
Quantitative Proteomics Analysis Reveals That Cyclooxygenase-2 Modulates Mitochondrial Respiratory Chain Complex IV in Cardiomyocytes

 , , ,
, , ,  ,
, 
Abstract
:1. Introduction
2. Results
2.1. Proteome Differences in Cardiomyocytes from hCOX-2-Tg Mice
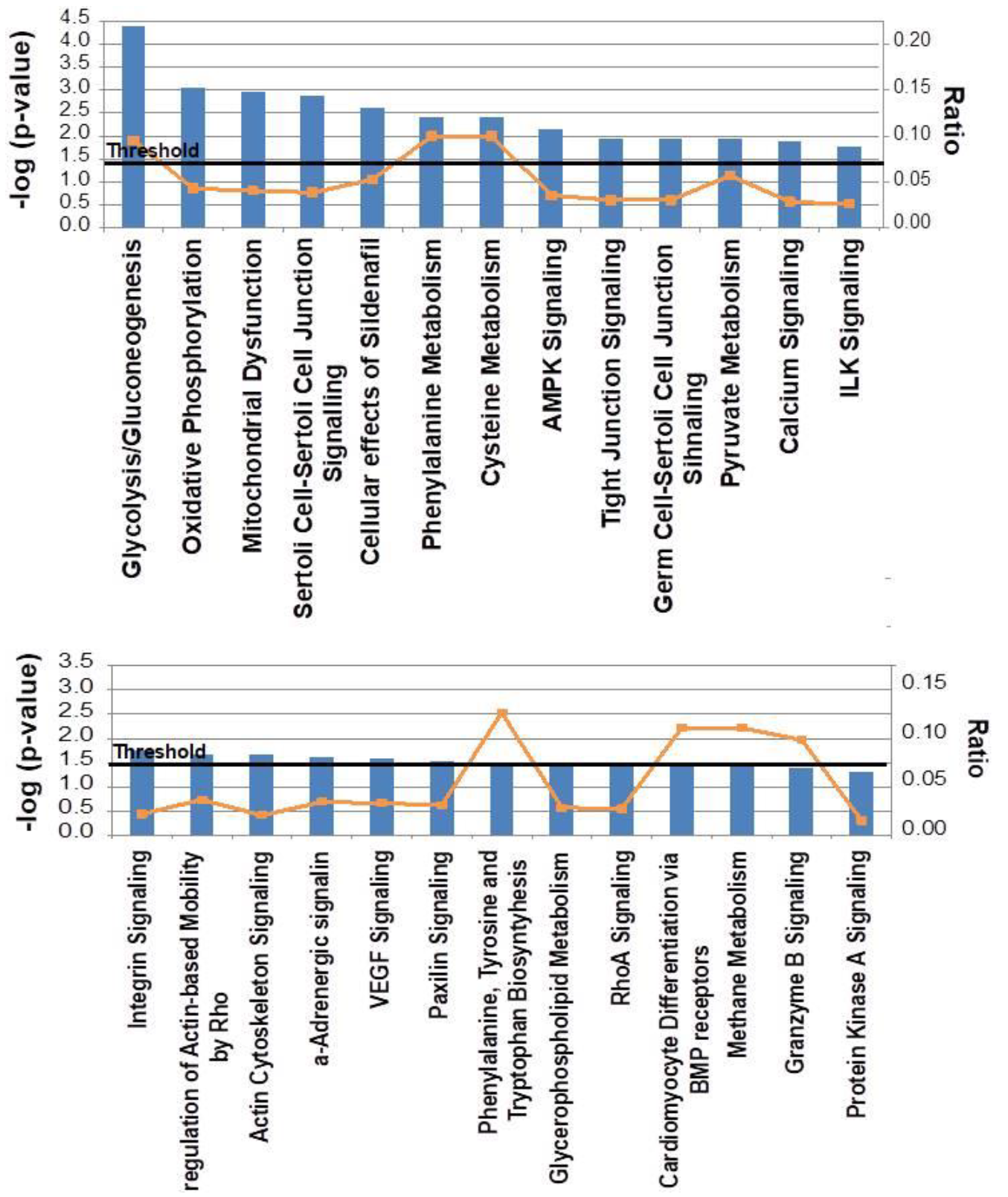
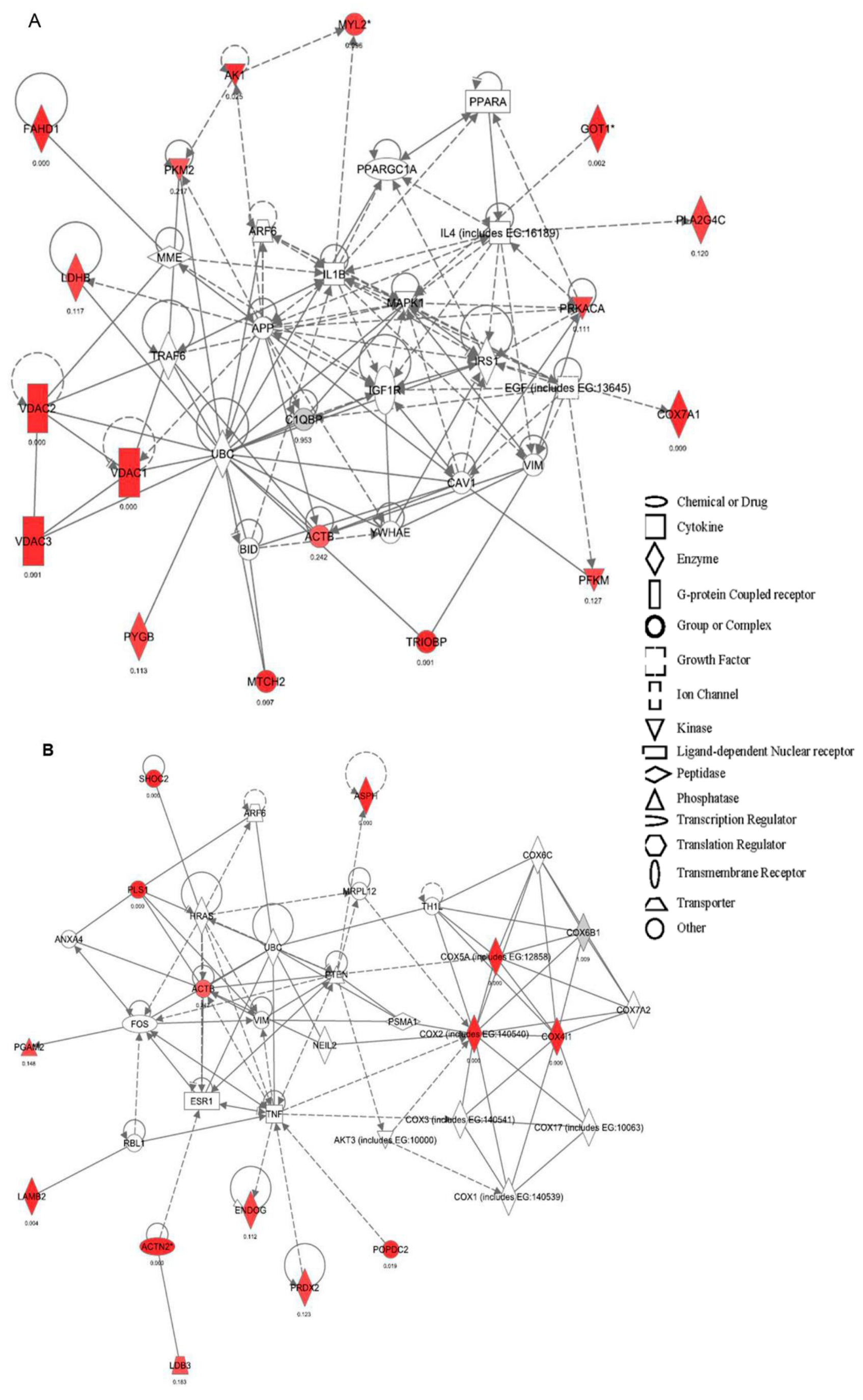
2.2. Canonical Pathways and Protein Network Analysis Altered in hCOX-2 Tg Animals
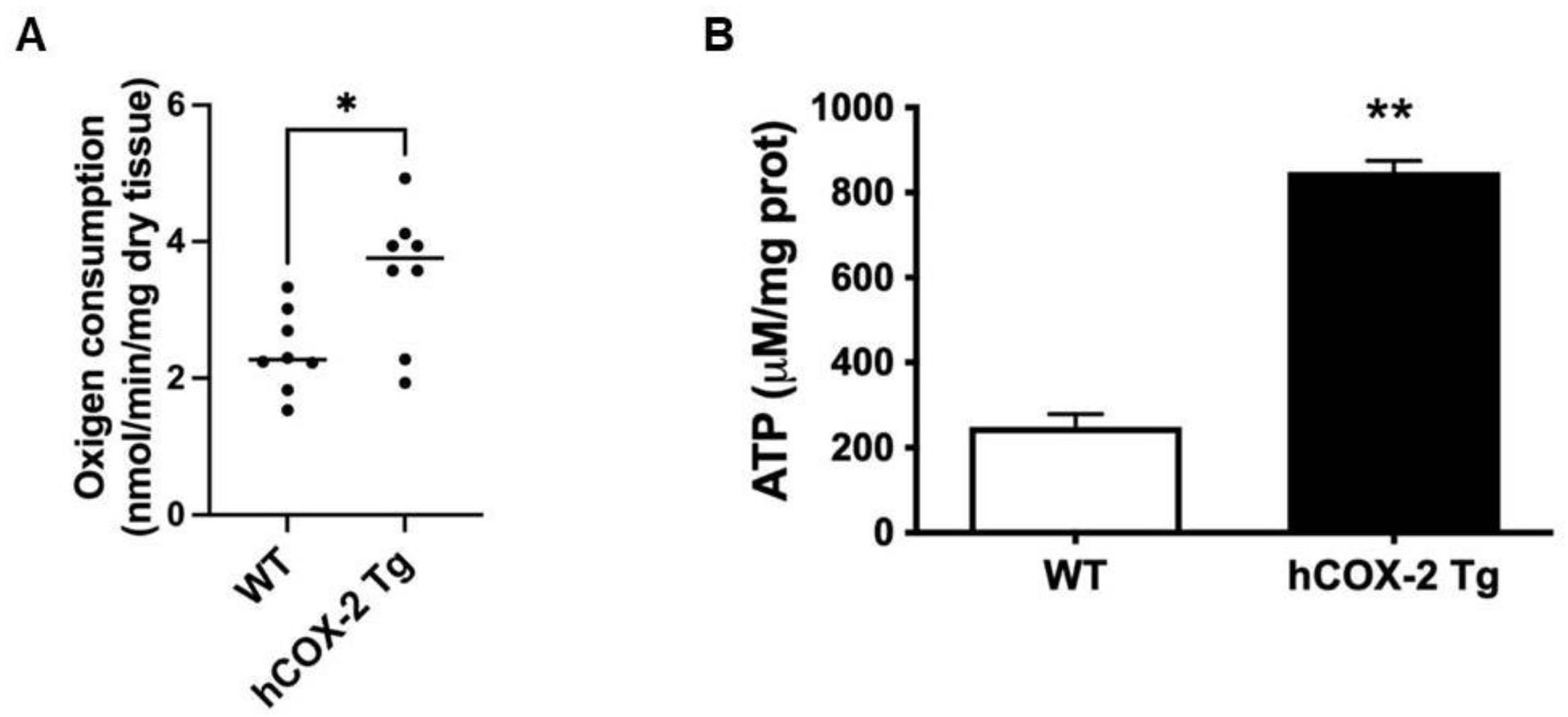
2.3. Oxygen Consumption Rate and ATP Generation Are Elevated in Heart Mitochondria from hCOX-2 Tg Mice
2.4. Constitutive COX-2 Expression in Cardiomyocytes Increases the Proportion of Complex IV Proteins
2.5. Complex IV Protein Enrichment Is Related to Increased Cytochrome c oxidase Activity in hCOX-2 Tg Mice
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Animal Model
4.3. Isolation of Cardiomyocytes
4.4. Quantitative High-Throughput Analysis of Proteome by 18O Labeling
4.5. Functional Classification
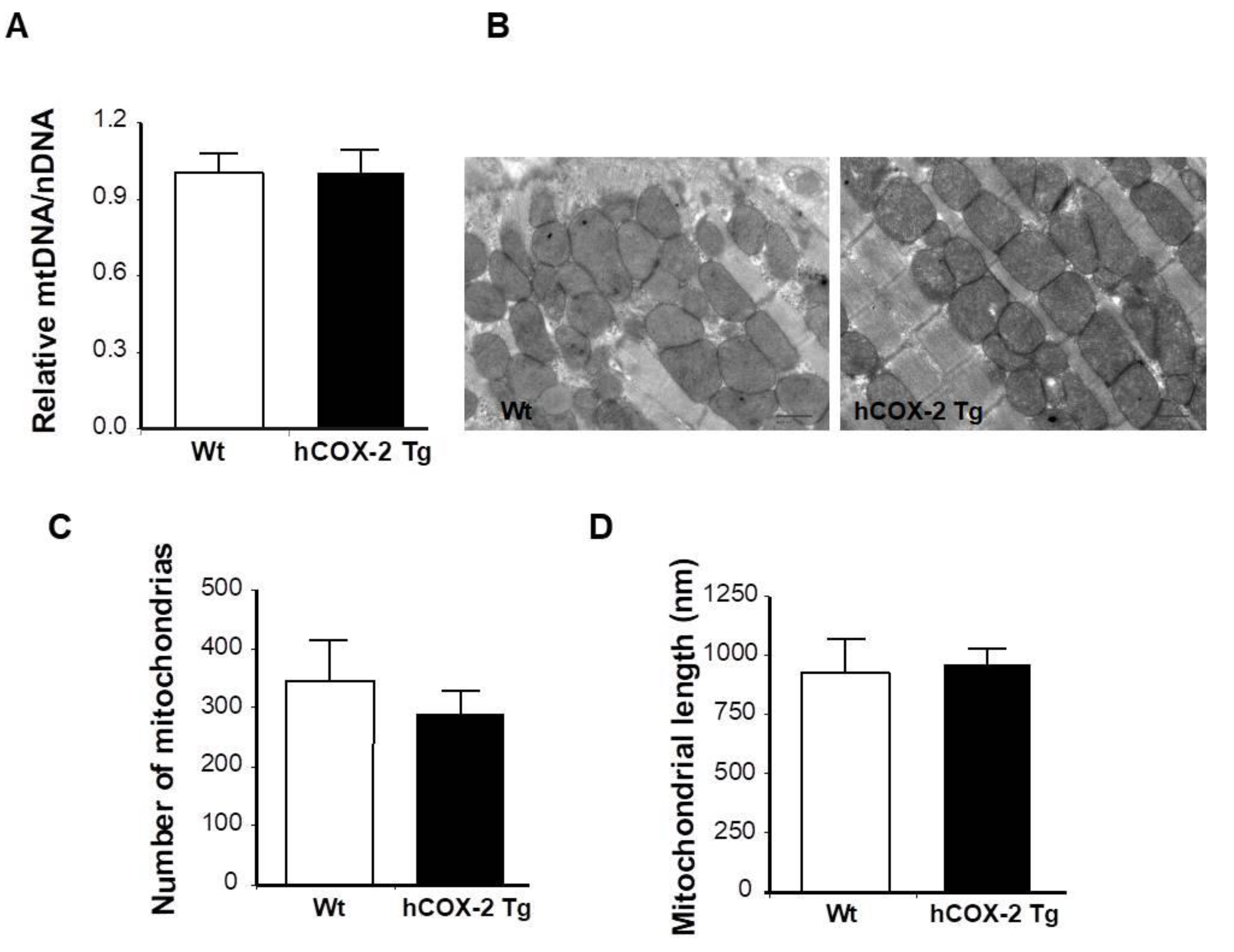
4.6. DNA Isolation and qPCR for Mitochondrial to Genomic DNA Ratio
4.7. Transmission Electron Microscopy (TEM)
4.8. RNA Isolation
4.9. Preparation of Mouse Heart Mitochondria
4.10. Analysis of the Respirasome Composition by Blue-Native Electrophoresis (BNE) and Western Blotting (BNGE)
4.11. Western Blot Analysis
4.12. Mitochondrial Function
4.13. Measurement of Complex IV Activity
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Finegold, J.A.; Asaria, P.; Francis, D.P. Mortality from ischaemic heart disease by country, region, and age: Statistics from World Health Organisation and United Nations. Int. J. Cardiol. 2013, 168, 934–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Cai, M.; Xu, Y.; Swartz, H.M.; He, G. Late phase ischemic preconditioning preserves mitochondrial oxygen metabolism and attenuates post-ischemic myocardial tissue hyperoxygenation. Life Sci. 2011, 88, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Napoli, P.; Taccardi, A.A.; De Caterina, R.; Barsotti, A. Pathophysiology of ischemia-reperfusion injury: Experimental data. Italy Heart J. 2002, 3 (Suppl. 4), 24S–28S. [Google Scholar]
- Balakumar, P.; Rohilla, A.; Singh, M. Pre-conditioning and postconditioning to limit ischemia-reperfusion-induced myocardial injury: What could be the next footstep? Pharmacol. Res. 2008, 57, 403–412. [Google Scholar] [CrossRef]
- Miyamoto, T.; Ogino, N.; Yamamoto, S.; Hayaishi, O. Purification of prostaglandin endoperoxide synthetase from bovine vesicular gland microsomes. J. Biol. Chem. 1976, 251, 2629–2636. [Google Scholar] [CrossRef]
- Williams, C.S.; Mann, M.; DuBois, R.N. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999, 18, 7908–7916. [Google Scholar] [CrossRef] [Green Version]
- Koki, A.; Khan, N.K.; Woerner, B.M.; Dannenberg, A.J.; Olson, L.; Seibert, K.; Edwards, D.; Hardy, M.; Isakson, P.; Masferrer, J.L. Cyclooxygenase-2 in human pathological disease. Adv. Exp. Med. Biol. 2002, 507, 177–184. [Google Scholar]
- Goren, N.; Cuenca, J.; Martin-Sanz, P.; Bosca, L. Attenuation of NF-kappaB signalling in rat cardiomyocytes at birth restricts the induction of inflammatory genes. Cardiovasc. Res. 2004, 64, 289–297. [Google Scholar] [CrossRef]
- Saito, T.; Rodger, I.W.; Hu, F.; Robinson, R.; Huynh, T.; Giaid, A. Inhibition of COX pathway in experimental myocardial infarction. J. Mol. Cell. Cardiol. 2004, 37, 71–77. [Google Scholar] [CrossRef]
- Bolli, R. Preconditioning: A paradigm shift in the biology of myocardial ischemia. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H19–H27. [Google Scholar] [CrossRef] [Green Version]
- Camitta, M.G.; Gabel, S.A.; Chulada, P.; Bradbury, J.A.; Langenbach, R.; Zeldin, D.C.; Murphy, E. Cyclooxygenase-1 and -2 knockout mice demonstrate increased cardiac ischemia/reperfusion injury but are protected by acute preconditioning. Circulation 2001, 104, 2453–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inserte, J.; Molla, B.; Aguilar, R.; Través, P.G.; Barba, I.; Martín-Sanz, P.; Boscá, L.; Casado, M.; Garcia-Dorado, D. Constitutive COX-2 activity in cardiomyocytes confers permanent cardioprotection. Constitutive COX-2 expression and cardioprotection. J. Mol. Cell. Cardiol. 2009, 46, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Jorge, I.; Navarro, P.; Martinez-Acedo, P.; Nunez, E.; Serrano, H.; Alfranca, A.; Redondo, J.M.; Vazquez, J. Statistical model to analyze quantitative proteomics data obtained by 18O/16O labeling and linear ion trap mass spectrometry: Application to the study of vascular endothelial growth factor-induced angiogenesis in endothelial cells. Mol. Cell. Proteom. 2009, 8, 1130–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Marqués, F.; Trevisan-Herraz, M.; Martínez-Martínez, S.; Camafeita, E.; Jorge, I.; Lopez, J.A.; Méndez-Barbero, N.; Méndez-Ferrer, S.; Del Pozo, M.A.; Ibáñez, B.; et al. A novel systems-biology algorithm for the analysis of coordinated protein responses using quantitative proteomics. Mol. Cell. Proteom. 2016, 15, 1740–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Takahashi, T.; Zhu, S.J.; Sumino, H.; Saegusa, S.; Nakahashi, T.; Iwai, K.; Morimoto, S.; Kanda, T. Inhibition of cyclooxygenase-2 enhances myocardial damage in a mouse model of viral myocarditis. Life Sci. 2005, 78, 195–204. [Google Scholar] [CrossRef]
- Degousee, N.; Martindale, J.; Stefanski, E.; Cieslak, M.; Lindsay, T.F.; Fish, J.E.; Marsden, P.A.; Thuerauf, D.J.; Glembotski, C.C.; Rubin, B.B. MAP kinase kinase 6-p38 MAP kinase signaling cascade regulates cyclooxygenase-2 expression in cardiac myocytes in vitro and in vivo. Circ. Res. 2003, 92, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.; Aponte, A.M.; Steenbergen, C.; Murphy, E. Cardioprotection leads to novel changes in the mitochondrial proteome. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H75–H91. [Google Scholar] [CrossRef] [Green Version]
- Fuertes-Agudo, M.; Luque-Tévar, M.; Cucarella, C.; Brea, R.; Boscá, L.; Quintana-Cabrera, R.; Martín-Sanz, P.; Casado, M. COX-2 Expression in Hepatocytes Improves Mitochondrial Function after Hepatic Ischemia-Reperfusion Injury. Antioxidants 2022, 11, 1724. [Google Scholar] [CrossRef]
- Benard, G.; Faustin, B.; Passerieux, E.; Galinier, A.; Rocher, C.; Bellance, N.; Delage, J.P.; Casteilla, L.; Letellier, T.; Rossignol, R. Physiological diversity of mitochondrial oxidative phosphorylation. Am. J. Physiol. Cell Physiol. 2006, 291, C1172–C1182. [Google Scholar] [CrossRef] [Green Version]
- Rossignol, R.; Malgat, M.; Mazat, J.P.; Letellier, T. Threshold effect and tissue specificity. Implication for mitochondrial cytopathies. J. Biol. Chem. 1999, 274, 33426–33432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Li, J.; Wang, Y.; Yang, T. Proteomic analysis of effluents from perfused human heart for transplantation: Identification of potential biomarkers for ischemic heart damage. Proteome Sci. 2012, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Jennings, R.B.; Sebbag, L.; Schwartz, L.M.; Crago, M.S.; Reimer, K.A. Metabolism of preconditioned myocardium: Effect of loss and reinstatement of cardioprotection. J. Mol. Cell. Cardiol. 2001, 33, 1571–1588. [Google Scholar] [CrossRef]
- de Albuquerque, C.P.; Gerstenblith, G.; Weiss, R.G. Importance of metabolic inhibition and cellular pH in mediating preconditioning contractile and metabolic effects in rat hearts. Circ. Res. 1994, 74, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paillard, M.; Tubbs, E.; Thiebaut, P.A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, C.M.; Cedars, A.; Gross, R.W. Eicosanoid signalling pathways in the heart. Cardiovasc. Res. 2009, 82, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Schagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schägger, H. Respiratory Chain Supercomplexes. IUBMB Life (Int. Union Biochem. Mol. Biol. Life) 2001, 52, 119–128. [Google Scholar] [CrossRef]
- Ogbi, M.; Johnson, J.A. Protein kinase Cepsilon interacts with cytochrome c oxidase subunit IV and enhances cytochrome c oxidase activity in neonatal cardiac myocyte preconditioning. Biochem. J. 2006, 393, 191–199. [Google Scholar] [CrossRef] [Green Version]
- McGuinness, J.; Neilan, T.G.; Cummins, R.; Sharkasi, A.; Bouchier-Hayes, D.; Redmond, J.M. Intravenous glutamine enhances COX-2 activity giving cardioprotection. J. Surg. Res. 2009, 152, 140–147. [Google Scholar] [CrossRef]
- Salabei, J.K.; Lorkiewicz, P.K.; Holden, C.R.; Li, Q.; Hong, K.U.; Bolli, R.; Bhatnagar, A.; Hill, B.G. Glutamine Regulates Cardiac Progenitor Cell Metabolism and Proliferation. Stem Cells 2015, 33, 2613–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLeod, C.J.; Jeyabalan, A.P.; Minners, J.O.; Clevenger, R.; Hoyt, R.F., Jr.; Sack, M.N. Delayed ischemic preconditioning activates nuclear-encoded electron-transfer-chain gene expression in parallel with enhanced postanoxic mitochondrial respiratory recovery. Circulation 2004, 110, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Riendeau, D.; Percival, M.D.; Boyce, S.; Brideau, C.; Charleson, S.; Cromlish, W.; Ethier, D.; Evans, J.; Falgueyret, J.P.; Ford-Hutchinson, A.W.; et al. Biochemical and pharmacological profile of a tetrasubstituted furanone as a highly selective COX-2 inhibitor. Br. J. Pharmacol. 1997, 121, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Shioya, T. A simple technique for isolating healthy heart cells from mouse models. J. Physiol. Sci. 2007, 57, 327–335. [Google Scholar] [CrossRef] [Green Version]
- Bonzon-Kulichenko, E.; Perez-Hernandez, D.; Nunez, E.; Martinez-Acedo, P.; Navarro, P.; Trevisan-Herraz, M.; Ramos Mdel, C.; Sierra, S.; Martinez-Martinez, S.; Ruiz-Meana, M.; et al. A robust method for quantitative high-throughput analysis of proteomes by 18O labeling. Mol. Cell. Proteom. 2011, 10, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Ferrer, D.; Ramos-Fernandez, A.; Martinez-Bartolome, S.; Garcia-Ruiz, P.; Vazquez, J. Quantitative proteomics using 16O/18O labeling and linear ion trap mass spectrometry. Proteomics 2006, 6 (Suppl. 1), S4–S11. [Google Scholar] [CrossRef]
- Martinez-Bartolome, S.; Navarro, P.; Martin-Maroto, F.; Lopez-Ferrer, D.; Ramos-Fernandez, A.; Villar, M.; Garcia-Ruiz, J.P.; Vazquez, J. Properties of average score distributions of SEQUEST: The probability ratio method. Mol. Cell. Proteom. 2008, 7, 1135–1145. [Google Scholar] [CrossRef] [Green Version]
- Navarro, P.; Vazquez, J. A refined method to calculate false discovery rates for peptide identification using decoy databases. J. Proteome Res. 2009, 8, 1792–1796. [Google Scholar] [CrossRef]
- Navarro, P.; Trevisan-Herraz, M.; Bonzon-Kulichenko, E.; Nunez, E.; Martinez-Acedo, P.; Perez-Hernandez, D.; Jorge, I.; Mesa, R.; Calvo, E.; Carrascal, M.; et al. General statistical framework for quantitative proteomics by stable isotope labeling. J. Proteome Res. 2014, 13, 1234–1247. [Google Scholar] [CrossRef]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.; Moorman, A.F. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Gene | Description | Subcellular Localization | Zq | FDRq | Total Peptide Count |
|---|---|---|---|---|---|---|
| Q7M6Y9 | Heatr7b2 | Uncharacterized protein | ↑10.31 | 0 | 1 | |
| P00405 | Mtco2 | Cytochrome c oxidase subunit 2 | Mitochondrion inner membrane | ↑6.53 | 7.70 × 10−9 | 6 |
| Q8R0F8 | Fahd1 | Fumaryl acetoacetate hydrolase domain-containing protein 1 | Mitochondrion | ↑6.16 | 8.33 × 10−8 | 1 |
| P19783 | Cox4i1 | Cytochrome c oxidase subunit 4 isoform 1 | Mitochondrion inner membrane | ↑6.03 | 1.29 × 10−7 | 6 |
| P12787 | Cox5a | Cytochrome c oxidase subunit 5A | Mitochondrion inner membrane | ↑5.96 | 1.95 × 10−7 | 4 |
| Q9D8B4 | Ndufa11 | NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 11 | Mitochondrion inner membrane | ↑5.91 | 1.99 × 10−7 | 1 |
| Q60932 | Vdac1 | Voltage-dependent anion-selective channel protein 1 | Mitochondrion outer membrane | ↑5.37 | 4.63 × 10−6 | 7 |
| Q6AYI5 | Gpatc2 | G patch domain containing 2 | ↑5.28 | 7.64 × 10−6 | 1 | |
| P19536 | Cox5b | Cytochrome c oxidase subunit 5B | Mitochondrion inner membrane | ↑5.20 | 8.58 × 10−6 | 1 |
| Q8R2U7 | Lrrc42 | Leucine-rich repeat-containing protein 42 | ↑4.97 | 2.89 × 10−5 | 1 | |
| P56392 | Cox7a1 | Cytochrome c oxidase subunit 7A1 | Mitochondrion inner membrane | ↑4.66 | 0.0001 | 1 |
| P00397 | Mtco1 | Cytochrome c oxidase subunit 1 | Mitochondrion inner membrane | ↑4.55 | 0.0002 | 1 |
| Q60930 | Vdac2 | Voltage-dependent anion-selective channel protein 2 | Mitochondrion outer membrane | ↑4.42 | 0.0003 | 4 |
| A2AL77 | Asph | Aspartate-beta-hydroxylase | ↑4.35 | 0.0004 | 1 | |
| Q99KW3 | Triobp | TRIO and F-actin-binding protein | Cytoplasm | ↑4.12 | 0.001 | 1 |
| Q60931 | Vdac3 | Voltage-dependent anion-selective channel protein 3 | Mitochondrion outer membrane | ↑4.11 | 0.001 | 3 |
| Q61292 | Lamb2 | Laminin subunit beta-2 | Extracellular matrix | ↑3.75 | 0.004 | 1 |
| Q791V5 | Mtch2 | Mitochondrial carrier homolog 2 | Mitochondrion inner membrane | ↑3.58 | 0.007 | 1 |
| Q9ES82 | Popdc2 | Popeye domain-containing protein 2 | Membrane | ↑3.33 | 0.018 | 1 |
| Q9CRY7 | Gdpd1 | Glycerophosphodiester phosphodiesterase domain-containing protein 1 | Membrane | ↑3.31 | 0.018 | 1 |
| Q9D1R1 | Tmem126b | Transmembrane protein 126B | Membrane | ↑3.29 | 0.019 | 1 |
| D3ZX78 | Vom2r16 | Protein Vom2r16 | Membrane | ↑3.28 | 0.019 | 1 |
| Q61941 | Nnt | NAD(P) transhydrogenase | Mitochondrion inner membrane | ↓2.99 | 0.046 | 12 |
| Q9R0Y5 | Ak1 | Adenylate kinase isoenzyme 1 | Cytoplasm | ↓3.20 | 0.025 | 4 |
| Q3UJH8 | Got1 | Aspartate aminotransferase | Cytoplasm | ↓3.94 | 0.002 | 1 |
| Q8K3Q4 | Actn2 | Actinin alpha 2 | ↓4.73 | 8.16 × 10−5 | 1 | |
| Q3V0K9 | Pls1 | Plastin-1 | Cytoplasm | ↓15.88 | 0 | 1 |
| Q6TUF7 | LRRGT00087 | ↓25.09 | 0 | 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez, M.S.; Núñez, E.; Fuertes-Agudo, M.; Cucarella, C.; Fernandez-Velasco, M.; Boscá, L.; Vázquez, J.; Rossignol, R.; Martin-Sanz, P.; Casado, M. Quantitative Proteomics Analysis Reveals That Cyclooxygenase-2 Modulates Mitochondrial Respiratory Chain Complex IV in Cardiomyocytes. Int. J. Mol. Sci. 2022, 23, 13476. https://doi.org/10.3390/ijms232113476
Alvarez MS, Núñez E, Fuertes-Agudo M, Cucarella C, Fernandez-Velasco M, Boscá L, Vázquez J, Rossignol R, Martin-Sanz P, Casado M. Quantitative Proteomics Analysis Reveals That Cyclooxygenase-2 Modulates Mitochondrial Respiratory Chain Complex IV in Cardiomyocytes. International Journal of Molecular Sciences. 2022; 23(21):13476. https://doi.org/10.3390/ijms232113476
Chicago/Turabian StyleAlvarez, Maria Soledad, Estefanía Núñez, Marina Fuertes-Agudo, Carme Cucarella, Maria Fernandez-Velasco, Lisardo Boscá, Jesús Vázquez, Rodrigue Rossignol, Paloma Martin-Sanz, and Marta Casado. 2022. "Quantitative Proteomics Analysis Reveals That Cyclooxygenase-2 Modulates Mitochondrial Respiratory Chain Complex IV in Cardiomyocytes" International Journal of Molecular Sciences 23, no. 21: 13476. https://doi.org/10.3390/ijms232113476