
Engineering of Bacillus Promoters Based on Interacting Motifs between UP Elements and RNA Polymerase (RNAP) α-Subunit

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. The Selected Promoters Vary in Strength and Stage-Specific Expression
2.2. PLan Was Predicted to Be Involved in Quorum Sensing (QS)
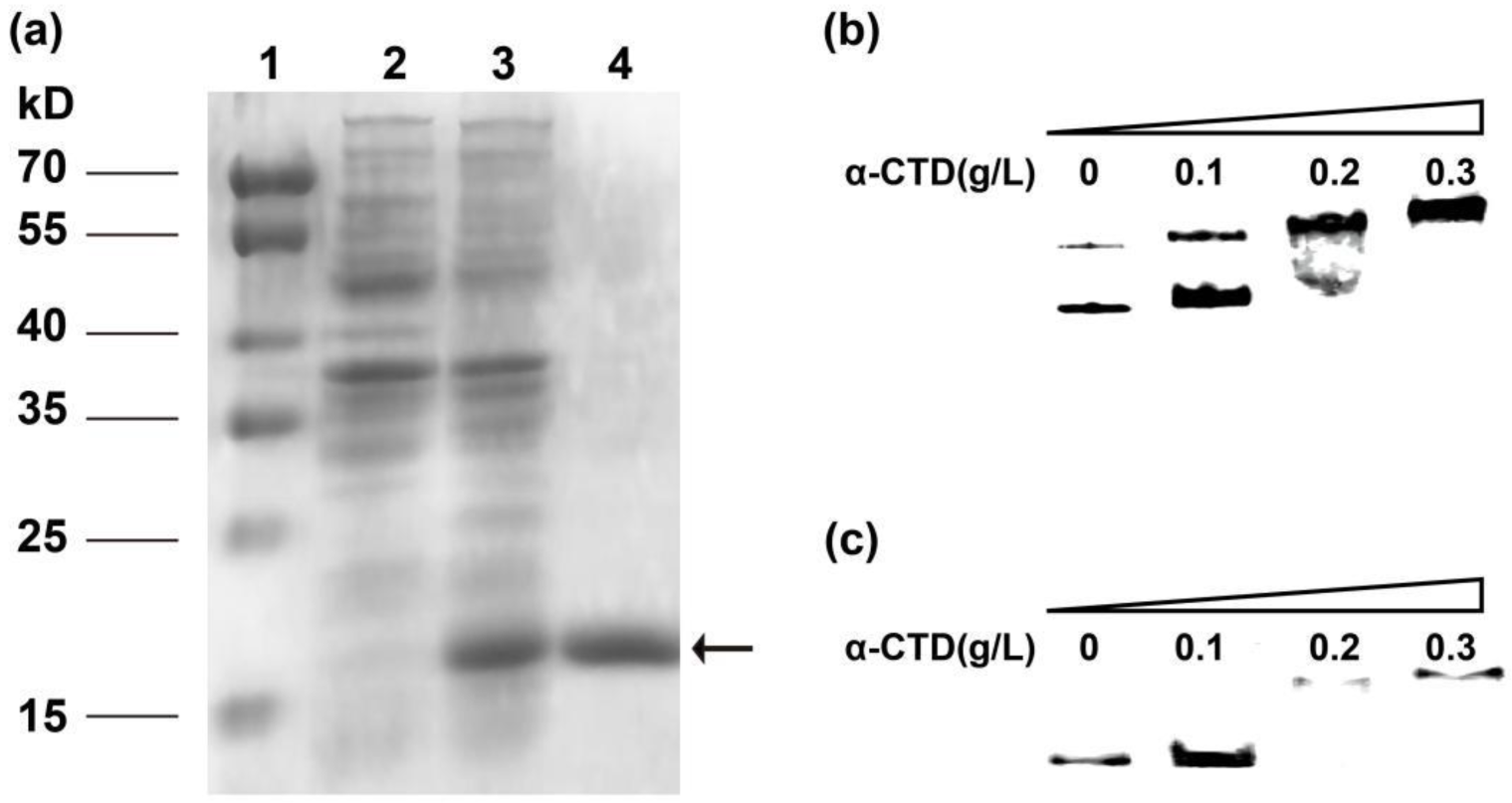
2.3. Heterologously Expressed α-CTD Directly Interacts with Both PtrxA and PLan In Vitro
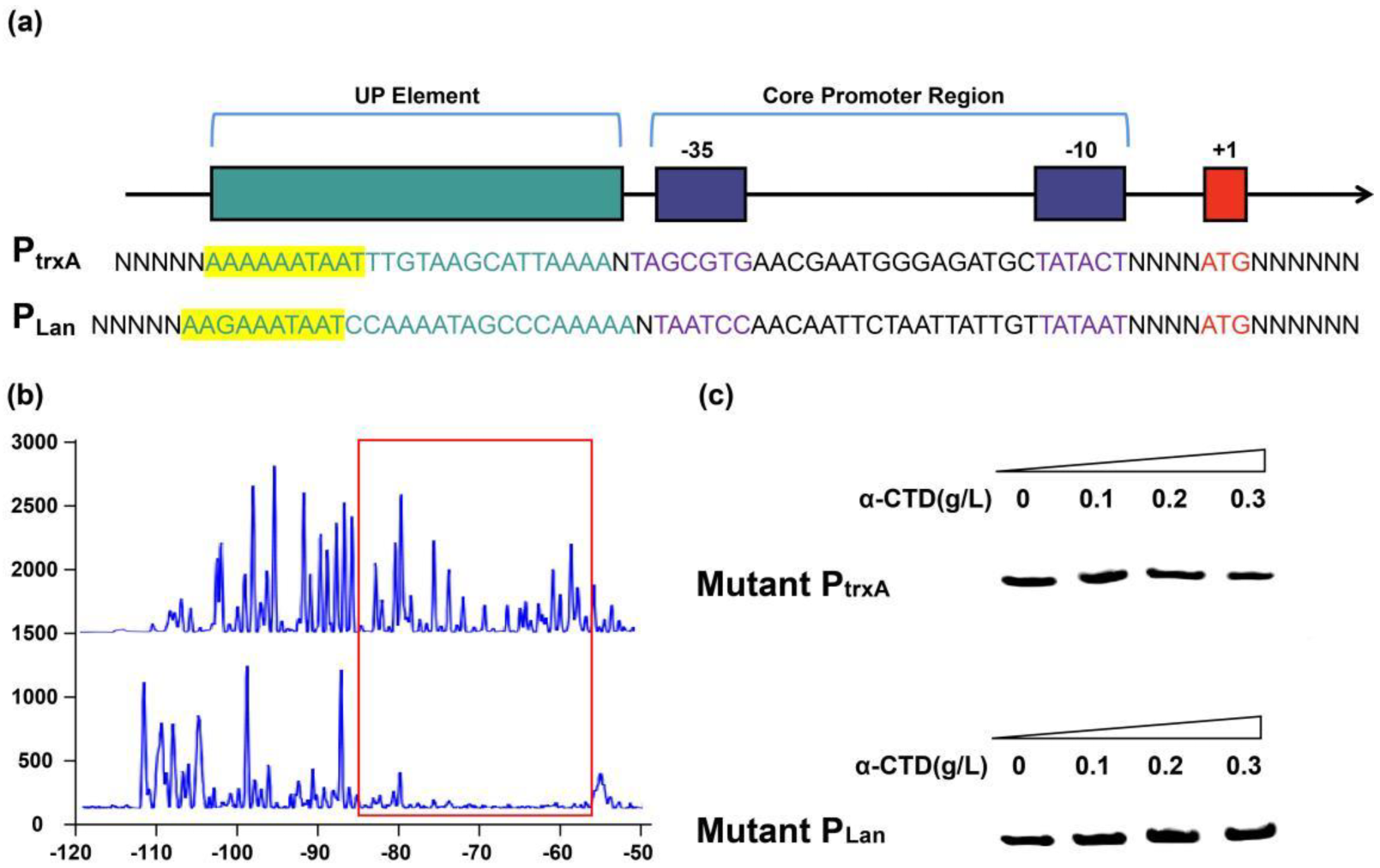
2.4. α-CTD Binds to an UP Element 50 bp Upstream of TSS
2.5. UP Elements with More Flanking Base Pairs Ensured Higher Promoter Activity
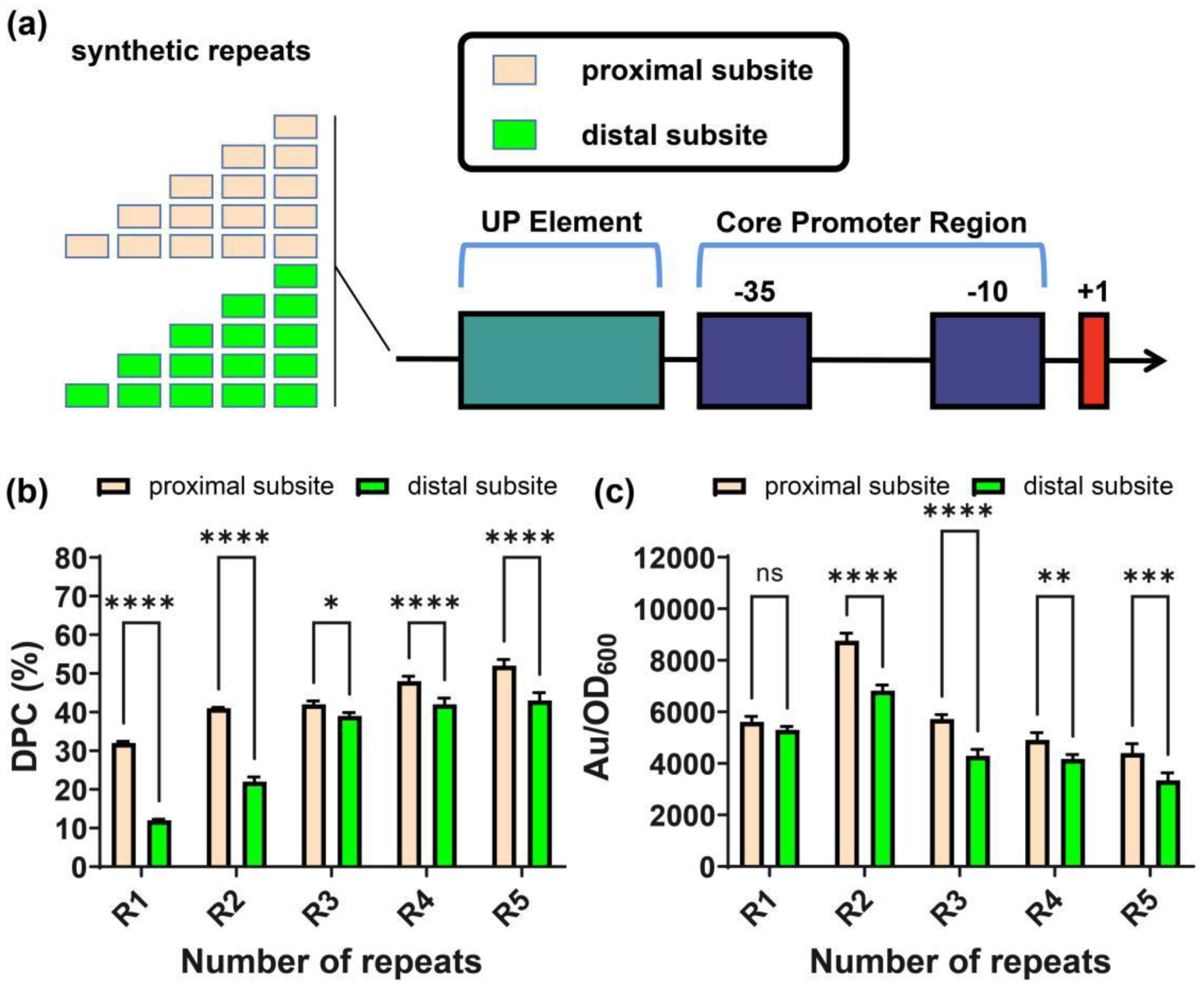
2.6. Repeated Specific Subsite in UP Elements Improved Promoter Activity
2.7. Synthetic UP Elements Improved Activity of Varied Core Promoters
3. Discussion
4. Materials and Methods
4.1. Media and Strain Cultivation
4.2. Plasmid Construction
4.3. Fluorescence Measurements
4.4. Purification of Recombinant α-CTD
4.5. Electrophoretic Mobility Shift Assays (EMSA)
4.6. Fluorescence Polarization (FP)
4.7. DNaseI Footprinting
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Segall-Shapiro, T.H.; Sontag, E.D.; Voigt, C.A. Engineered promoters enable constant gene expression at any copy number in bacteria. Nat. Biotechnol. 2018, 36, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Guiziou, S.; Sauveplane, V.; Chang, H.J.; Clerte, C.; Declerck, N.; Jules, M.; Bonnet, J. A part toolbox to tune genetic expression in Bacillus subtilis. Nucleic Acids Res. 2016, 44, 7495–7508. [Google Scholar] [PubMed] [Green Version]
- Kedzierska-Mieszkowska, S.; Potrykus, K.; Arent, Z.; Krajewska, J. Identification of sigma(E)-dependent promoter upstream of clpb from the pathogenic Spirochaete Leptospira interrogans by applying an E. coli two-plasmid system. Int. J. Mol. Sci. 2019, 20, 6325. [Google Scholar]
- Hauk, P.; Stephens, K.; McKay, R.; Virgile, C.R.; Ueda, H.; Ostermeier, M.; Ryu, K.S.; Sintim, H.O.; Bentley, W.E. Insightful directed evolution of Escherichia coli quorum sensing promoter region of the lsrACDBFG operon: A tool for synthetic biology systems and protein expression. Nucleic Acids Res. 2016, 44, 10515–10525. [Google Scholar]
- Liu, X.Y.; Gupta, S.T.P.; Bhimsaria, D.; Reed, J.L.; Rodriguez-Martinez, J.A.; Ansari, A.Z.; Raman, S. De novo design of programmable inducible promoters. Nucleic Acids Res. 2019, 47, 10452–10463. [Google Scholar] [PubMed]
- Presnell, K.V.; Flexer-Harrison, M.; Alper, H.S. Design and synthesis of synthetic UP elements for modulation of gene expression in Escherichia coli. Synth. Syst. Biotechnol. 2019, 4, 99–106. [Google Scholar] [CrossRef]
- Cazier, A.P.; Blazeck, J. Advances in promoter engineering: Novel applications and predefined transcriptional control. Biotechnol. J. 2021, 16, 2100239. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, H.C.; Wei, L.; Li, S.L.; Liu, L.Y.; Wang, X.W. Synthetic promoter design in Escherichia coli based on a deep generative network. Nucleic Acids Res. 2020, 48, 6403–6412. [Google Scholar] [CrossRef]
- Feng, X.F.; Marchisio, M.A.; Novel, S. cerevisiae Hybrid synthetic promoters based on foreign core promoter sequences. Int. J. Mol. Sci. 2021, 22, 5704. [Google Scholar] [CrossRef]
- Rangel-Chavez, C.P.; Galan-Vasquez, E.; Pescador-Tapia, A.; Delaye, L.; Martinez-Antonio, A. RNA polymerases in strict endosymbiont bacteria with extreme genome reduction show distinct erosions that might result in limited and differential promoter recognition. PLoS ONE 2021, 16, e0239350. [Google Scholar] [CrossRef]
- Shin, Y.; Hedglin, M.; Murakami, K.S. Structural basis of reiterative transcription from the pyrG and pyrBI promoters by bacterial RNA polymerase. Nucleic Acids Res. 2020, 48, 2144–2155. [Google Scholar] [PubMed] [Green Version]
- Wang, Y.; Shi, Y.; Hu, L.; Du, G.; Chen, J.; Kang, Z. Engineering strong and stress-responsive promoters in Bacillus subtilis by interlocking sigma factor binding motifs. Synth. Syst. Biotechnol. 2019, 4, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Nakano, M.M.; Lin, A.; Zuber, C.S.; Newberry, K.J.; Brennan, R.G.; Zuber, P. Promoter recognition by a complex of spx and the c-terminal domain of the rna polymerase alpha subunit. PLoS ONE 2010, 5, e8664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Ma, Y.; Zhao, Y.; Shen, W.; Chen, X. Systematic engineering of transport and transcription to boost alkaline alpha-amylase production in Bacillus subtilis. Appl. Microbiol. Biot. 2020, 104, 2973–2985. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, H.; Zheng, J.; Ye, Y.; Pan, L. Identification of strong promoters based on the transcriptome of Bacillus licheniformis. Biotechnol. Lett. 2017, 39, 873–881. [Google Scholar] [PubMed]
- Carter, H.L., 3rd; Moran, C.P., Jr. New RNA polymerase sigma factor under spo0 control in Bacillus subtilis. Proc. Natl. Acad. Sci. USA 1986, 83, 9438–9442. [Google Scholar] [CrossRef] [Green Version]
- Meinhardt, F.; Stahl, U.; Ebeling, W. Highly efficient expression of homologous and heterologous genes in Bacillus megaterium. Appl. Microbiol. Biot. 1989, 30, 343–350. [Google Scholar] [CrossRef]
- Yang, H.; Qu, J.; Zou, W.; Shen, W.; Chen, X. An overview and future prospects of recombinant protein production in Bacillus subtilis. Appl. Microbiol. Biot. 2021, 105, 6607–6626. [Google Scholar] [CrossRef]
- Yu, X.; Xu, J.; Liu, X.; Chu, X.; Wang, P.; Tian, J.; Wu, N.; Fan, Y. Identification of a highly efficient stationary phase promoter in Bacillus subtilis. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wang, H.; Wang, B.; Pan, L. Efficient production of extracellular pullulanase in Bacillus subtilis ATCC6051 using the host strain construction and promoter optimization expression system. Microb. Cell Factories 2018, 17, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Shi, C.; Li, D.; Chen, X.; Li, J.; Zhang, Y.; Yuan, H.; Li, Y.; Lu, F. Engineering a highly efficient expression system to produce BcaPRO protease in Bacillus subtilis by an optimized promoter and signal peptide. Int. J. Biol. Macromol. 2019, 138, 903–911. [Google Scholar] [PubMed]
- Knerr, P.J.; van der Donk, W.A. Discovery, biosynthesis, and engineering of lantipeptides. Annu. Rev. Biochem. 2012, 81, 479–505. [Google Scholar] [CrossRef] [PubMed]
- Dischinger, J.; Josten, M.; Szekat, C.; Sahl, H.-G.; Bierbaum, G. Production of the novel two-peptide lantibiotic lichenicidin by Bacillus licheniformis DSM 13. PLoS ONE 2009, 4, e6788. [Google Scholar] [CrossRef] [PubMed]
- George, E.A.; Muir, T.W. Molecular mechanisms of agr quorum sensing in virulent staphylococci. Chembiochem 2007, 8, 847–855. [Google Scholar] [CrossRef]
- Ji, G.Y.; Beavis, R.C.; Novick, R.P. Cell density control of staphylococcal virulence mediated by an octapeptide pheromone. Proc. Natl. Acad. Sci. USA 1995, 92, 12055–12059. [Google Scholar] [CrossRef] [Green Version]
- Geisinger, E.; George, E.A.; Muir, T.W.; Novick, R.P. Identification of ligand specificity determinants in AgrC, the Staphylococcus aureus quorum-sensing receptor. J. Biol. Chem. 2008, 283, 8930–8938. [Google Scholar] [CrossRef] [Green Version]
- Finney, A.H.; Blick, R.J.; Murakami, K.; Ishihama, A.; Stevens, A.M. Role of the C-terminal domain of the alpha subunit of RNA polymerase in LuxR-dependent transcriptional activation of the lux operon during quorum sensing. J. Bacteriol. 2002, 184, 4520–4528. [Google Scholar] [CrossRef] [Green Version]
- Ball, A.S.; van Kessel, J.C. The master quorum-sensing regulators LuxR/HapR directly interact with the alpha subunit of RNA polymerase to drive transcription activation in Vibrio harveyi and Vibrio cholerae. Mol. Microbiol. 2019, 111, 1317–1334. [Google Scholar]
- Birch, C.A.; Davis, M.J.; Mbengi, L.; Zuber, P. Exploring the amino acid residue requirements of the rna polymerase (RNAP) alpha subunit c-terminal domain for productive interaction between spx and RNAP of Bacillus subtilis. J. Bacteriol. 2017, 199, e00124–17. [Google Scholar]
- Chen, H.; Tang, H.; Ebright, R.H. Functional interaction between RNA polymerase alpha subunit C-terminal domain and sigma(70) in UP-element- and activator-dependent transcription. Mol. Cell 2003, 11, 1621–1633. [Google Scholar] [CrossRef]
- Rao, L.; Ross, W.; Appleman, J.A.; Gaal, T.; Leirmo, S.; Schlax, P.J.; Record, M.T.; Gourse, R.L. Factor-independent activation of rrnb p1–an extended promoter with an upstream element that dramatically increases promoter strength. J. Mol. Biol. 1994, 235, 1421–1435. [Google Scholar] [CrossRef] [PubMed]
- Estrem, S.T.; Gaal, T.; Ross, W.; Gourse, R.L. Identification of an UP element consensus sequence for bacterial promoters. Proc. Natl. Acad. Sci. USA 1998, 95, 9761–9766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.R.; Jin, K.; Zhang, L.; Ding, Z.Y.; Gu, Z.H.; Shi, G.Y. Development of an inducible secretory expression system in Bacillus licheniformis based on an engineered xylose operon. J. Agric. Food Chem. 2018, 66, 9456–9464. [Google Scholar] [PubMed]
- Yang, M.M.; Zhang, W.W.; Ji, S.Y.; Cao, P.H.; Chen, Y.L.; Zhao, X. Generation of an artificial double promoter for protein expression in Bacillus subtilis through a promoter trap system. PLoS ONE 2013, 8, e56321. [Google Scholar]
- Li, Y.R.; Liu, X.; Zhang, L.; Ding, Z.Y.; Xu, S.; Gu, Z.H.; Shi, G.Y. Transcriptional changes in the xylose operon in Bacillus licheniformis and their use in fermentation optimization. Int. J. Mol. Sci. 2019, 20, 4615. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.M.; Cai, X.; Huang, Z.H.; Liu, Z.Q.; Zheng, Y.G. Construction of a highly active secretory expression system in Bacillus subtilis of a recombinant amidase by promoter and signal peptide engineering. Int. J. Biol. Macromol. 2020, 143, 833–841. [Google Scholar] [CrossRef]
- Einav, T.; Phillips, R. How the avidity of polymerase binding to the-35/-10 promoter sites affects gene expression. Proc. Natl. Acad. Sci. USA 2019, 116, 13340–13345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Ma, C.L.; Xu, L.D.; Tian, P.F. Exploiting tandem repetitive promoters for high-level production of 3-hydroxypropionic acid. Appl. Microbiol. Biotechnol. 2019, 103, 4017–4031. [Google Scholar] [CrossRef]
- Urtecho, G.; Tripp, A.D.; Insigne, K.D.; Kim, H.; Kosuri, S. Systematic Dissection of Sequence Elements controlling sigma 70 promoters using a genomically encoded multiplexed reporter assay in Escherichia coli. Biochemistry 2019, 58, 1539–1551. [Google Scholar] [CrossRef]
- Xiao, F.X.; Li, Y.R.; Zhang, Y.P.; Wang, H.R.; Zhang, L.; Ding, Z.Y.; Gu, Z.H.; Xu, S.; Shi, G.Y. A new CcpA binding site plays a bidirectional role in carbon catabolism in Bacillus licheniformis. Iscience 2021, 24, 102400. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W.; Sambrook, J. The Condensed Protocols: From Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2006. [Google Scholar]
- Li, Y.R.; Wang, H.R.; Zhang, L.; Ding, Z.Y.; Xu, S.; Gu, Z.H.; Shi, G.Y. Efficient genome editing in Bacillus licheniformis mediated by a conditional CRISPR/Cas9 system. Microorganisms 2020, 8, 754. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.P.; Li, Y.R.; Xiao, F.X.; Wang, H.R.; Zhang, L.; Ding, Z.Y.; Xu, S.; Gu, Z.H.; Shi, G.Y. Engineering of a biosensor in response to malate in Bacillus licheniformis. Acs Synth. Biol. 2021, 10, 1775–1784. [Google Scholar] [CrossRef] [PubMed]
- Zannoni, A.; Pelliciari, S.; Musiani, F.; Chiappori, F.; Roncarati, D.; Scarlato, V. Definition of the binding architecture to a target promoter of hp1043, the essential master regulator of Helicobacter pylori. Int. J. Mol. Sci. 2021, 22, 7848. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Promoters | Description | Reference |
|---|---|---|
| PHpaII | Strong constitutive promoter in B. subtilis | [14] |
| PglvA | Upstream of ynfA in B. licheniformis | [15] |
| PspovG | Upstream of spovG in B. subtilis | [16] |
| Pacpp | Upstream of acpP in B. megaterium | [17] |
| PydzA | Upstream of ydzA in B. subtilis | [18] |
| P43 | σ55 and σ37 promoter in B. subtilis | [19] |
| PLan | Upstream of lanC in B. licheniformis | This study |
| PTrxA | Upstream of trxA in B. subtilis | [13] |
| PDS | Upstream of QGI43659.1 in B. licheniformis | This study |
| Pr2 | σw promoter in B. amyloliquefaciens | [20] |
| Pshuttle-09 | Artificial promoter in B. subtilis | [21] |
| Strain or Plasmid | Description | Source |
|---|---|---|
| Strains | ||
| Escherichia coli JM109 | F′, traD36, proAB +. lacIq,△(lacZ), M15/△(lac-proAB), glnV44, e14−, gyrA96, recA1, relA1, endA1, thi, hsdR17 | Our lab |
| E. coli BL21(DE3) | F-ompT gal dcm lon hsdSB (rB- mB) λ(DE3) | Our lab |
| Bacillus licheniformis CICIM B1391 | Wild-type | Our lab |
| EcCTD | BL21, harboring pCTD | This work |
| BlHpaIIG | B. licheniformis CICIM B1391 harboring pHpaIIGFP | This work |
| BlSpovGG | B. licheniformis CICIM B1391 harboring pSpovGGFP | This work |
| BlYdzAG | B. licheniformis CICIM B1391 harboring pYdzAGFP | This work |
| Bl43G | B. licheniformis CICIM B1391 harboring p43GFP | This work |
| BlTrxAG | B. licheniformis CICIM B1391 harboring pTrxAGFP | This work |
| BlGlvAG | B. licheniformis CICIM B1391 harboring pGlvAGFP | This work |
| BlLanG | B. licheniformis CICIM B1391 harboring pLanGFP | This work |
| BlDSG | B. licheniformis CICIM B1391 harboring pDSGFP | This work |
| Bl09G | B. licheniformis CICIM B1391 harboring p09GFP | This work |
| BlAcppG | B. licheniformis CICIM B1391 harboring pAcppGFP | This work |
| BlR2G | B. licheniformis CICIM B1391 harboring pR2GFP | This work |
| BlTrxAGUP0 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP0 | This work |
| BlTrxAGUP1 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP1 | This work |
| BlTrxAGUP2 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP2 | This work |
| BlTrxAGUP3 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP3 | This work |
| BlTrxAGUP4 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP4 | This work |
| BlTrxAGUP5 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP5 | This work |
| BlTrxAGUP5P1 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP5P1 | This work |
| BlTrxAGUP5P2 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP5P2 | This work |
| BlTrxAGUP5P3 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP5P3 | This work |
| BlTrxAGUP5P4 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP5P4 | This work |
| BlTrxAGUP5P5 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP5P5 | This work |
| BlTrxAGUP5D1 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP5D1 | This work |
| BlTrxAGUP5D2 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP5D2 | This work |
| BlTrxAGUP5D3 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP5D3 | This work |
| BlTrxAGUP5D4 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP5D4 | This work |
| BlTrxAGUP5D5 | B. licheniformis CICIM B1391 harboring pTrxAGFPUP5D5 | This work |
| BlHpaIIGUP5P2 | B. licheniformis CICIM B1391 harboring pHpaIIUP5P2GFP | This work |
| BlSpovGGUP5P2 | B. licheniformis CICIM B1391 harboring pSpovGUP5P2GFP | This work |
| BlYdzAGUP5P2 | B. licheniformis CICIM B1391 harboring pYdzAUP5P2GFP | This work |
| Bl43GUP5P2 | B. licheniformis CICIM B1391 harboring p43UP5P2GFP | This work |
| BlGlvAGUP5P2 | B. licheniformis CICIM B1391 harboring pGlvAUP5P2GFP | This work |
| BlDSGUP5P2 | B. licheniformis CICIM B1391 harboring pDSUP5P2GFP | This work |
| Bl09GUP5P2 | B. licheniformis CICIM B1391 harboring p09UP5P2GFP | This work |
| BlAcppGUP5P2 | B. licheniformis CICIM B1391 harboring pAcppUP5P2GFP | This work |
| BlR2GUP5P2 | B. licheniformis CICIM B1391 harboring pR2UP5P2GFP | This work |
| Plasmids | ||
| pMD19-T | E. coli cloning vector, ApR | TaKaRa |
| pMA5 | E. coli/Bacillus shuttle vector, NeoR/ApR, PHpaII | Our lab |
| pHY300-PLK | E. coli/Bacillus shuttle vector, ApR/TetR | Our lab |
| pET-28(a) | E. coli expression vector, KanR | TaKaRa |
| pCTD | pET-28a derivative with α-CTD encoding gene in B. licheniformis | This work |
| pHpaIIGFP | pGFP derivative with promoter PHpaII | This work |
| pSpovGGFP | pGFP derivative with promoter PSpovG | This work |
| pYdzAGFP | pGFP derivative with promoter PYdzA | This work |
| p43GFP | pGFP derivative with promoter P43 | This work |
| pTrxAGFP | pGFP derivative with promoter PTrxA | This work |
| pGlvAGFP | pGFP derivative with promoter PGlvA | This work |
| pLanGFP | pGFP derivative with promoter PLan | This work |
| pDSGFP | pGFP derivative with promoter PDS | This work |
| p09GFP | pGFP derivative with promoter Pshuttle-09 | This work |
| pAcppGFP | pGFP derivative with promoter Pacpp | This work |
| pR2GFP | pGFP derivative with promoter Pr2 | This work |
| pTrxAGFPUP0 | pTrxAGFP derivative with UP0 inserted | This work |
| pTrxAGFPUP1 | pTrxAGFP derivative with UP1 inserted | This work |
| pTrxAGFPUP2 | pTrxAGFP derivative with UP2 inserted | This work |
| pTrxAGFPUP3 | pTrxAGFP derivative with UP3 inserted | This work |
| pTrxAGFPUP4 | pTrxAGFP derivative with UP4 inserted | This work |
| pTrxAGFPUP5 | pTrxAGFP derivative with UP5 inserted | This work |
| pTrxAGFPUP5P1 | pTrxAGFPUP5 derivative with 1 proximal subsite inserted | This work |
| pTrxAGFPUP5P2 | pTrxAGFPUP5 derivative with 2 proximal subsites inserted | This work |
| pTrxAGFPUP5P3 | pTrxAGFPUP5 derivative with 3 proximal subsites inserted | This work |
| pTrxAGFPUP5P4 | pTrxAGFPUP5 derivative with 4 proximal subsites inserted | This work |
| pTrxAGFPUP5P5 | pTrxAGFPUP5 derivative with 5 proximal subsites inserted | This work |
| pTrxAGFPUP5D1 | pTrxAGFPUP5 derivative with 1 distal subsite inserted | This work |
| pTrxAGFPUP5D2 | pTrxAGFPUP5 derivative with 2 distal subsites inserted | This work |
| pTrxAGFPUP5D3 | pTrxAGFPUP5 derivative with 3 distal subsites inserted | This work |
| pTrxAGFPUP5D4 | pTrxAGFPUP5 derivative with 4 distal subsites inserted | This work |
| pTrxAGFPUP5D5 | pTrxAGFPUP5 derivative with 5 distal subsites inserted | This work |
| pHpaIIUP5P2GFP | pGFP derivative with promoter PHpaII-UP5-2P | This work |
| pSpovGUP5P2GFP | pGFP derivative with promoter PSpovG-UP5-2P | This work |
| pYdzAUP5P2GFP | pGFP derivative with promoter PYdzA-UP5-2P | This work |
| p43UP5P2GFP | pGFP derivative with promoter P43-UP5-2P | This work |
| pGlvAUP5P2GFP | pGFP derivative with promoter PGlvA-UP5-2P | This work |
| pDSUP5P2GFP | pGFP derivative with promoter PDS-UP5-2P | This work |
| p09UP5P2GFP | pGFP derivative with promoter Pshuttle-09-UP5-2P | This work |
| pAcppUP5P2GFP | pGFP derivative with promoter Pacpp-UP5-2P | This work |
| pR2UP5P2GFP | pGFP derivative with promoter Pr2-UP5-2P | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Ma, X.; Zhang, L.; Ding, Z.; Xu, S.; Gu, Z.; Shi, G. Engineering of Bacillus Promoters Based on Interacting Motifs between UP Elements and RNA Polymerase (RNAP) α-Subunit. Int. J. Mol. Sci. 2022, 23, 13480. https://doi.org/10.3390/ijms232113480
Li Y, Ma X, Zhang L, Ding Z, Xu S, Gu Z, Shi G. Engineering of Bacillus Promoters Based on Interacting Motifs between UP Elements and RNA Polymerase (RNAP) α-Subunit. International Journal of Molecular Sciences. 2022; 23(21):13480. https://doi.org/10.3390/ijms232113480
Chicago/Turabian StyleLi, Youran, Xufan Ma, Liang Zhang, Zhongyang Ding, Sha Xu, Zhenghua Gu, and Guiyang Shi. 2022. "Engineering of Bacillus Promoters Based on Interacting Motifs between UP Elements and RNA Polymerase (RNAP) α-Subunit" International Journal of Molecular Sciences 23, no. 21: 13480. https://doi.org/10.3390/ijms232113480
APA StyleLi, Y., Ma, X., Zhang, L., Ding, Z., Xu, S., Gu, Z., & Shi, G. (2022). Engineering of Bacillus Promoters Based on Interacting Motifs between UP Elements and RNA Polymerase (RNAP) α-Subunit. International Journal of Molecular Sciences, 23(21), 13480. https://doi.org/10.3390/ijms232113480