
Counteracting Immunosuppression in the Tumor Microenvironment by Oncolytic Newcastle Disease Virus and Cellular Immunotherapy



Abstract
:1. Introduction
2. Spontaneous Anti-Tumor T Cell Responses and Cellular Immunotherapy Studies
2.1. Evidence for Spontaneous Anti-Tumor T Cell Responses from a Mouse tumor Model
2.2. Spontaneous Anti-Tumor Immune Responses in Cancer Patients
2.3. Therapy of Human Tumors in NOD/SCID Mice with Patient-Derived Reactivated MTCs from BM
2.4. Therapeutic Potential of Cancer-Reactive MTCs from BM in Cancer Patients
2.5. Cellular Immunotherapy Counteracting Advanced Metastasized Cancer
- (i)
- reversion of tumor tissue pH from acid to neutral after 3–4 days as a first sign of the immunotherapeutic effect,
- (ii)
- donor CD4 T cell infiltration in the tumors 6 days after cell transfer,
- (iii)
- formation of a broad capsule of fibrous tissue between the tumor area and the skin,
- (iv)
- tumor rejection and long-term survival,
- (v)
- wound healing and scar tissue formation at sites of primary tumor rejection (skin) and at sites of metastases (liver and kidney),
- (vi)
- reconstitution of normal fur at the site of rejected primary tumor,
- (vii)
- cellular interactions: donor CD4+ and CD8+ immune T-T cell interactions, donor T cell-host macrophage interactions around liver metastases, vß6 donor T cells recognizing a tumor-associated viral superantigen (vSAG-7) interacting with tumor cells and APCs [29],
- (viii)
- reversibility of a state of cachexia,
- (ix)
- disproval of the hypothesis that a tumor is a never healing wound [30].
3. The Tumor Microenvironment
3.1. BM TME
3.2. CAFs in the TME
3.3. TME “Cold”, Barriers to T Cell Infiltration, Tumor Cell Defense Mechanisms against CTL Attack
3.4. MDSCs and Immunosuppression
3.5. MDSC, CAF and Neutrophil Involvement in the Pre-Metastatic Niche
3.6. M2 TAMs
3.7. Tolerogenic DCs
3.8. Anergic T Cells and Treg Cells
3.9. Dysfunctional NK Cells
3.10. Tumor-Derived Factors
3.11. Metabolic Barrier, T Cells, Hypoxia and Tumor Dormancy
3.12. TGF-ß and EMT
3.13. Therapy Resistance and Cancer Hallmarks
4. Counteracting Immunosuppression via Oncolytic Newcastle Disease Virus
4.1. Counteracting CAFs
4.2. Turning Cold into Hot Tumors
4.2.1. ICI Treatment
4.2.2. OV Treatment
4.2.3. OV Treatment Combined with ICI and Role of DCs
4.3. Downregulation of MDSCs
4.4. Macrophage Activation and Polarization
4.5. DC Activation and Polarization
4.6. T Cell Activation and Polarization
4.7. NK Cell Activation
4.8. Targeting Rac1 and Spread of NDV in Tumors
4.8.1. Targeting Rac1
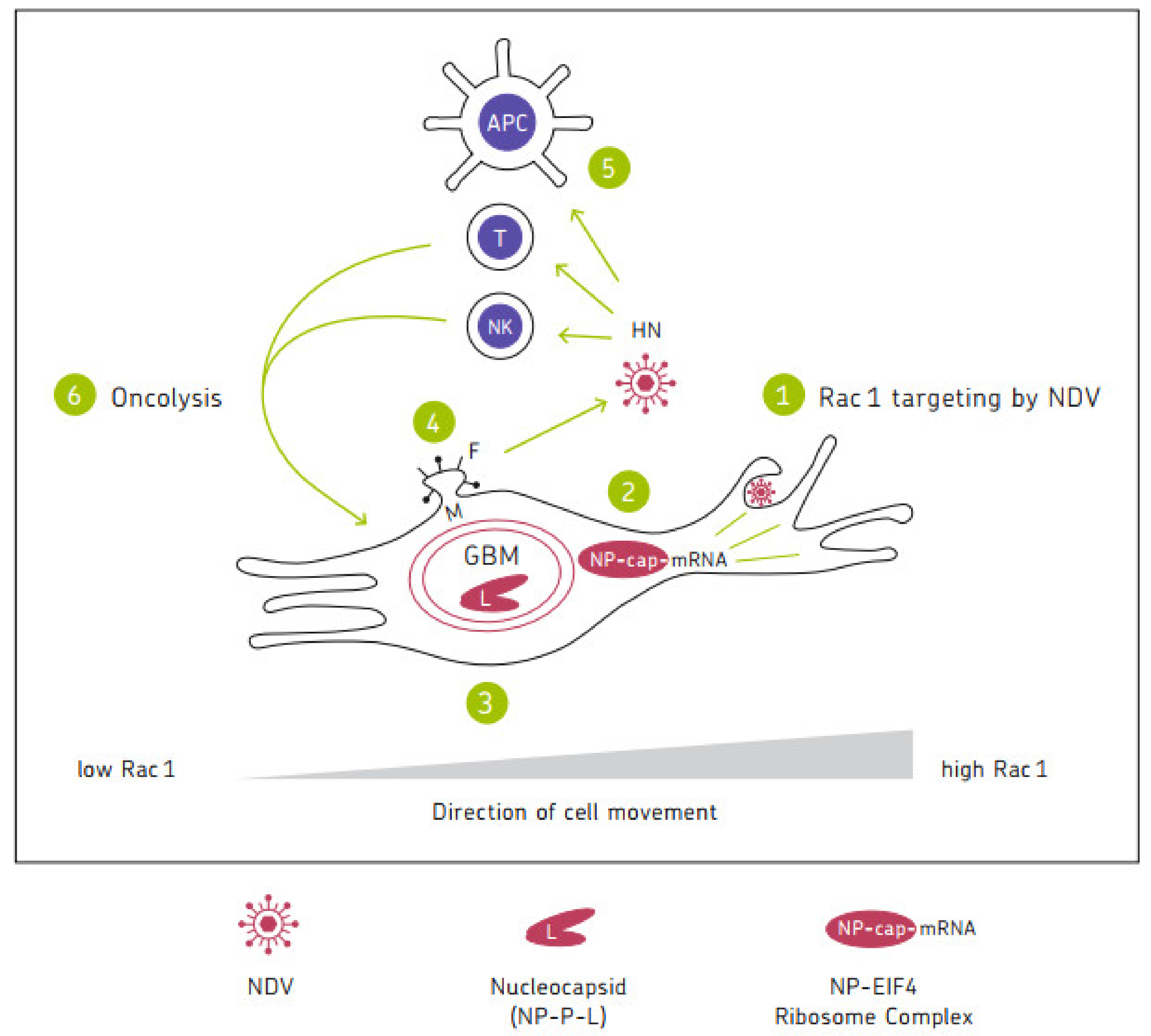
4.8.2. Spread of Oncolytic NDV in Tumors
4.9. Suppression of the Metabolic Barrier
4.10. Breaking Cancer Therapy Resistance and Immune Resistance
4.10.1. Breaking Resistance to Chemo- or Radiotherapy
4.10.2. Breaking Resistance to Hypoxia, Apoptosis, TRAIL, Drugs and Small Molecule Inhibitors (SMIs)
4.10.3. Breaking of T Cell Tolerance to TAA-Expressing Tumor Cells
4.10.4. Breaking Resistance to Oncolysis, to ICI and to Anti-Viral Immunity
4.11. Recruitment of Cancer-Reactive MTCs
4.12. In Situ Vaccination
4.12.1. Photothermal Treatment Inducing ICD
4.12.2. IMI Strategy
4.12.3. Use of GM-CSF Modified NDV
4.13. Use of Antibody Modified NDV
4.14. Altering the TME by Sytemic Transfer of OV-Loaded Carrier Cells
4.15. Active-Specific Immunization (ASI)
4.16. Adoptive Cellular Immunotherapy (ACT)
4.17. Tumor-Suppressing Functions of Immune Cells within the TME
5. Post-Operative Vaccination with or without NDV of Glioblastoma Patients
5.1. Vaccination Studies with NDV
5.2. Vaccination Studies without NDV

{kind=link}
| Feature | Vaccine Type/Patients | Year | Comments | References |
|---|---|---|---|---|
| ASI | ATV-NDV Adults with primary GBM (n = 23) | 2004 | Two-year survival rate 39% compared to 11% in control group | [149] |
| IMI | IO-VACR Retrospective analysis of adults with primary GBM (n = 10) | 2017 | Combining NDV with DC vaccination and mEHT | [127] |
| ICD | IO-VACR Adults with primary GBM (n = 60) | 2018 2021 2022 | Combining IMI with chemotherapy, IDH1 wt (n = 25) | [150] [152] [151] |
| IMI ICD Peptides+ | IO-VACR + peptides (n = 1) | 2021 | A case of CR and specific T cell response | [152] |
| IMI | IO-VACR Children with DIPG (n = 41) | 2020 | A single institution experience | [153] |
| Meta analysis | TAA pulsed DC vaccine without NDV | 2014 | Clinical efficacy | [154] |
| Th peptide vaccine | IDH1-vac, Adults with astrocytomas grade 3 (n = 22) and 4 (GBM) (n = 11), Targeting mutant IDH1 | 2021 | Three-year survival rate 84%, no control group | [155] |
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Burman, B.; Pesci, G.; Zamarin, D. Newcastle disease virus at the forefront of cancer immunotherapy. Cancers 2020, 12, 3552. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xie, F.; Zhang, L.; Zhou, X.; Huang, J.; Wang, F.; Jin, J.; Zhang, L.; Zeng, L.; Zhou, F. Targeted anti-tumor immunotherapy using tumor infiltrating cells. Adv. Sci. 2021, 8, 2101672. [Google Scholar] [CrossRef] [PubMed]
- Khazaie, K.; Prifti, S.; Beckhove, P.; Griesbach, A.; Russel, S.; Collins, M.; Schirrmacher, V. Persistence of dormant tumor cells in the bone marrow of tumor cell-vaccinated mice correlates with long-term immunological protection. Proc. Natl. Acad. Sci. USA 1994, 91, 7430–7434. [Google Scholar] [CrossRef] [Green Version]
- Feuerer, M.; Beckhove, P.; Garbi, N.; Mahnke, Y.; Limmer, A.; Hommel, M.; Hämmerling, G.J.; Kyewski, B.; Hamann, A.; Umansky, V.; et al. Bone marrow as a priming site for T-cell responses to blood-borne antigen. Nat. Med. 2003, 9, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Mahnke, Y.D.; Schirrmacher, V. A novel tumour model system for the study of long-term protective immunity and immune T cell memory. Cell. Immunol. 2003, 221, 89–99. [Google Scholar] [CrossRef]
- Mahnke, Y.D.; Schwendemann, J.; Beckhove, P.; Schirrmacher, V. Maintenance of long-term tumour-specific T-cell memory by residual dormant tumour cells. Immunology 2005, 115, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Di Rosa, F.; Gebhardt, T. Bone marrow T cells and the integrated functions of recirculating and tissue-resident memory T cells. Front. Immunol. 2016, 7, 51. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.B.; Carpenter, B.; Santos e Sousa, P.; Pospori, C.; Khorshed, R.; Griffin, J.; Velica, P.; Zech, M.; Ghorashian, S.; Forrest, C.; et al. Redirection to the bone marrow improves T cell persistence and antitumor functions. J. Clin. Investig. 2018, 128, 2010–2024. [Google Scholar] [CrossRef]
- Macallan, D.C.; Borghans, J.A.M.; Asquith, B. Human T cell memory: A dynamic view. Vaccines 2017, 5, 5. [Google Scholar] [CrossRef]
- Feuerer, M.; Beckhove, P.; Mahnke, Y.; Hommel, M.; Kyewski, B.; Hamann, A.; Umansky, V.; Schirrmacher, V. Bone marrow microenvironment facilitating dendritic cell: CD4 T cell interactions and maintenance of CD4 memory. Int. J. Oncol. 2004, 25, 867–876. [Google Scholar]
- Schirrmacher, V.; Feuerer, M.; Fournier, P.; Ahlert, T.; Umansky, V.; Beckhove, P. T-cell priming in bone marrow: The potential for long-lasting protective anti-tumor immunity. Trends Mol. Med. 2003, 8, 526–634. [Google Scholar] [CrossRef] [PubMed]
- Murao, A.; Oka, Y.; Tsuboi, A.; Elisseeva, O.A.; Tanaka-Haranda, Y.; Fujiki, F.; Nakajima, H.; Nishida, S.; Hosen, N.; Shirakata, T.; et al. High frequencies of less differentiated and more proliferative WT1-specific CD8+ T cells in bone marrow in tumor-bearing patients: An important role of bone marrow as a secondary lymphoid organ. Cancer Sci. 2010, 101, 848–854. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. New insights into mechanisms of long-term protective anti-tumor immunity induced by cancer vaccines modified by virus infection. Biomedicines 2020, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radbruch, A.; McGrath, M.A.; Siracusa, F.; Hoffmann, U.; Sercan-Alp, Ö.; Hutloff, A.; Tokoyoda, K.; Chang, H.-D.; Dong, J. Homeostasis and durability of T-cell memory- The resting and the restless T-cell memory. Cold Spring Harb. Perspect. Biol. 2021, 13, a038083. [Google Scholar] [CrossRef]
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-tumor immunity controlled through mRNA m6A and YTHDF1 in dendritic cells. Nature 2019, 566, 270–274. [Google Scholar] [CrossRef]
- Duong, E.; Fessenden, T.B.; Lutz, E.; Dinter, T.; Yim, L.; Blatt, S.; Bhutkar, A.; Wittrup, K.D.; Spranger, S. Type I interferon activates MHC class I-dressed CD11b+ conventional dendritic cells to promote protective anti-tumor CD8+ T cell immunity. Immunity 2022, 55, 308–323.e9. [Google Scholar] [CrossRef]
- Alspach, E.; Lussier, D.M.; Micelli, A.P.; Kizhvatov, I.; DuPage, M.; Luoma, A.M.; Meng, W.; Lichti, C.F.; Esaulova, E.; Vomund, A.N.; et al. MHC-II neoantigens shape tumor immunity and response to immunotherapy. Nature 2019, 574, 696–701. [Google Scholar] [CrossRef]
- Beckhove, P.; Schirrmacher, V. Local tumor growth and spontaneous systemic T cell responses in cancer patients: A paradox and puzzle. In Innate and Adaptive Immunity in the Tumor Microenvironment; Yefenof, E., Witz, I., Eds.; The Tumor Microenvironment 1 Series; Springer Science + Business Media: New York, NY, USA, 2008; pp. 53–76. [Google Scholar]
- Sommerfeldt, N.; Schütz, F.; Sohn, C.; Förster, J.; Schirrmacher, V.; Beckhove, P. The shaping of a polyvalent and highly individual T cell repertoire in the bone marrow of breast cancer patients. Cancer Res. 2006, 66, 8258–8265. [Google Scholar] [CrossRef] [Green Version]
- Feuerer, M.; Rocha, M.; Bai, L.; Umansky, V.; Solomayer, E.F.; Bastert, G.; Diel, I.J.; Schirrmacher, V. Enrichment of memory T cells and other profound immunological changes in the bone marrow from untreated breast cancer patients. Int. J. Cancer 2001, 92, 96–105. [Google Scholar] [CrossRef]
- Schmitz-Winnenthal, F.; Volk, C.; Z`graggen, K.; Galindo, L.; Nummer, D.; Ziouta, Y.; Bucur, M.; Weitz, J.; Schirrmacher, V.; Büchler, M.W.; et al. High frequencies of functional tumor-reactive T cells in bone marrow and blood of pancreatic cancer patients. Cancer Res. 2005, 65, 10079–10087. [Google Scholar] [CrossRef] [Green Version]
- Bai, L.; Beckhove, P.; Feuerer, M.; Umansky, V.; Choi, C.; Schütz, F.; Solomayer, E.; Diel, I.J.; Schirrmacher, V. Cognate interactions between memory T cells and tumor antigen-presenting dendritic cells from bone marrow of breast cancer patients: Bidirectional cell stimulation, survival and antitumor activity in vivo. Int. J. Cancer 2003, 103, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Beckhove, P.; Bai, L.; Solomayer, E.F.; Bastert, G.; Diel, I.J.; Pedain, C.; Oberniedermayr, M.; Schirrmacher, V.; Umansky, V. Therapy of human tumors in NOD/SCID mice with patient-derived reactivated memory T cells from bone marrow. Nat. Med. 2001, 7, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Beckhove, P.; Feuerer, M.; Dolenc, M.; Schuetz, F.; Choi, C.; Sommerfeld, N.; Schwendemann, J.; Ehlert, K.; Altevogt, P.; Bastert, G.; et al. Specifically activated memory T cell subsets from cancer patients recognize and reject xenotransplanted autologous tumors. J. Clin. Investig. 2004, 114, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Schirrmacher, V. Cancer-reactive memory T cells from bone marrow: Spontaneous induction and therapeutic potential (Review). Int. J. Oncol. 2015, 47, 2005–2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuetz, F.; Ehlert, K.; Ge, Y.; Schneeweiss, A.; Rom, J.; Inzkirweli, N.; Sohn, C.; Schirrmacher, V.; Beckhove, P. Treatment of advanced metastasized breast cancer with bone marrow-derived tumour-reactive memory T cells: A pilot clinical study. Cancer Immunol. Immunother. 2009, 58, 887–900. [Google Scholar] [CrossRef]
- Domschke, C.; Ge, Y.; Bernhardt, I.; Schott, S.; Keim, S.; Juenger, S.; Bucur, M.; Mayer, L.; Blumenstein, M.; Rom, J.; et al. Long-term survival after adoptive bone marrow T cell therapy of advanced metastasized breast cancer: Follow-up analysis of a clinical pilot trial. Cancer Immunol. Immunother. 2013, 62, 1053–1060. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Beckhove, P.; Krüger, A.; Rocha, M.; Umansky, V.; Fichtner, K.-P.; Hull, W.E.; Zangemeister-Wittke, U.; Griesbach, A.; Jurianz, K.; et al. Effective immune rejection of advanced metastasized cancer. Int. J. Oncol. 1995, 6, 505–521. [Google Scholar] [CrossRef]
- Schirrmacher, V. Complete remission of cancer in late-stage disease by radiation and transfer of allogeneic MHC-matched immune T cells: Lessons from GvL studies in animals. Cancer Immunol. Immunother. 2014, 63, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650. [Google Scholar]
- Bhome, R.; Bullock, M.D.; Al Saihati, H.A.; Goh, R.W.; Primrose, J.N.; Sayan, A.E.; Mirnezami, A.H. A top-down view of the tumor microenvironment: Structure, cells and signaling. Front. Cell Dev. Biol. 2015, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Sethakorn, N.; Henninger, E.; Sanchez-de-Diego, C.; Ding, A.B.; Yada, R.C.; Kerr, S.C.; Kosoff, D.; Beebe, D.J.; Lang, J.M. Advancing treatment of bone metastases through novel translational approaches targeting the bone microenvironment. Cancers 2022, 14, 757. [Google Scholar] [CrossRef] [PubMed]
- Stefanovic, S.; Schuetz, F.; Sohn, C.; Beckhove, P.; Domschke, C. Bone marrow microenvironment in cancer patients: Immunological aspects and clinical implications. Cancer Met. Rev. 2013, 32, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Kochetkova, M.; Samuel, M.S. Differentiation of the tumor microenvironment: Are CAFs the organizers ? Trends Cell Biol. 2022, 32, 285–294. [Google Scholar] [CrossRef]
- Ghahremanifard, P.; Chanda, A.; Bonni, S.; Bose, P. TGF-ß mediated immune evasion in cancer–spotlight on cancer-associated fibroblasts. Cancers 2020, 12, 3650. [Google Scholar] [CrossRef]
- Sperb, N.; Tsesmelis, M.; Wirth, T. Crosstalk between tumor and stromal cells in pancreatic ductal adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 5486. [Google Scholar] [CrossRef]
- Czekay, R.; Cheon, D.; Samarakoon, R.; Kutz, S.M.; Higgins, P.J. Cancer-associated fibroblasts: Mechanisms of tumor progression and novel therapeutic targets. Cancers 2022, 14, 1231. [Google Scholar] [CrossRef]
- Yeung, T.; Leung, C.S.; Wong, K.; Samimi, G.; Thompson, M.S.; Liu, J.; Zaid, T.M.; Ghosh, S.; Birrer, M.J.; Mok, C.M. TGF-ß modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013, 73, 5016–5028. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Huang, D.; Saw, P.E.; Song, E. Turning cold tumors hot: From molecular mechanisms to clinical applications. Trends Immunol. 2022, 43, 523–545. [Google Scholar] [CrossRef]
- McKenzie, B.; Khazen, R.; Valitutti, S. Greek fire, poison arrows, and scorpion bombs: How tumor cells defend against the siege weapons of cytotoxic T lymphocytes. Front. Immunol. 2022, 13, 894306. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat. Rev. 2021, 21, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Gumberger, P.; Bjornsson, B.; Sandström, P.; Bojmar, L.; Zambirinis, C. The liver pre-metastatic niche in pancreatic cancer: A potential opportunity for intervention. Cancers 2022, 14, 3028. [Google Scholar] [CrossRef] [PubMed]
- Schoeps, B.; Eckfeld, C.; Prokopchuk, O.; Böttcher, J.; Häußler, D.; Steiger, K.; Demir, I.E.; Knolle, P.; Soehnlein, O.; Jenne, D.E.; et al. TIMP1 triggers neutrophil extracellular trap formation in pancreatic cancer. Caner Res. 2021, 81, 3568–3579. [Google Scholar] [CrossRef]
- Li, M.; He, L.; Zhu, J.; Zhang, P.; Liang, S. Targeting tumor-associated macrophages for cancer treatment. Cell Biosci. 2022, 12, 85. [Google Scholar] [CrossRef]
- Xu, Z.; Chen, Y.; Ma, L.; Chen, Y.; Liu, J.; Guo, Y.; Yu, T.; Zhang, L.; Zhu, L.; Shu, Y. Role of exosomal non-coding RNAs from tumor cells and tumor-associated macrophages in the tumor microenvironment. Mol. Ther. 2022, 30, 3133–3154. [Google Scholar] [CrossRef]
- Cavalcante, R.S.; Ishikawa, U.; Silva, E.S.; Silva-Junior, A.A.; Araujo, A.; Cruz, L.J.; Chan, A.B.; de Araujo Junior, R.F. STAT3/NF-kB signalling disruption in M2 tumor-associated macrophages is a major target of PLGA nanocarriers/PD-L1 antibody immunomodulatory therapy in breast cancer. Br. J. Pharmacol. 2021, 178, 2284–2304. [Google Scholar] [CrossRef]
- Gardner, A.; Pulido, A.M.; Ruffell, B. Dendritic cells and their role in immunotherapy. Front. Immunol. 2020, 11, 924. [Google Scholar] [CrossRef]
- Hedge, S.; Krisnawan, V.E.; Herzog, B.H.; Zuo, C.; Breden, M.A.; Knolhoff, B.L.; Hogg, G.D.; Tang, J.P.; Baer, J.M.; Mpoy, C.; et al. Dendritic cell paucity leads to dysfunctional immune surveillance in pancreatic cancer. Cancer Cell 2020, 37, 289–307.c9. [Google Scholar] [CrossRef]
- Jarnicki, A.A.G.; Lysaght, J.; Todryk, S.; Mills, K.H.G. Suppression of antitumor immunity by IL-10 and TGFß producing T cell infiltrating the growing tumour: Influence of tumour microenvironment on the induction of CD4+ and CD8+ regulatory T cells. J. Immunol. 2006, 177, 896–904. [Google Scholar] [CrossRef] [Green Version]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing type-2 dendritic cells to drive protective antitumor CD4(+) T cell immunity. Cell 2019, 177, 556–571. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.-H.; Lee, J.-H.; Choi, S.-Y.; Jung, N.-C.; Song, J.-Y.; Seo, H.-G.; Lim, D.-S. Functional ambivalence of dendritic cells: Tolerogenicity and immunogenicity. Int. J. Mol. Sci. 2021, 22, 4430. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Acharya, N.; Subramanian, A.; Purohit, V.; Tabaka, M.; Hou, Y.; He, D.; Dixon, K.O.; Lambden, C.; Xia, J.; et al. Tim-3 adapter protein Bat3 acts as an endogenous regulator of tolerogenic dendritic cell function. Sci. Immunol. 2022, 7, eabm0631. [Google Scholar] [CrossRef] [PubMed]
- Lei, Q.; Wang, D.; Sun, K.; Wang, L.; Zhang, Y. Resistance mechanisms of anti-PD1/PDL1 therapy in solid tumors. Front. Cell Dev. Biol. 2020, 8, 672. [Google Scholar] [CrossRef]
- Baldanzi, G. Immune checkpoint receptors signaling in T cells. Int. J. Mol. Sci. 2022, 23, 3529. [Google Scholar] [CrossRef]
- Mokhtar, A.M.; Salikin, N.H.; Haron, A.S.; Amin-Nordin, S.; Hashim, I.F.; Makhtar, M.M.Z.; Zulfigar, S.B.; Ismail, N.I. RhoG’s role in T cell activation and function. Front. Immunol. 2022, 13, 845064. [Google Scholar] [CrossRef]
- Noyes, D.; Bag, A.; Oseni, S.; Semidey-Hurtado, J.; Cen, L.; Sarnaik, A.A.; Sondak, V.K.; Adeegbe, D. Tumor-associated Tregs obstruct antitumor immunity by promoting T cell dysfunction and restricting clonal diversity in tumor-infiltrating CD8+ T cells. J. Immunother. Cancer 2022, 10, e004605. [Google Scholar] [CrossRef]
- Riggan, L.; Shah, S.; O’Sullivan, T.E. Arrested development: Suppression of NK cell function in the tumor microenvironment. Clin. Transl. Immunol. 2021, 10, e1238. [Google Scholar] [CrossRef]
- Tumino, N.; Besi, F.; Martini, S.; Di Pace, A.L.; Monari, E.; Quatrini, L.; Pelosi, A.; Fiore, P.F.; Fiscon, G.; Paci, P.; et al. Polymorphonuclear myeloid-derived suppressor cells are abundant in peripheral blood of cancer patients and suppress Natural killer cell anti-tumor activity. Front. Immunol. 2022, 12, 803014. [Google Scholar] [CrossRef]
- Guimaraes, F.S.; Rossi, G.R.; Dagley, L.F.; Foroutan, M.; McCulloch, T.R.; Yousef, J.; Park, H.; Gunter, J.H.; Beavis, P.A.; Lin, C.; et al. TGF-ß and CIS overcomes NK cell suppression to restore anti-tumor immunity. Cancer Immunol. Res. 2022, 10, 1047–1054. [Google Scholar] [CrossRef]
- Shurin, M.R.; Shurin, G.V.; Lokshin, A.; Yurkovetsky, Z.R.; Gutkin, D.W.; Chatta, G.; Zhong, H.; Han, B.; Ferris, R.L. Intratumoral cytokines, chemokines/growth factors and tumour infiltrating dendritic cells: Friends or enemies? Cancer Metastasis Rev. 2006, 25, 333–356. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.R.; Rathmell, W.K.; Rathmell, J.C. The tumor microenvironment as a metabolic barrier to effector T cells and immunotherapy. eLife 2020, 9, e55185. [Google Scholar] [CrossRef]
- Tirpe, A.A.; Gulei, D.; Ciortea, S.M.; Crivii, C.; Berindan-Neagoe, I. Hypoxia: Overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int. J. Mol. Sci. 2019, 20, 6140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hope, H.C.; Pickersgill, G.; Ginefra, P.; Vannini, N.; Cook, G.P.; Salmond, R.J. TGFß limits Myc-dependent TCR-induced metabolic reprogramming in CD8+ T cells. Front. Immunol. 2022, 13, 913184. [Google Scholar] [CrossRef] [PubMed]
- Baldominos, P.; Barbera-Mourelle, A.; Berreiro, O.; Huang, Y.; Wight, A.; Cho, J.-W.; Zhao, X.; Estivilli, G.; Adam, I.; Sanchez, X.; et al. Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell 2022, 185, 1694–1708.e19. [Google Scholar] [CrossRef] [PubMed]
- Rentschler, M.; Braumüller, H.; Briquez, P.S.; Wieder, T. Cytokine-induced senescence in the tumor microenvironment and its effects on anti-tumor immune responses. Cancers 2022, 14, 1364. [Google Scholar] [CrossRef]
- Schirrmacher, V. Shifts in tumor cell phenotypes induced by signals from the microenvironment. Relevance for the immunobiology of cancer metastasis. Immunobiology 1980, 157, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L.; Brunson, K.W.; Fidler, I.J. Specificity of arrest, survival, and growth of selected metastatic variant cell lines. Cancer Res. 1978, 38 Pt 2, 4105–4111. [Google Scholar]
- Balayan, V.; Guddan, A.K. Tumor dormancy: Biologic and therapeutic implications. World J. Oncol. 2022, 13, 8–19. [Google Scholar] [CrossRef]
- Tu, S.-M.; Estecio, M.R.; Lin, S.-H.; Zacharias, N.M. Stem cell theory of cancer: Rude awakening or bad dream from cancer dormancy? Cancers 2022, 14, 655. [Google Scholar] [CrossRef]
- Hoerster, R.; Muether, P.S.; Vierkotten, S.; Hermann, M.M.; Kirchhof, B.; Fauser, S. Upregulation of TGF-ß1 in experimental proliferative vitreretinopathy is accompanied by epithelial to mesenchymal transition. Graefes Arch. Clin. Exp. Ophthalmol. 2014, 252, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Senga, S.S.; Grose, R.P. Hallmarks of cancer—The new testament. Open Biol. 2021, 11, 200358. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of cancer: New dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Ilkow, C.S.; Marguerie, M.; Batenchuk, C.; Mayer, J.; Neriah, D.B.; Cousineau, S.; Falls, T.; Jennings, V.A.; Boileau, M.; Bellamy, D.; et al. Reciprocal cellular cross-talk within the tumor microenvironment promotes oncolytic virus activity. Nat. Med. 2015, 21, 530–536. [Google Scholar] [CrossRef]
- Wilden, H.; Fournier, P.; Zawatzky, R.; Schirrmacher, V. Expression of RIG-I, IRF3, IFN-beta and IRF7 determines resistance or susceptibility of cells to infection by Newcastle disease virus. Int. J. Oncol. 2009, 34, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Hemminki, O.; dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef]
- Schirrmacher, V. Molecular mechanisms of anti-neoplastic and immune stimulatory properties of oncolytic Newcastle disease virus. Biomedicines 2022, 10, 562. [Google Scholar] [CrossRef]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nat. Rev. Immunol. 2018, 18, 498–513. [Google Scholar] [CrossRef]
- Sitta, J.; Claudio, P.P.; Howard, C.M. Virus-based immuno-oncology models. Biomedicines 2022, 10, 1441. [Google Scholar] [CrossRef]
- Fournier, P.; Schirrmacher, V. Bispecific antibodies and trispecific immunocytokines for targeting the immune system against cancer: Preparing for the future. BioDrugs 2013, 27, 35–53. [Google Scholar] [CrossRef]
- Guo, Z.S.; Lotze, M.T.; Zhu, Z.; Storkus, W.J.; Song, X.-T. Bi- and tri-specific T cell engager-armed oncolytic viruses: Next-generation cancer immunotherapy. Biomedicines 2020, 8, 204. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, U.; Hassain, N.A.; Bann, S.; Mathew, S. Oncolytic viruses as nanomedicines against the tumor microenvironment. Biointerface Res. Appl. Chem. 2021, 11, 14825–14852. [Google Scholar] [CrossRef]
- Lan, Q.; Xia, S.; Wang, Q.; Xu, W.; Huang, H.; Jiang, S.; Lu, L. Development of oncolytic virotherapy: From genetic modification to combination therapy. Front. Med. 2020, 14, 160–184. [Google Scholar] [CrossRef] [Green Version]
- Ribas, A.; Dummer, R.; Puzanov, I.; VanderWalde, A.; Andtbacka, R.H.I.; Michielin, O.; Olszanski, A.J.; Malvehy, J.; Cebon, J.; Fernandez, E.; et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 2017, 170, 1109–1119.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilsed, C.M.; Casey, T.H.; de Jong, E.; Bosco, A.; Zemek, R.M.; Salmons, J.; Wan, G.; Millward, M.J.; Nowak, A.K.; Lake, R.A.; et al. Retinoic acid induces an IFN-driven inflammatory tumor microenvironment, sensitizing to immune checkpoint therapy. Front. Oncol. 2022, 12, 849793. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD-L1 and BRAF inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Ni, J.; Galani, I.E.; Cerwenka, A.; Schirrmacher, V.; Fournier, P. Antitumor vaccination by Newcastle disease virus hemagglutinin-neuraminidase plasmid DNA application: Changes in tumor microenvironment and activation of innate anti-tumor immunity. Vaccine 2011, 29, 1185–1193. [Google Scholar] [CrossRef]
- Umansky, V.; Shatrov, V.A.; Lehmann, V.; Schirrmacher, V. Induction of NO synthesis in macrophages by Newcastle disease virus is associated with activation of nuclear factor-kappa B. Int. Immunol. 1996, 8, 491–498. [Google Scholar] [CrossRef]
- Washburn, B.; Weigand, M.A.; Grosse-Wilde, A.; Janke, M.; Stahl, H.; Rieser, E.; Sprick, M.R.; Schirrmacher, V.; Walczak, H. TNF-related apoptosis-inducing ligand mediates tumoricidal activity of human monocytes stimulated by Newcastle disease virus. J. Immunol. 2003, 170, 1814–1821. [Google Scholar] [CrossRef] [Green Version]
- Schirrmacher, V. Signaling through RIG-I and type I interferon receptor: Immune activation by Newcastle disease virus in man versus immune evasion by Ebola virus (Review). Int. J. Mol. Sci. 2015, 36, 3–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaslavsky, E.; Hershberg, U.; Seto, J.; Pham, A.M.; Marquez, S.; Duke, J.L.; Wetmur, J.C.; Tenowever, B.R.; Sealfron, S.C.; Kleinstein, S.H. Antiviral response dictated by a choreographed cascade of transcription factors. J. Immunol. 2010, 184, 2908–2917. [Google Scholar] [CrossRef] [PubMed]
- Termeer, C.C.; Schirrmacher, V.; Bröcker, E.B.; Becker, J.C. Newcastle disease virus infection induces B7-1/b7-2-independent T-cell costimulatory activity in human melanoma cells. Cancer Gene Ther. 2000, 7, 316–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ertel, C.; Millar, N.S.; Emmerson, P.T.; Schirrmacher, V.; von Hoegen, P. Viral hemagglutinin augments peptide-specific cytotoxic T cell response. Eur. J. Immunol. 1993, 23, 2592–2596. [Google Scholar] [CrossRef] [PubMed]
- Von Hoegen, P.; Zawatzky, R.; Schirrmacher, V. Modification of tumor cells by a low dose of Newcastle disease virus: III. Potentiation of tumor-specific cytolytic T cell activation via induction of interferon a,ß. Cell. Immunol. 1990, 126, 80–90. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Valenzuela, J.O.; Agarwal, P.; Lins, D.; Mescher, M.F. Type I IFNs provide a third signal to CD8+ T cells to stimulate clonal expansion and differentiation. J. Immunol. 2005, 174, 4465–4469. [Google Scholar] [CrossRef] [Green Version]
- Jergovic, M.; Coplen, C.; Uhrlaub, J.; Besselsen, D.G.; Cheng, S.; Smithey, M.J.; Nikolich-Zugich, J. Infection-induced type I interferons critically modulate the homeostasis and function of CD8+ naïve T cells. Nat. Commun. 2021, 12, 5303. [Google Scholar] [CrossRef]
- Shao, X.; Wang, X.; Guo, X.; Jiang, K.; Ye, T.; Chen, J.; Fang, J.; Gu, L.; Wang, S.; Zhang, G.; et al. STAT3 contributes to oncolytic Newcastle disease virus-induced immunogenic cell death in melanoma cells. Front. Oncol. 2019, 9, 436. [Google Scholar] [CrossRef] [Green Version]
- Schirrmacher, V.; Schlude, C.; Weitz, J.; Beckhove, P. Strong T-cell costimulation can reactivate tumor-antigen-specific T cells in late-stage metastasized colorectal carcinoma patients: Results from a phase I clinical study. Int. J. Oncol. 2015, 46, 71–77. [Google Scholar] [CrossRef] [Green Version]
- Jarahian, M.; Watzl, C.; Fournier, P.; Arnold, A.; Djandji, D.; Zahedi, S.; Cerwenka, A.; Paschen, A.; Schirrmacher, V.; Momburg, F. Activation of natural killer cells by Newcastle disease virus hemagglutinin-neuraminidase. J. Virol. 2009, 83, 8108–8121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Song, D.Z.; Liang, S.; Zhang, Z.F.; Gao, L.X.; Fan, X.H. The hemagglutinin-neuraminidase protein of Newcastle disease virus upregulates expression of the TRAIL gene in murine natural killer cells through the activation of Syk and NF-kB. PLoS ONE 2017, 12, e0178746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puhlmann, J.; Puehler, F.; Mumberg, D.; Boukamp, P.; Beier, R. Rac1 is required for oncolytic NDV replication in human cancer cells and establishes a link between tumorigenesis and sensitivity to oncolytic virus. Oncogene 2010, 29, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Sun, S.; Wang, T.; Li, Y.; Jiang, K.; Lin, G.; Ma, Y.; Barr, M.P.; Song, F.; Zhang, G.; et al. Oncolytic newcastle disease virus triggers cell death of lung cancer spheroids and is enhanced by pharmacological inhibition of autophagy. Am. J. Cancer Res. 2015, 5, 3612–3623. [Google Scholar]
- Gorji-Bahri, G.; Moghimi, H.R.; Hashemi, A. RAB5A is associated with genes involved in exosome secretion: Integration of bioinformatics analysis and experimental validation. J. Cell. Biochem. 2021, 122, 425–441. [Google Scholar] [CrossRef]
- Xu, X.; Qian, J.; Ding, J.; Li, J.; Nan, F.; Wang, W.; Qin, Q.; Fei, Y.; Xue, C.; Wang, J.; et al. Detection of viral components in exosomes derived from NDV-infected DF-1 cells and their promoting ability in virus replication. Microb. Pathol. 2019, 128, 414–422. [Google Scholar] [CrossRef]
- Zhou, C.; Tan, L.; Sun, Y.; Qiu, X.; Meng, C.; Liao, Y.; Song, C.; Liu, W.; Nair, V.; Ding, C.; et al. Exosomes carry microRNAs into neighboring cells to promote diffusive infection of Newcastle disease virus. Viruses 2019, 11, 527. [Google Scholar] [CrossRef] [Green Version]
- Al-Ziaydi, A.G.; Al-Shammari, A.M.; Hamzah, M.I.; Kadhim, H.S.; Jabir, M.S. Newcastle disease virus suppress glycolysis pathway and induce breast cancer cells death. Virusdisease 2020, 31, 341–348. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Shin, D.H.; Sohoni, S.; Singh, S.K.; Rivera-Molina, Y.; Jiang, H.; Fan, X.; Gumin, J.; Lang, F.F.; Alvarez-Breckenridge, C.; et al. Reshaping the tumor microenvironment with oncolytic viruses, positive regulation of the immune synapse, and blockade of the immunoduppressive oncometabolic circuitry. J. Immunother. Cancer 2022, 10, e004935. [Google Scholar] [CrossRef]
- Ju, F.; Luo, Y.; Lin, C.; Jia, X.; Xu, Z.; Tian, R.; Lin, Y.; Zhao, M.; Chang, Y.; Huang, X.; et al. Oncolytic virus expressing PD-1 inhibitors activates a collaborative intratumoral immune response to control tumor and synergizes with CTLA-4 or TIM-3 blockade. J. Immunother. Cancer 2022, 10, e004762. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Van Gool, S.; Stuecker, W. Breaking therapy resistance: An update on oncolytic Newcastle disease virus for improvements of cancer therapy. Biomedicines 2019, 7, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ch’ng, W.C.; Stanbridge, E.J.; Yusoff, K.; Shafee, N. The oncolytic activity of Newcastle disease virus infection in clear cell renal carcinoma cells in normoxic and hypoxic conditions: The interplay between von Hippel-Lindau and interferon-ß signaling. J. Interferon Cytokine Res. 2013, 33, 346–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mansour, M.; Palese, P.; Zamarin, D. Oncolytic specificity of Newcastle disease virus is mediated by selectivity for apoptosis-resitant cells. J. Virol. 2011, 85, 6015–6023. [Google Scholar] [CrossRef] [PubMed]
- Lazar, I.; Yaacov, B.; Shiloach, T. The oncolytic activity of Newcastle disease virus NDV-HUJ on chemoresistant primary melanoma cells is dependent on the proapoptotic activity of the inhibitor of apoptosis protein Livin. J. Virol. 2010, 84, 639–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Long, Y.; Liu, B.; Yang, D.; Li, C.; Chen, T.; Wang, X.; Liu, C.; Zhu, H. ISG12a mediates cell response to Newcastle disease viral infection. Virology 2014, 462–463, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Ji, Y.R.; Lee, Y.M. Crosstalk between angiogenesis and immune regulation in the tumor microenvironment. Arch. Pharm. Res. 2022, 45, 401–416. [Google Scholar] [CrossRef]
- Cardama, G.A.; Alonso, D.F.; Gonzales, N.; Maggio, J.; Gomez, D.E.; Rolfo, C.; Menna, P.L. Relevance of small GTPase Rac1 pathway in drug- and radio-resistance mechanisms: Opportunities in cancer therapeutics. Crit. Rev. Oncol./Hematol. 2018, 124, 29–36. [Google Scholar] [CrossRef]
- Oseledchyk, A.; Ricca, J.M.; Gigoux, M.; Ko, B.; Redelman-Sidi, G.; Walther, T.; Liu, C.; Iyer, G.; Merghoub, T.; Wolchok, J.D.; et al. Lysis-independent potentiation of immune checkpoint blockade by oncolytic virus. Oncotarget 2018, 9, 28702–28716. [Google Scholar] [CrossRef] [Green Version]
- Ricca, J.M.; Oseledchyk, A.; Walther, T.; Liu, C.; Mangarin, L.; Merghoub, T.; Wolchok, J.D.; Zamarin, D. Pre-existing immunity to oncolytic virus potentiates its immunotherapeutic efficacy. Mol. Ther. 2018, 26, 1008–1019. [Google Scholar] [CrossRef] [Green Version]
- Schulze, T.; Kemmner, W.; Weitz, J.; Wernecke, K.-D.; Schirrmacher, V.; Schlag, P.M. Efficiency of adjuvant active-specific immunization with Newcastle disease virus modified tumor cells in colorectal cancer patients following resection of liver metastases: Results of a prospective randomized trial. Cancer Immunol. Immunother. 2009, 58, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V.; Fournier, P.; Schlag, P. Autologous tumor cell vaccines for post-operative active-specific immunotherapy of colorectal carcinoma: Long-term patient survival and mechanism of function. Expert Rev. Vaccines 2014, 13, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Versteven, M.; Flumens, D.; Campillo-Davo, D.; De Reu, H.; Van Bruggen, L.; Peeters, S.; Tendeloo, V.V.; Berneman, Z.; Dolstra, H.; Anguille, S.; et al. Anti-tumor potency of short-term interleukin-15 dendritic cells is potentiated by in situ silencing of programmed-death ligands. Front. Immunol. 2022, 13, 734256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Du, J.; Song, Q.; Zhang, C.; Wu, X. A novel in situ dendritic cell vaccine triggered by Rose Bengal enhances adaptive antitumor immunity. J. Immunol. Res. 2022, 1178874. [Google Scholar] [CrossRef]
- Chen, A.; Wu, L.; Luo, Y.; Lu, S.; Wang, Y.; Zhou, Z.; Zhou, D.; Xie, Z.; Yue, J. Deep tumor penetrating gold nano-adjuvant for NIR-II-triggered in situ tumor vaccination. Small 2022, e2200993. [Google Scholar] [CrossRef]
- Schirrmacher, V.; Lorenzen, D.; Van Gool, S.W.; Stuecker, W. A new strategy of cancer immunotherapy combining hyperthermia/oncolytic virus pretreatment with specific autologous anti-tumor vaccination—A review. Austin Oncol. Case Rep. 2017, 2, 1006. [Google Scholar]
- Janke, M.; Peeters, B.; de Leeuw, O.; Moormann, R.; Arnold, A.; Fournier, P.; Schirrmacher, V. Recombinant Newcastle disease virus (NDV) with inserted gene coding for GM-CSF as a new vector for cancer immunogene therapy. Gene Ther. 2007, 14, 1639–1649. [Google Scholar] [CrossRef]
- Harper, J.; Burke, S.; Travers, J.; Rath, N.; Leinster, A.; Navarro, C.; Franks, R.; Leyland, R.; Mulgrew, K.; McGlinchey, K.; et al. Recombinant Newcastle disease virus immunotherapy drives oncolytic effects and durable systemic antitumor immunity. Mol. Cancer Ther. 2021, 20, 1723–1734. [Google Scholar] [CrossRef]
- Pühler, F.; Willuda, J.; Mumberg, D.; Römer-Oberdörfer, A.; Beier, R. Generation of a recombinant Newcastle disease virus and expression of a full IgG antibody from two transgenes. Gene Ther. 2008, 15, 371–383. [Google Scholar] [CrossRef]
- Wei, D.; Li, X.-L.; Wang, Y.; Xu, J.; Feng, F.; Nan, G.; Wang, B.; Li, C.; Guo, T.; Chen, Z.-N.; et al. Oncolytic Newcastle disease virus expressing chimeric antibody enhanced anti-tumor efficacy in orthotopic hepatoma-bearing mice. J. Exp. Clin. Cancer Res. 2015, 34, 153. [Google Scholar] [CrossRef] [Green Version]
- Pfirschke, C.; Schirrmacher, V. Cross-infection of tumor cells by contact with T lymphocytes loaded with Newcastle disease virus. Int. J. Oncol. 2009, 34, 951–962. [Google Scholar] [CrossRef] [Green Version]
- Keshavarz, M.; Ebrahimzadeh, M.S.; Miri, S.M.; Dianat-Moghadam, H.; Ghorbanhosseini, S.S.; Mohebbi, S.R.; Keyvani, H.; Ghaemi, A. Oncolytic Newcastle disease virus delivered by mesenchymal stem cell-engineered system enhances the therapeutic effects altering tumor microenvironment. Virol. J. 2020, 17, 64. [Google Scholar] [CrossRef] [PubMed]
- Heicappell, R.; Schirrmacher, V.; von Hoegen, P.; Ahlert, T.; Appelhans, B. Prevention of metastatic spread by postoperative immunotherapy with virally modified autologous tumor cells. I. Parameters for optimal therapeutic effects. Int. J. Cancer 1986, 37, 569–577. [Google Scholar] [CrossRef]
- Schild, H.; von Hoegen, P.; Schirrmacher, V. Modification of tumor cells by a low dose of Newcastle disease virus. II. Augmented tumor-specific T cell response as a result of CD4+ and CD8+ immune T cell cooperation. Cancer Immunol. Immunother. 1989, 28, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Zangemeister-Wittke, U.; Kyewski, B.; Schirrmacher, V. Recruitment and activation of tumor-specific immune T cells in situ. CD8+ cells predominate the secondary response in sponge matrices and exert both delayed-type hypersensitivity-like and cytotoxic T lymphocyte activity. J. Immunol. 1989, 143, 379–385. [Google Scholar] [PubMed]
- Delfanti, G.; Dellabona, P.; Casorati, G.; Fedeli, M. Adoptive immunotherapy with engineered iNKT cells to target cancer cells and the suppressive microenvironment. Front. Med. 2022, 9, 897750. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-R.; Zeng, S.; Dunn, Z.S.; Zhou, Y.; Li, Z.; Yu, J.; Wang, Y.-C.; Ku, J.; Cook, N.; Kramer, A.; et al. Off-the-shelf third-party HSC-engineered iNKT cells for ameliorating GvHD while preserving GvL effect in the treatment of blood cancers. iScience 2022, 25, 104859. [Google Scholar] [CrossRef]
- Zhou, Y.; Husman, T.; Cen, X.; Tsao, T.; Brown, J.; Bajpai, A.; Li, M.; Zhou, K.; Yang, L. Interleukin 15 in cell-based cancer immunotherapy. Int. J. Mol. Sci. 2022, 23, 7311. [Google Scholar] [CrossRef]
- Li, Q.; Hu, W.; Liao, B.; Song, C.; Li, L. Natural high-avidity T-cell receptor efficiently mediates regression of cancer/testis antigen 83 positive common solid cancers. J. Immunother. Cancer 2022, 10, e004713. [Google Scholar] [CrossRef]
- Evgin, L.; Kottke, T.; Tonne, J.; Thompson, J.; Huff, A.L.; van Vloten, J.; Moore, M.; Michael, J.; Driscoli, C.; Pulido, J.; et al. Oncolytic virus-mediated expansion of dual-specific CAR T cells improves efficacy against solid tumors in mice. Sci. Transl. Med. 2022, 14, eabn2231. [Google Scholar] [CrossRef]
- Wildes, T.J.; Grippin, A.; Dyson, K.A.; Wummer, B.M.; Damiani, D.J.; Abraham, R.S.; Flores, C.T.; Mitchell, D.A. Cross-talk between T cells and hematopoietic stem cells during adoptive cellular therapy for malignant glioma. Clin. Cancer Res. 2018, 24, 3955–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, Z.; Xing, D.; Zhang, T.; Ding, N.; Xiang, D.; Zhao, Z.; Qu, J.; Hu, C.; Shen, X.; Xue, X.; et al. Tumor-infiltrating B cell is associated with the control of progression of gastric cancer. Immunol. Res. 2021, 69, 43–52. [Google Scholar] [CrossRef]
- Cordell, E.C.; Alghamri, M.S.; Castro, M.G.; Gutmann, D.H. T lymphocytes as dynamic regulators of glioma pathobiology. Neuro Oncol. 2022, noac055. [Google Scholar] [CrossRef] [PubMed]
- Mohme, M.; Schliffke, S.; Maire, C.L.; Rünger, A.; Glau, L.; Mende, K.C.; Matschke, J.; Gehbauer, C.; Akyüz, N.; Zapf, S.; et al. Immunophenotyping of newly diagnosed and recurrent glioblastoma defines distinct immune exhaustion profiles in peripheral and tumor-infiltrating lymphocytes. Clin. Cancer Res. 2018, 24, 4187–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollis, P.M.; Ebert, L.M.; Toubia, J.; Bastow, C.R.; Ormsky, R.J.; Poonnoose, S.I.; Lenin, S.; Tea, M.N.; Pitson, S.M.; Gomez, G.A.; et al. Characterising distinct migratory profiles of infiltrating T-cell subsets in human glioblastoma. Front. Immunol. 2022, 13, 850226. [Google Scholar] [CrossRef]
- Csatary, L.K.; Gosztonyi, G.; Szeberenyi, J.; Fabian, Z.; Liszka, V.; Bodey, B.; Csatary, C.M. MTH-68/H oncolytic virus treatment in hig-grade gliomas. J. Neurooncol. 2004, 67, 83–93. [Google Scholar] [CrossRef]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef]
- Steiner, H.H.; Bonsanto, M.M.; Beckhove, P.; Brysch, M.; Geletneky, K.; Ahmadi, R.; Schuele-Freyer, R.; Kremer, P.; Ranaie, C.; Matejic, D.; et al. Antitumor vaccination of patients with glioblastoma multiforme: A pilot study to assess feasibility, safety, and clinical benefit. J. Clin. Oncol. 2004, 22, 4272–4281. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Feyen, O.; Prix, L.; Schirrmacher, V.; Stuecker, W. The induction of immunogenic cell death (ICD) during maintenance chemotherapy and susequent multimodal immunotherapy for glioblastoma (GBM). Austin Oncol. Case Rep. 2018, 3, 1010. [Google Scholar]
- Van Gool, S.W.; Makalowski, J.; Bitar, M.; Van de Vliet, P.; Schirrmacher, V.; Stuecker, W. Synergy between TMZ and individualized multimodal immunotherapy to improve overall survival of IDH1 wild-type MGMT promoter-unmethylated GBM patients. Genes Immun. 2022. [Google Scholar] [CrossRef]
- Van Gool, S.W.; Makalowski, J.; Fiore, S.; Sprenger, T.; Prix, L.; Schirrmacher, V.; Stuecker, W. Randomized controlled immunotherapy clinical trials for GBM challenged. Cancers 2021, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Van Gool, S.W.; Makalowski, J.; Bonner, E.R.; Feyen, O.; Domogalla, M.P.; Prix, L.; Schirrmacher, V.; Nazarian, J.; Stuecker, W. Addition of multimodal immunotherapy to combination treatment strategies for children with DIPG: A single institution experience. Medicines 2020, 7, 29. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.X.; Zhang, X.Y.; Liu, J.L.; Li, D.; Li, J.L.; Liu, Y.S.; Wang, M.; Xu, B.L.; Wang, H.B.; Wang, Z.X. Clinical efficacy of tumor antigen pulsed DC treatment for high grade glioma patients: Evidence from a meta-analysis. PLoS ONE 2014, 9, e107173. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef] [PubMed]
| Feature | Mol Determinant | Year | Comment | References |
|---|---|---|---|---|
| MHC-I TAA cross-presentation | mRNA m6A, YTHDF1 protein | 2019 | CD8+ T cell response after immunoediting in DCs | [15] |
| MHC-II neoantigens | hmMHC binding predictor | 2019 | CD4+ T helper cell responses, a key function in therapy | [17] |
| DC1 | APC function IFN-I BM DCs | 2022 2008 | ISG+ CD11b+ DCs Resident, APC function | [16] [4] |
| BM as priming site against blood-borne TAAs | ß-galactosidase (lacZ) and ovalbumin as surrogate TAAs | 2003 2003 | CD8+ T cell response in BM, potential for long-lasting protective anti-tumor immunity | [4] [11] |
| Dormant tumor cells from BM | Maintenance of TAA-specific MTCs | 2003 2005 | Adoptive MTC transfer in nude mice | [5] [6] |
| Breast cancer | BM MTC repertoire | 2006 | Polyvalent and highly individual | [19] |
| Pancreatic cancer | Functional BM MTCs | 2005 | High frequencies | [21] |
| Solid tumor patients | WT1-specific CD8+ T cells | 2010 | High frequencies BM as secondary lymphoid organ | [12] |
| Therapy of human tumors | Re-activated BM MTCs from breast cancer, Clinical study | 2001 2003 2004 2009 2013 | Adoptive MTC transfer in NOD/SCID mice Expansion capacity Overall survival | [23] [22] [24] [26] [27] |
| Rejection of advanced metastasized cancer | Vß6 donor T cells recognizing tumor-associated vSAG7 | 1995 | Adoptive transfer of allogeneic MHC identical immune T cells | [28] |
| Feature | Mol Determinant | Year | Comment | References |
|---|---|---|---|---|
| 3.1. BM TME | Targeting bone metastases | 2022 | Novel approaches | [33] |
| 3.2. CAF | ECM, cytokines, TGF-ß, PAI-1, EMT | 2022 | Increase in cancer cell motility, proinvasion and proangiogenic factors | [38] |
| 3.3. TME “cold” Barriers to T cell infiltration Tumor defence | TGF-ß1, IL10, IDO Versican Signaling CTL attack | 2006 2013 2015 2022 2022 | Induction of Treg Ovarian cancer invasion Structure and cells Mol mechanisms Tumor defense mechanisms | [51] [39] [31] [40] [41] |
| 3.4. MDSC | IRF8, COX2, STAT3 | 2019 2021 | Immunosuppression | [42] [43] |
| 3.5. Pre-metastatic niche | VEGF, GM-CSF, exosomes (TGF-ß), TIMP1 | 2022 2021 | Stromal reprogramming | [44] [45] |
| 3.6. M2 TAM | IL10, TGF-ß1, MIF, PD-L1, ncRNAs | 2022 2022 | Inhibition of Th1, FGFs Exosomes | [46] [47] |
| 3.7. Tolerogenic DC | Th17 responses DC | 2020 2020 | Pancreatic cancer Role in immunotherapy | [50] [49] |
| 3.8. Anergic T cells | Low TCR, perforin, Fas-L, PD1 | 2020 2015 | Checkpoint inhibition of CTL TILs, negative receptor signaling | [55] [31] |
| T reg cells | Secreting TGF-ß and IL-10 | 2022 | Promotion of T cell dysfunction | [58] |
| 3.9. Dysfunctional NK cells | Exosomes from PMN-MDSC TGF-ß and CIS | 2021 2022 2022 | Suppression of NK cell function in the TME Inhibition of IL-15 signaling | [59 [60] [61] |
| 3.10. Tumor-derived factors | TGF-ß, chemokines, prostaglandins, lactic acid | 2006 | Recruitment of immune inhibitory cells | [62] |
| 3.11. Metabolic barrier, hypoxia, T cell exhaustion | PD1 Dysfunctional mitochondria VEGF, bFGF HIF | 2020 1971 2019 | Depletion of nutrients, Accumulation of waste products Angiogenesis Role of HIF genes | [63] [32] [64] |
| 3.12. EMT | TGF-ß1 | 2014 | Retinopathy | [72] |
| 3.13. Therapy resistance | Genome instability, Tumor promoting inflammation | 2000 2021 2022 | Hallmarks of cancer Hallmarks of cancer Hallmarks of cancer | [73] [74] [75] |
| Feature | Mol Determinant | Year | Comment | References |
|---|---|---|---|---|
| 4.1. Counteracting CAF | FGF2 RIG-I, IRF3/7, IFN-ß | 2015 2009 | Reduced RIG-I expression, Promotion of OV activity NDV susceptibility linked to reduced expression | [76] [77] |
| 4.2. Turning cold Into hot tumors | Retinoic acid NDV induced ICD DC maturation + ICI OV treatment: TVEC + ICI NDV + ICI | 2022 2022 2016 2017 2014 | Increased T cell priming, trafficking and effectorfunction Immunotherapy response Combination cancer Immunotherapy Abscopal effect | [88] [79] [89] [78] [87] |
| 4.3. Downregulating MDSC | pHN DNA plasmid vaccination | 2011 | HN as vaccine adjuvant, systemic function | [90] |
| 4.4. Macrophage activation and polarization | NO, NFkB TRAIL upregulation | 1996 2003 | Macrophage cytotoxicity | [91] [92] |
| 4.5. DC activation and polarization | 24 TFs NFkB, Th1 cytokines | 2010 2022 | 779 upregulated genes TAA cross-presentation | [94] [79] |
| 4.6. T cell activation and polarization | Costimulation via NDV HN effect Induction of IFN-I IFN-I ATV-NDV-aHN-aCD28 | 2000 1993 1990 2005 2015 | CD4 Th1 cell activation CD8 CTL activation Peptide-spec. CTL Counteracting Th2 Third signal Reactivation of anergic T cells in clinical study | [95] [96] [97] [98] [101] |
| 4.7. NK cell activation | HN-NKp46 interaction TRAIL up | 2009 2017 | Stimuating NK cell cytotoxicity | [102] [103] |
| 4.8. Targeting Rac1 | Oncolytic NDV | 2010 2022 | Link to tumorigenesis | [104] [79] |
| 4.9. Suppression of the metabolic barrier | Suppression by NDV of the glycolysis pathway, Delta-24-RGDOX plus IDO inhibitors, YST-OVH plus PD1 inhibitors | 2020 2022 2022 | Suppression of energy source for cell growth and proliferation, OV effect on Immune synapse, Localized delivery of PD-1 inhibitors | [110] [111] [112] |
| 4.10. Breaking cancer therapy resistance and immune resistance | ICD, autophagic cell death, necroptosis, pyroptosis, ferroptosis T cell tolerance ICI Oncolysis Anti-viral immunity | 2019 2018 2011 2013 2022 2011 2014 2018 2018 | Breaking resistance to chemo/radiotherapy Hypoxia, SMI Angiogenesis Apoptosis and TRAIL Murine melanoma Bladder cancer Potentiation of efficacy | [113] [119] [115] [114] [118] [115] [87] [120] [121] |
| 4.11. Recruitment of cancer-reactive MTCs from BM | Tumor cell vaccine ATV-NDV | 2009 2014 | Improvement of long-term survival of colon carcinoma in RCT study | [122] [123] |
| 4.12. In situ vaccination | IO-VACR rNDV-GM-CSF | 2017 2007 2021 | Pretreatment with NDV + mEHT, OV effect, recruitment of DCs | [127] [128] [129] |
| 4.13. Antibody modified NDV | rNDV-vL-vH rNDV-CD147 | 2008 2015 | Anti-angiogenesis Anti-hepatoma | [130] [131] |
| 4.14. NDV-loaded carrier cells | Activated T cells, Mesenchymal stem cells | 2009 2020 | Cross-infection of tumor cells, Altering the TME | [132] [133] |
| 4.15. Active specific immunization | ATV-NDV post-operative vaccination, T-T cooperation | 1986 1989 | Prevention of metastatic spread, Augmented T cell response | [134] [135] |
| 4.16. Adoptive cellular immunotherapy | BM MTCs GvL counteracting advanced cancer | 2001 1995 2014 | Therapy of human tumor xenografts, A role of vSAG-7 reactive vß6 T cells | [23] [28] [29] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schirrmacher, V.; van Gool, S.; Stuecker, W. Counteracting Immunosuppression in the Tumor Microenvironment by Oncolytic Newcastle Disease Virus and Cellular Immunotherapy. Int. J. Mol. Sci. 2022, 23, 13050. https://doi.org/10.3390/ijms232113050
Schirrmacher V, van Gool S, Stuecker W. Counteracting Immunosuppression in the Tumor Microenvironment by Oncolytic Newcastle Disease Virus and Cellular Immunotherapy. International Journal of Molecular Sciences. 2022; 23(21):13050. https://doi.org/10.3390/ijms232113050
Chicago/Turabian StyleSchirrmacher, Volker, Stefaan van Gool, and Wilfried Stuecker. 2022. "Counteracting Immunosuppression in the Tumor Microenvironment by Oncolytic Newcastle Disease Virus and Cellular Immunotherapy" International Journal of Molecular Sciences 23, no. 21: 13050. https://doi.org/10.3390/ijms232113050
APA StyleSchirrmacher, V., van Gool, S., & Stuecker, W. (2022). Counteracting Immunosuppression in the Tumor Microenvironment by Oncolytic Newcastle Disease Virus and Cellular Immunotherapy. International Journal of Molecular Sciences, 23(21), 13050. https://doi.org/10.3390/ijms232113050