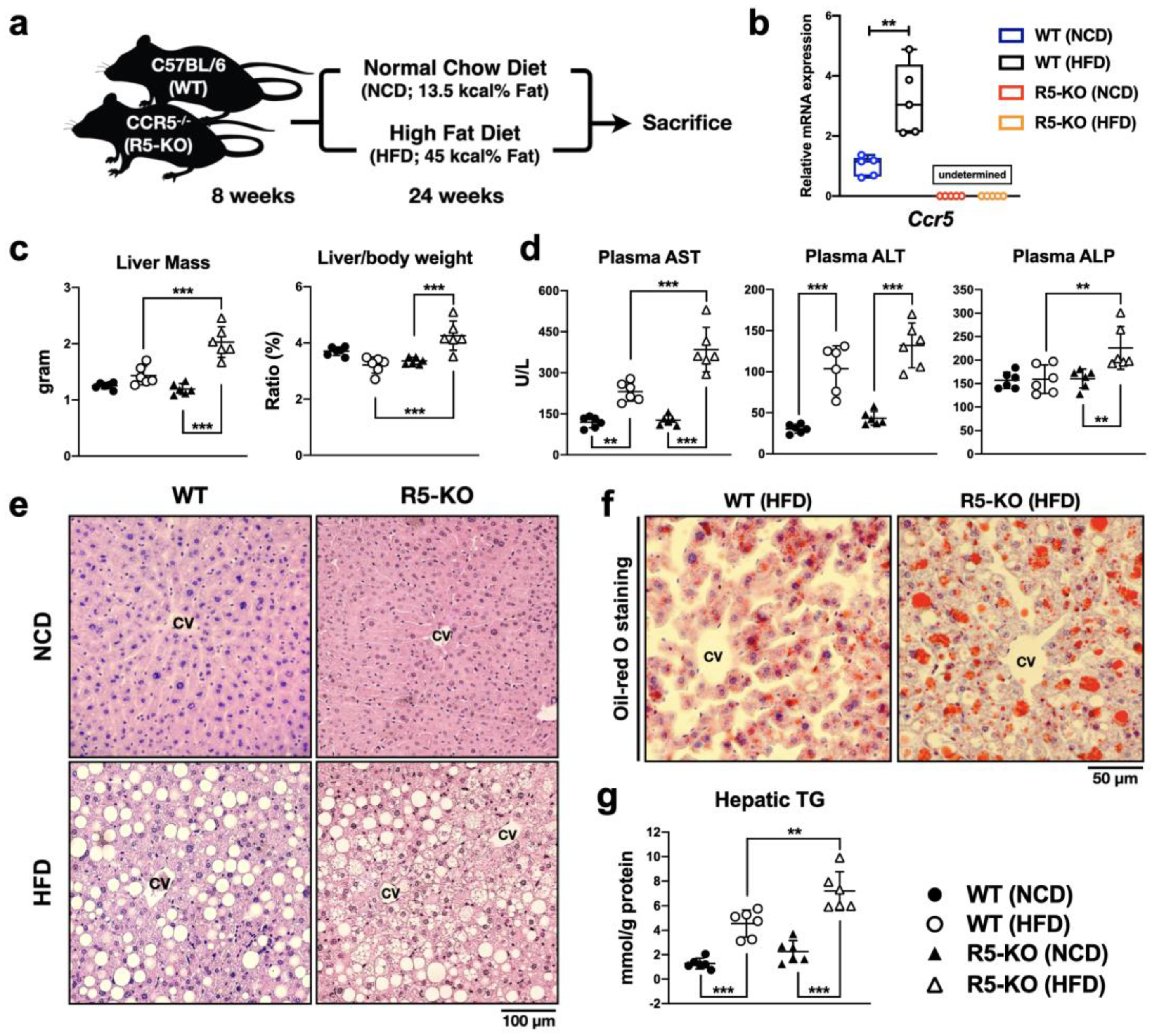
2.1. Global Deficiency of CCR5 Exacerbates Liver Damage and Steatosis in NAFLD
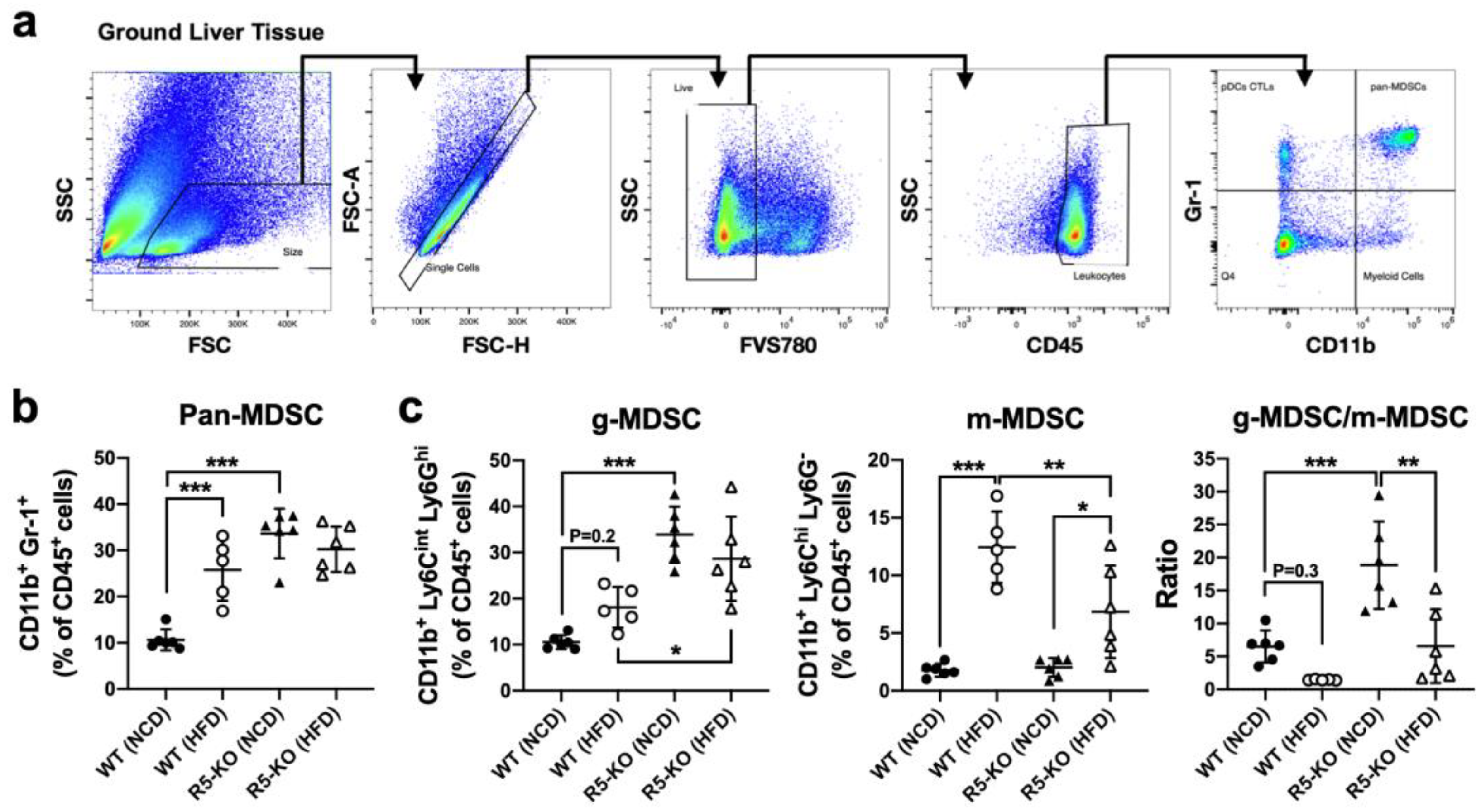
2.2. Global Deficiency of CCR5 Elevates Hepatic g-MDSC in Both Lean and Obese Mice
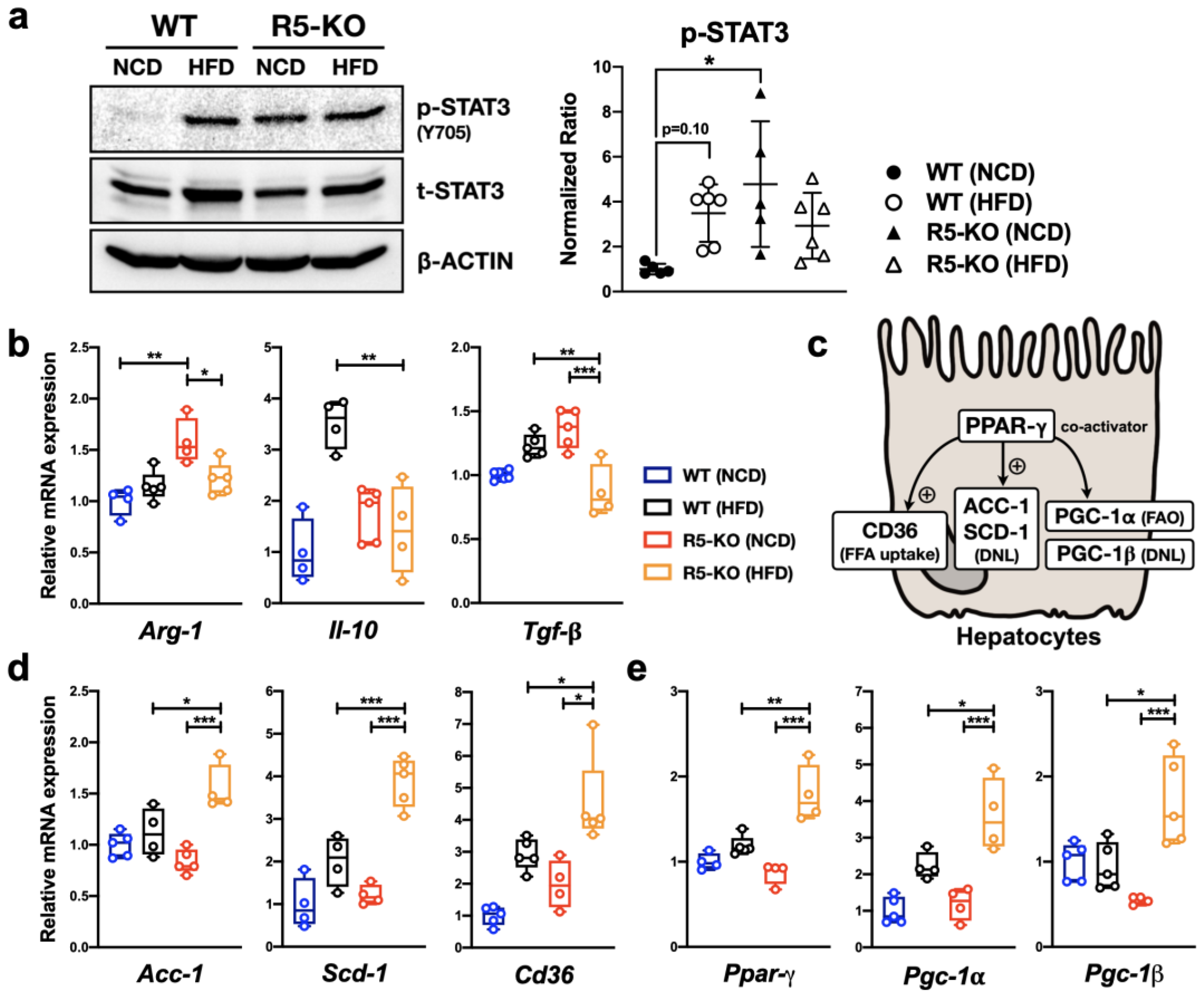
2.3. Global Deficiency of CCR5 and HFD Both Activate Hepatic STAT3
2.4. Global Deficiency of CCR5 Has Distinct Effects on Cytokine Gene Expression
2.5. Global Deficiency of CCR5 Synergizes with HFD in Regulating Lipid-Metabolic Genes
2.6. CCR5 Signaling Modulates Anti-Inflammatory Profile of g-MDSC in the Hepatic Microenvironment
2.7. CCR5 Regulates STAT3 Signaling Differently in Primary Hepatocytes
2.8. CCR5 Regulates Hepatocyte Lipid Metabolism through STAT3
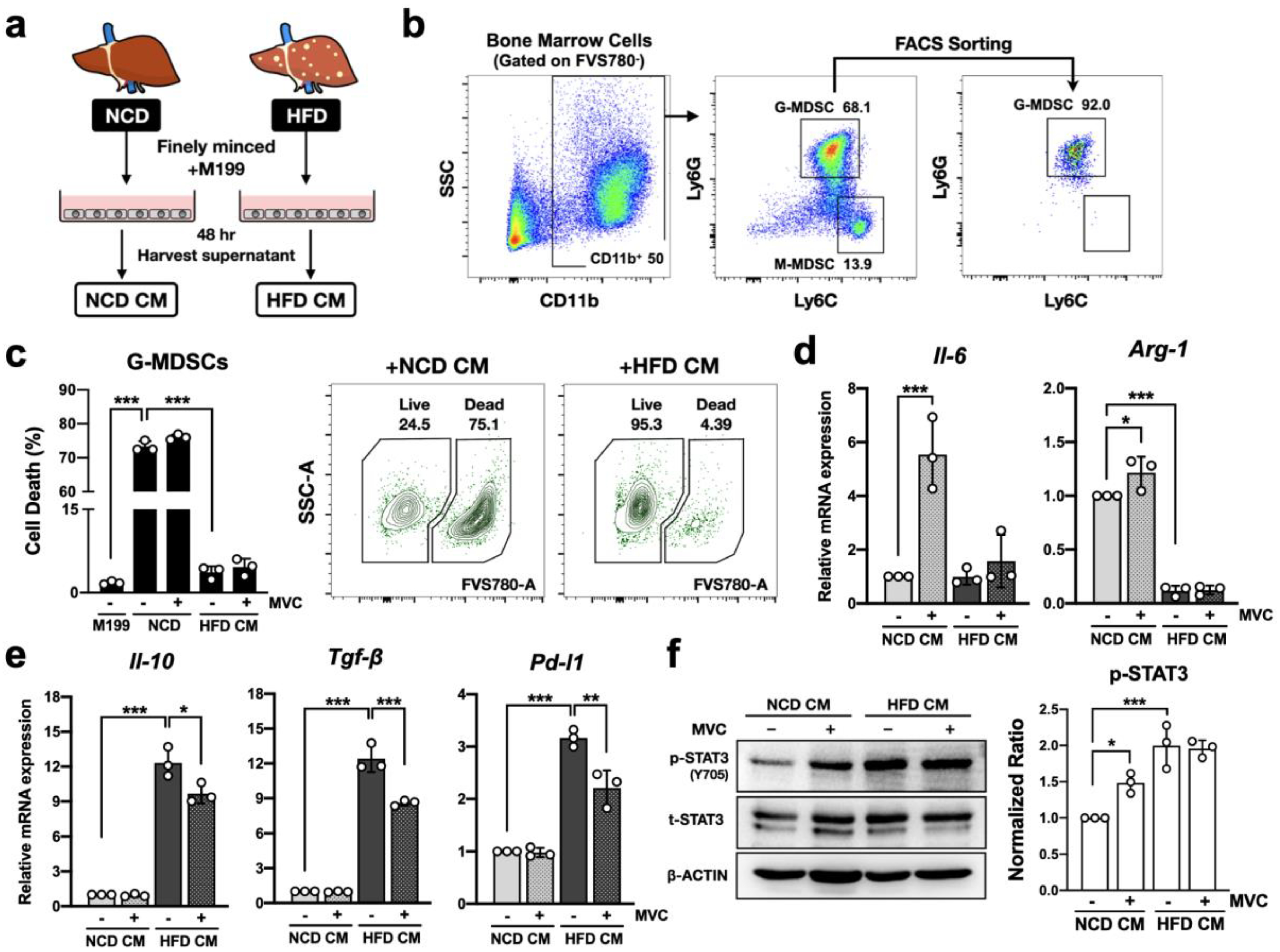
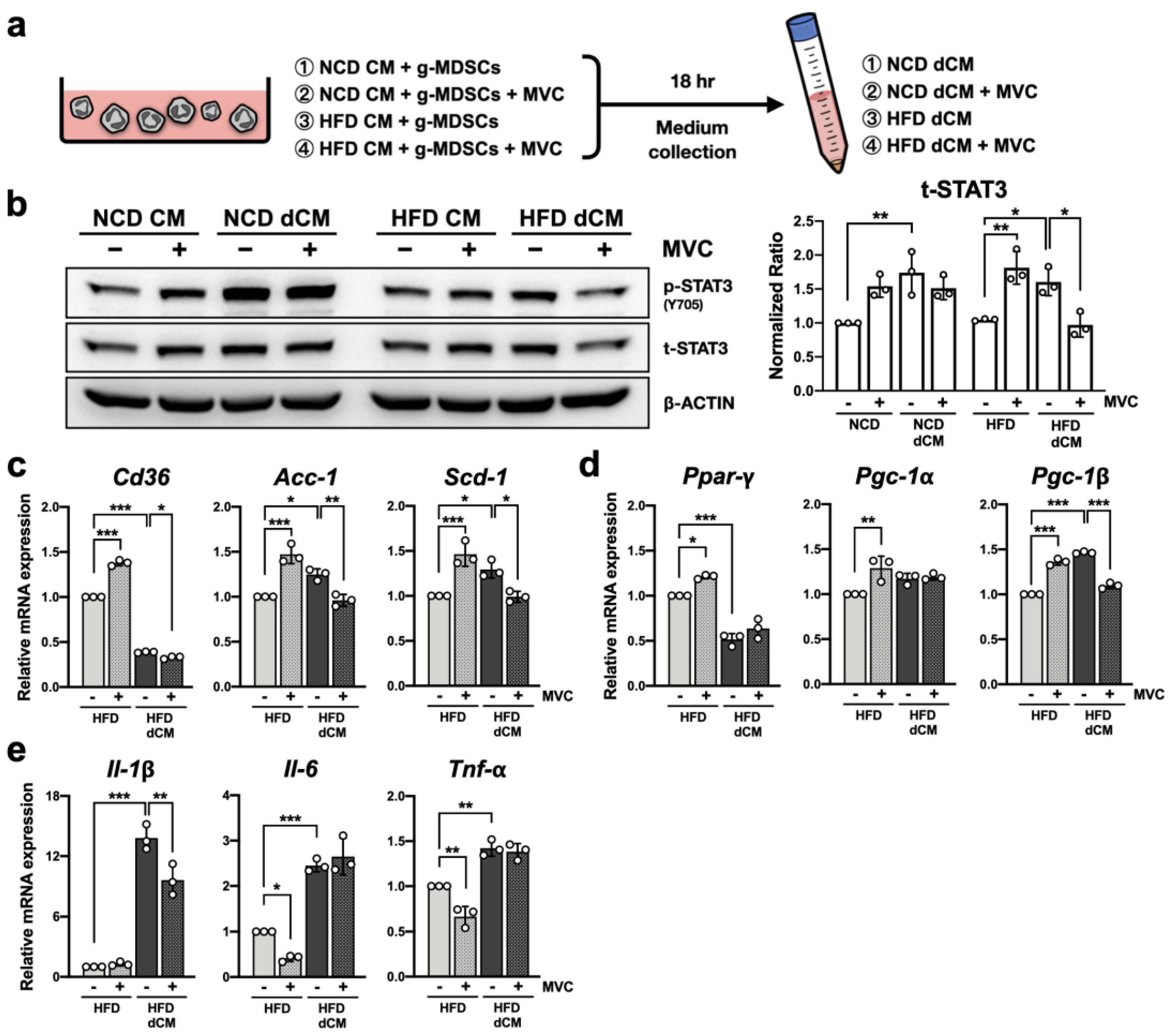
2.9. Blockade of CCR5 Signaling in g-MDSCs Reduces Hepatocyte STAT3 Signaling under the Fatty Liver Microenvironment
2.10. Blockade of CCR5 Signaling in g-MDSCs Reduces Lipogenic Gene Expression in Hepatocytes under the Fatty Liver Microenvironment
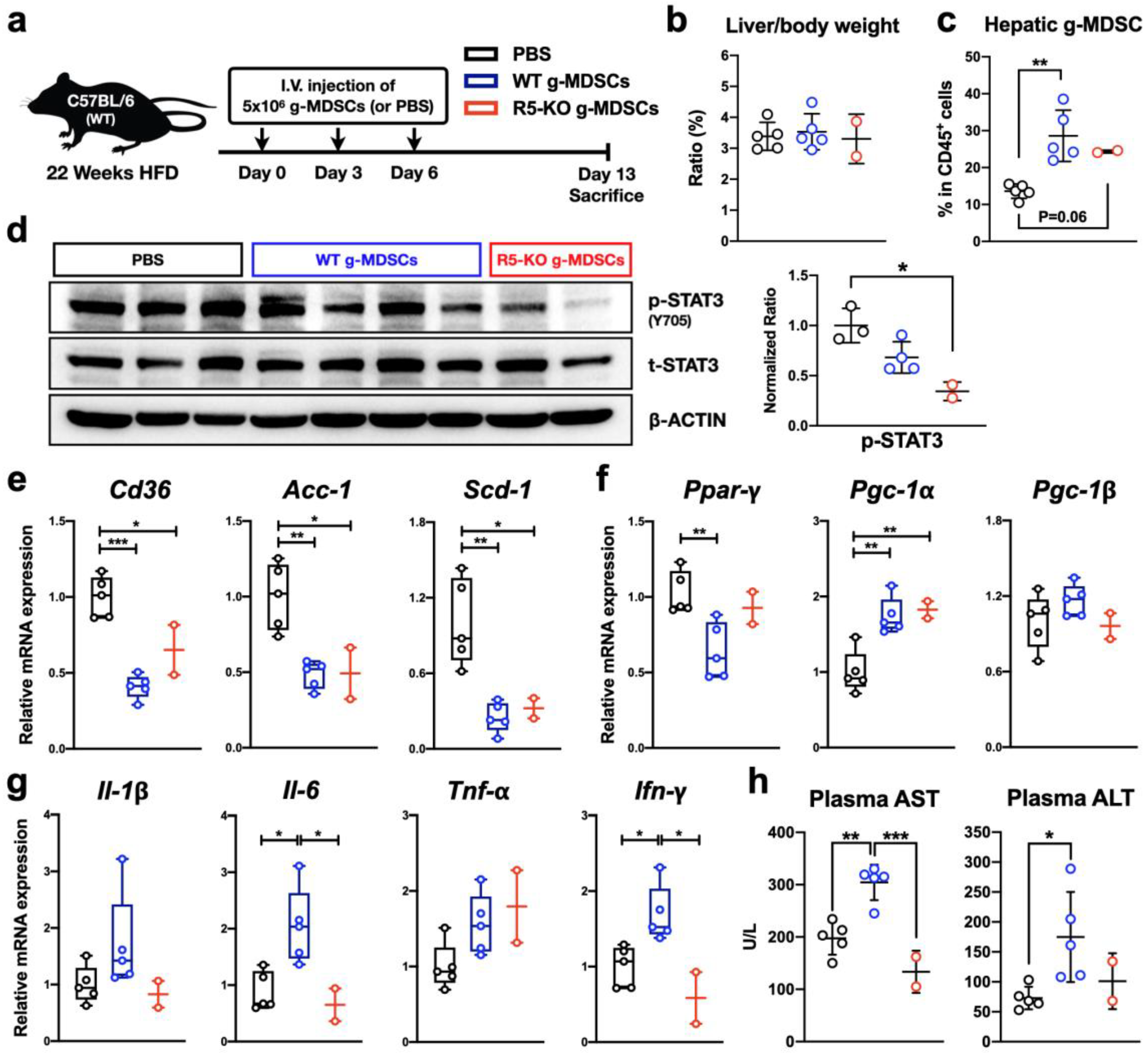
2.11. CCR5-Deficient g-MDSCs Reduce Hepatic STAT3 Activation and Lipogenic Gene Expression in NAFLD
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Biochemical Analysis of Blood and Liver Samples
4.3. Liver Sectioning and Staining
4.4. Profiling of Immune Cells Infiltrated in Hepatic Tissue
4.5. Liver-Conditioned Medium (CM) for Ex Vivo Experiments
4.6. Isolation and Culturing of Primary Hepatocytes and g-MDSCs
4.7. Immunoblotting
4.8. Tissue RNA Analysis by Quantitative RT-PCR
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rinella, M.E. Nonalcoholic fatty liver disease. A systematic review. JAMA 2015, 313, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Tacke, F.; Arrese, M.; Sharma, B.C.; Mostafa, I.; Bugianesi, E.; Wong, V.W.-S.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrese, M.; Feldstein, A.E. NASH-Related cirrhosis: An occult liver disease burden. Hepatol. Commun. 2017, 1, 84–86. [Google Scholar] [CrossRef]
- Chen, W.; Zhang, J.; Fan, H.N.; Zhu, J.S. Function and therapeutic advances of chemokine and its receptor in nonalcoholic fatty liver disease. Ther. Adv. Gastroenterol. 2018, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hübscher, S.G. Histological assessment of non-alcoholic fatty liver disease. Histopathology 2006, 49, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Nomiyama, H.; Hieshima, K.; Nakayama, T.; Sakaguchi, T.; Fujisawa, R.; Tanase, S.; Nishiura, H.; Matsuno, K.; Takamori, H.; Tabira, Y.; et al. Human CC chemokine liver-expressed chemokine/CCL16 is a functional ligand for CCR1, CCR2 and CCR5, and constitutively expressed by hepatocytes. Int. Immunol. 2001, 13, 1021–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwabe, R.F.; Bataller, R.; Brenner, D.A. Human hepatic stellate cells express CCR5 and RANTES to induce proliferation and migration. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G949–G958. [Google Scholar] [CrossRef] [Green Version]
- Ota, T. Chemokine systems link obesity to insulin resistance. Diabetes Metab. J. 2013, 37, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.-M.; Abdelfattah, A.M.; Vasan, R.; Fuchs, B.C.; Choi, M.Y. Hepatic stellate cells secrete Ccl5 to induce hepatocyte steatosis. Sci. Rep. 2018, 8, 7499. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Guo, C.-J.; Li, Y.; Douglas, S.D.; Qi, X.-X.; Song, L.; Ho, W.-Z. Interleukin-1beta induces macrophage inflammatory protein-1beta expression in human hepatocytes. Cell. Immunol. 2003, 226, 45–53. [Google Scholar] [CrossRef]
- Seki, E.; De Minicis, S.; Gwak, G.-Y.; Kluwe, J.; Inokuchi, S.; Bursill, C.A.; Llovet, J.M.; Brenner, D.A.; Schwabe, R.F. CCR1 and CCR5 promote hepatic fibrosis in mice. J. Clin. Investig. 2009, 119, 1858–1870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartneck, M.; Koppe, C.; Fech, V.; Warzecha, K.T.; Kohlhepp, M.; Huss, S.; Weiskirchen, R.; Trautwein, C.; Luedde, T.; Tacke, F. Roles of CCR2 and CCR5 for hepatic macrophage polarization in mice with liver parenchymal cell-specific NEMO deletion. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Martínez, L.; Pérez-Matute, P.; Aguilera-Lizarraga, J.; Rubio-Mediavilla, S.; Narro, J.; Recio, E.; Ochoa-Callejero, L.; Oteo, J.A.; Blanco, J.R. Maraviroc, a CCR5 antagonist, ameliorates the development of hepatic steatosis in a mouse model of non-alcoholic fatty liver disease (NAFLD). J. Antimicrob. Chemother. 2014, 69, 1903–1910. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Martínez, L.; Ochoa-Callejero, L.; Rubio-Mediavilla, S.; Narro, J.; Bernardo, I.; Oteo, J.A.; Blanco, J.R. Maraviroc improves hepatic triglyceride content but not inflammation in a murine nonalcoholic fatty liver disease model induced by a chronic exposure to high-fat diet. Transl. Res. 2018, 196, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Lee, D.H.; Park, M.S.; Jung, Y.S.; Hong, J.T. C-C chemokine receptor type 5 deficiency exacerbates alcoholic fatty liver disease through pro-inflammatory cytokines and chemokines-induced hepatic inflammation. J. Gastroenterol. Hepatol. 2017, 32, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Werfel, T.A.; Elion, D.L.; Rahman, B.; Hicks, D.J.; Sanchez, V.; Gonzales-Ericsson, P.I.; Nixon, M.J.; James, J.L.; Balko, J.M.; Scherle, P.A.; et al. Treatment-induced tumor cell apoptosis and secondary necrosis drive tumor progression in the residual tumor microenvironment through MerTK and IDO1. Cancer Res. 2019, 79, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Han, Y.; Guo, Q.; Zhang, M.; Cao, X. Cancer-expanded myeloid-derived suppressor cells induce anergy of NK cells through membrane-bound TGF-beta1. J. Immunol. 2009, 182, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Movahedi, K.; Guilliams, M.; van den Bossche, J.; van den Bergh, R.; Gysemans, C.; Beschin, A.; Baetselier, P.D.; van Ginderachter, J.A. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell–suppressive activity. Blood 2008, 111, 4233–4244. [Google Scholar] [CrossRef]
- Youn, J.-I.; Gabrilovich, D.I. The biology of myeloid-derived suppressor cells: The blessing and the curse of morphological and functional heterogeneity. Eur. J. Immunol. 2010, 40, 2969–2975. [Google Scholar] [CrossRef]
- Youn, J.-I.; Nagaraj, S.; Collazo, M.; Gabrilovich, D.I. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J. Immunol. 2008, 181, 5791–5802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nefedova, Y.; Huang, M.; Kusmartsev, S.; Bhattacharya, R.; Cheng, P.; Salup, R.; Jove, R.; Gabrilovich, D.I. Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J. Immunol. 2004, 172, 464–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abad, C.; Nobuta, H.; Li, J.; Kasai, A.; Yong, W.H.; Waschek, J.A. Targeted STAT3 disruption in myeloid cells alters immunosuppressor cell abundance in a murine model of spontaneous medulloblastoma. J. Leukoc. Biol. 2014, 95, 357–367. [Google Scholar] [CrossRef]
- Ban, Y.; Mai, J.; Li, X.; Mitchell-Flack, M.; Zhang, T.; Zhang, L.; Chouchane, L.; Ferrari, M.; Shen, H.; Ma, X. Targeting autocrine CCL5–CCR5 axis reprograms immunosuppressive myeloid cells and reinvigorates antitumor immunity. Cancer Res. 2017, 77, 2857–2868. [Google Scholar] [CrossRef] [Green Version]
- Weber, R.; Riester, Z.; Hüser, L.; Sticht, C.; Siebenmorgen, A.; Groth, C.; Hu, X.; Altevogt, P.; Utikal, J.S.; Umansky, V. IL-6 regulates CCR5 expression and immunosuppressive capacity of MDSC in murine melanoma. J. Immunother. Cancer 2020, 8, e000949. [Google Scholar] [CrossRef]
- Hawila, E.; Razon, H.; Wildbaum, G.; Blattner, C.; Sapir, Y.; Shaked, Y.; Umansky, V.; Karin, N. CCR5 directs the mobilization of CD11b+Gr1+Ly6Clow polymorphonuclear myeloid cells from the bone marrow to the blood to support tumor development. Cell Rep. 2017, 21, 2212–2222. [Google Scholar] [CrossRef] [Green Version]
- Blattner, C.; Fleming, V.; Weber, R.; Himmelhan, B.; Altevogt, P.; Gebhardt, C.; Schulze, T.J.; Razon, H.; Hawila, E.; Wildbaum, G.; et al. CCR5+ myeloid-derived suppressor cells are enriched and activated in melanoma lesions. Cancer Res. 2017, 78, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Tang, W.J.; Yin, J.J.; Zhou, B.J. Signal transductions and nonalcoholic fatty liver: A mini-review. Int. J. Clin. Exp. Med. 2014, 7, 1624–1631. [Google Scholar]
- Skat-Rørdam, J.; Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. A role of peroxisome proliferator-activated receptor γ in non-alcoholic fatty liver disease. Basic Clin. Pharm. Toxicol. 2018, 124, 528–537. [Google Scholar] [CrossRef]
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 160–174. [Google Scholar] [CrossRef]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARg-coactivator 1a (PGC1a) and mitochondrial biogenesis in mice. Lab. Investig. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, N. TGF-β in hepatic stellate cell activation and liver fibrogenesis—Updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-R.; Kwak, Y.; Yang, T.; Han, J.H.; Park, S.-H.; Ye, M.B.; Lee, W.; Sim, K.-Y.; Kang, J.-A.; Kim, Y.-C.; et al. Myeloid-derived suppressor cells are controlled by regulatory T cells via TGF-b during murine colitis. Cell Rep. 2016, 17, 3219–3232. [Google Scholar] [CrossRef] [Green Version]
- Charni-Natan, M.; Goldstein, I. Protocol for primary mouse hepatocyte isolation. STAR Protoc. 2020, 1, 100086. [Google Scholar] [CrossRef]
- Pérez-Matute, P.; Pérez-Martínez, L.; Aguilera-Lizarraga, J.; Blanco, J.R.; Oteo, J.A. Maraviroc modifies gut microbiota composition in a mouse model of obesity: A plausible therapeutic option to prevent metabolic disorders in HIV-infected patients. Rev. Española Quimioter. 2015, 28, 200–206. [Google Scholar]
- Kennedy, A.; Webb, C.D.; Hill, A.A.; Gruen, M.L.; Jackson, L.G.; Hasty, A.H. Loss of CCR5 results in glucose intolerance in diet-induced obese mice. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E897–E906. [Google Scholar] [CrossRef] [Green Version]
- Kitade, H.; Sawamoto, K.; Nagashimada, M.; Inoue, H.; Yamamoto, Y.; Sai, Y.; Takamura, T.; Yamamoto, H.; Miyamoto, K.; Ginsberg, H.N.; et al. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes 2012, 61, 1680–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cave, M.C.; Clair, H.B.; Hardesty, J.E.; Falkner, K.C.; Feng, W.; Clark, B.J.; Sidey, J.; Shi, H.; Aqel, B.A.; McClain, C.J.; et al. Nuclear receptors and nonalcoholic fatty liver disease. Biochim. Biophys. Acta 2016, 1859, 1083–1099. [Google Scholar] [CrossRef] [Green Version]
- Moran-Salvador, E.; Lopez-Parra, M.; García-Alonso, V.; Titos, E.; Martínez-Clemente, M.; González-Périz, A.; Lopez-Vicario, C.; Barak, Y.; Arroyo, V.; Claria, J. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011, 25, 2538–2550. [Google Scholar] [CrossRef]
- Cuff, A.O.; Sillito, F.; Dertschnig, S.; Hall, A.; Luong, T.V.; Chakraverty, R.; Male, V. The obese liver environment mediates conversion of NK cells to a less cytotoxic ILC1-like phenotype. Front. Immunol. 2019, 10, 2180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Lafdil, F.; Kong, X.; Gao, B. Signal transducer and activator of transcription 3 in liver diseases: A novel therapeutic target. Int. J. Biol. Sci. 2011, 7, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Jiang, J.; Liu, J. CCR5 blockade suppresses melanoma development through inhibition of IL-6-Stat3 pathway via upregulation of SOCS3. Inflammation 2015, 38, 2049–2056. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Nefedova, Y.; Lei, A.; Gabrilovich, D.I. Neutrophils and PMN-MDSC: Their biological role and interaction with stromal cells. Semin. Immunol. 2018, 35, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.-Y.; Tsai, H.-T.; Su, L.-J.; Chuang, H.-C.; Shiu, L.-Y.; Huang, C.-C.; Fang, F.-M.; Yu, C.-C.; Su, H.-T.; Chen, C.-H. Aurora-A signaling is activated in advanced stage of squamous cell carcinoma of head and neck cancer and requires osteopontin to stimulate invasive behavior. Oncotarget 2014, 5, 2243–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-derived suppressor cells as a therapeutic target for cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zheng, L.; Mo, J.; Yao, X.; Fan, C.; Bao, Y. Protective effects of mitoTEMPO on nonalcoholic fatty liver disease via regulating myeloid-derived suppressor cells and inflammation in mice. BioMed Res. Int. 2020, 2020, 9329427. [Google Scholar] [CrossRef]
- Rensen, S.S.; Slaats, Y.; Nijhuis, J.; Jans, A.; Bieghs, V.; Driessen, A.; Malle, E.; Greve, J.W.; Buurman, W.A. Increased hepatic myeloperoxidase activity in obese subjects with nonalcoholic steatohepatitis. Am. J. Pathol. 2009, 175, 1473–1482. [Google Scholar] [CrossRef] [Green Version]
- Youn, J.-I.; Collazo, M.; Shalova, I.N.; Biswas, S.K.; Gabrilovich, D.I. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol. 2012, 91, 167–181. [Google Scholar] [CrossRef] [Green Version]
- Riehle, K.J.; Campbell, J.S.; McMahan, R.S.; Johnson, M.M.; Beyer, R.P.; Bammler, T.K.; Fausto, N. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J. Exp. Med. 2008, 205, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Wilkerson, A.; Kim, J.; Huang, A.Y.; Zhang, M. Nanoparticle systems modulating myeloid-derived suppressor cells for cancer immunotherapy. Curr. Top. Med. Chem. 2017, 17, 1843. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| 36b4 | CGACCTGGAAGTCCAACTAC | ATCTGCTGCATCTGCTTG |
| Ccr5 | TTTTCAAGGGTCAGTTCCGAC | GGAAGACCATCATGTTACCCAC |
| Arg-1 | AACACGGCAGTGGCTTTAACC | GGTTTTCATGTGGCGCATTC |
| Tgf-β | TGACGTCACTGGAGTTGTACGG | GGTTCATGTCATGGATGGTGC |
| Pd-l1 | GACCAGCTTTTGAAGGGAAATG | CTGGTTGATTTTGCGGTATGG |
| Il-10 | CAGAGCCACATGCTCCTAGA | TGTCCAGCTGGTCCTTTGTT |
| Il-6 | TACCACTTCACAAGTCGGAGGC | CTGCAAGTGCATCATCGTTGTTC |
| Tnf-α | GCCTCTTCTCATTCCTGCTTG | CTGATGAGAGGGAGGCCATT |
| Il-1β | CCTTCCAGGATGAGGACATGA | TGAGTCACAGAGGATGGGCTC |
| Ifn-γ | GCCATCAGCAACAACATAAGCGTC | CCACTCGGATGAGCTCATTGAATG |
| Cd36 | TCCTCTGACATTTGCAGGTCTATC | AAAGGCATTGGCTGGAAGAA |
| Acc-1 | AGGAAGATGGCGTCCGCTCTG | GGTGAGATGTGCTGGGTCAT |
| Scd-1 | CCGGAGACCCTTAGATCGA | TAGCCTGTAAAAGATTTCTGCAAA |
| Ppar-γ | CGGTTTCAGAAGTGCCTTG | GGTTCAGCTGGTCGATATCAC |
| Pgc-1α | ACTGAGCTACCCTTGGGATG | TAAGGATTTCGGTGGTGACA |
| Pgc-1β | TCCAGAAGTCAGCGGCCTTGTGTCA | CTCTGGGACAGGGCAGCACCGA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liao, T.-C.; Huang, J.-P.; Tsai, Y.-T.; Shih, W.-C.; Juan, C.-C.; Hsieh, P.-S.; Hung, L.-M.; Yu, C.-L. Granulocytic MDSC with Deficient CCR5 Alleviates Lipogenesis and Inflammation in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 13048. https://doi.org/10.3390/ijms232113048
Liao T-C, Huang J-P, Tsai Y-T, Shih W-C, Juan C-C, Hsieh P-S, Hung L-M, Yu C-L. Granulocytic MDSC with Deficient CCR5 Alleviates Lipogenesis and Inflammation in Nonalcoholic Fatty Liver Disease. International Journal of Molecular Sciences. 2022; 23(21):13048. https://doi.org/10.3390/ijms232113048
Chicago/Turabian StyleLiao, Tzu-Chieh, Jiung-Pang Huang, Yu-Ting Tsai, Wei-Ching Shih, Chi-Chang Juan, Po-Shiuan Hsieh, Li-Man Hung, and Chao-Lan Yu. 2022. "Granulocytic MDSC with Deficient CCR5 Alleviates Lipogenesis and Inflammation in Nonalcoholic Fatty Liver Disease" International Journal of Molecular Sciences 23, no. 21: 13048. https://doi.org/10.3390/ijms232113048