
Shotgun Metagenomics Study Suggests Alteration in Sulfur Metabolism and Oxidative Stress in Children with Autism and Improvement after Microbiota Transfer Therapy
 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
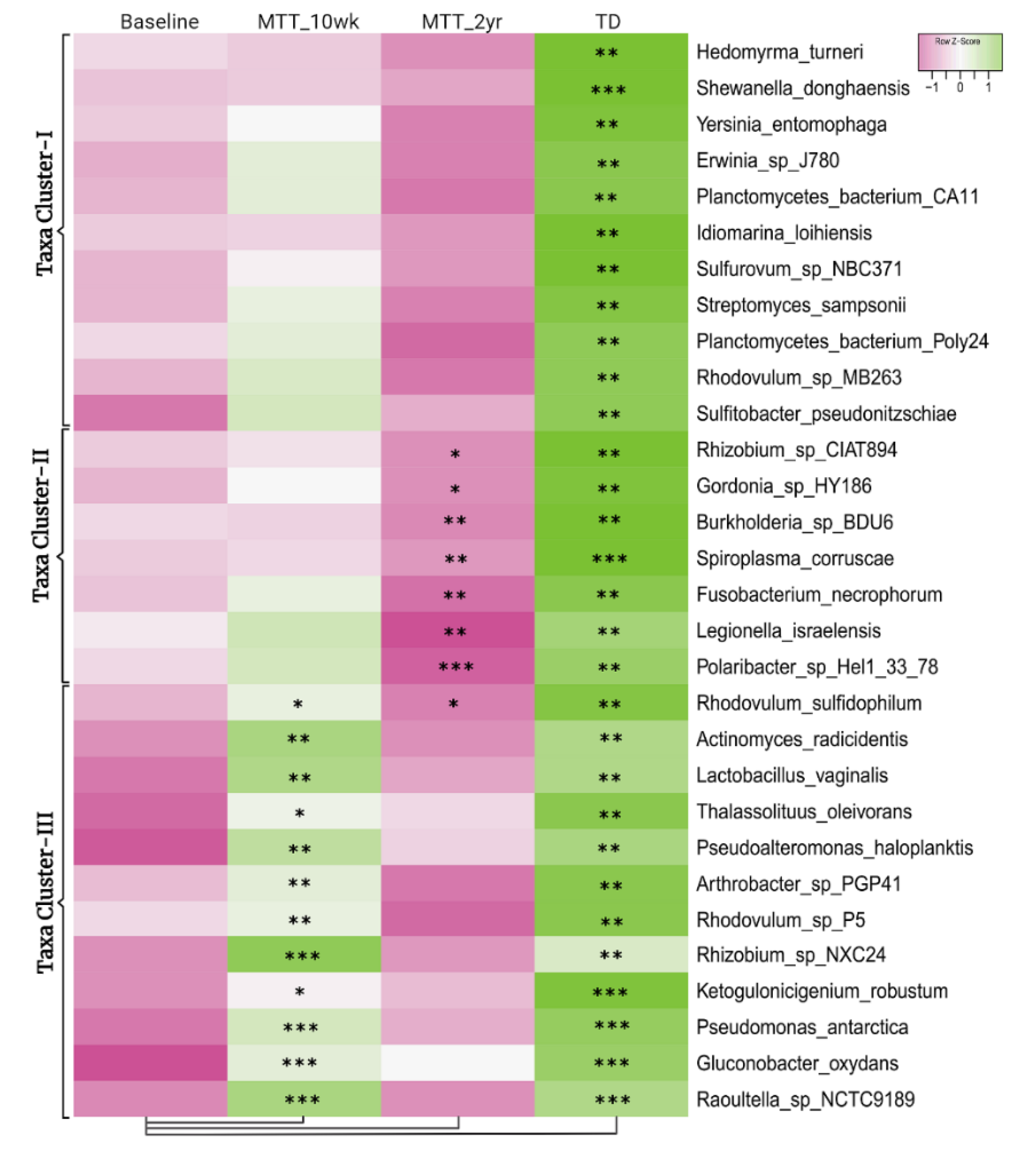
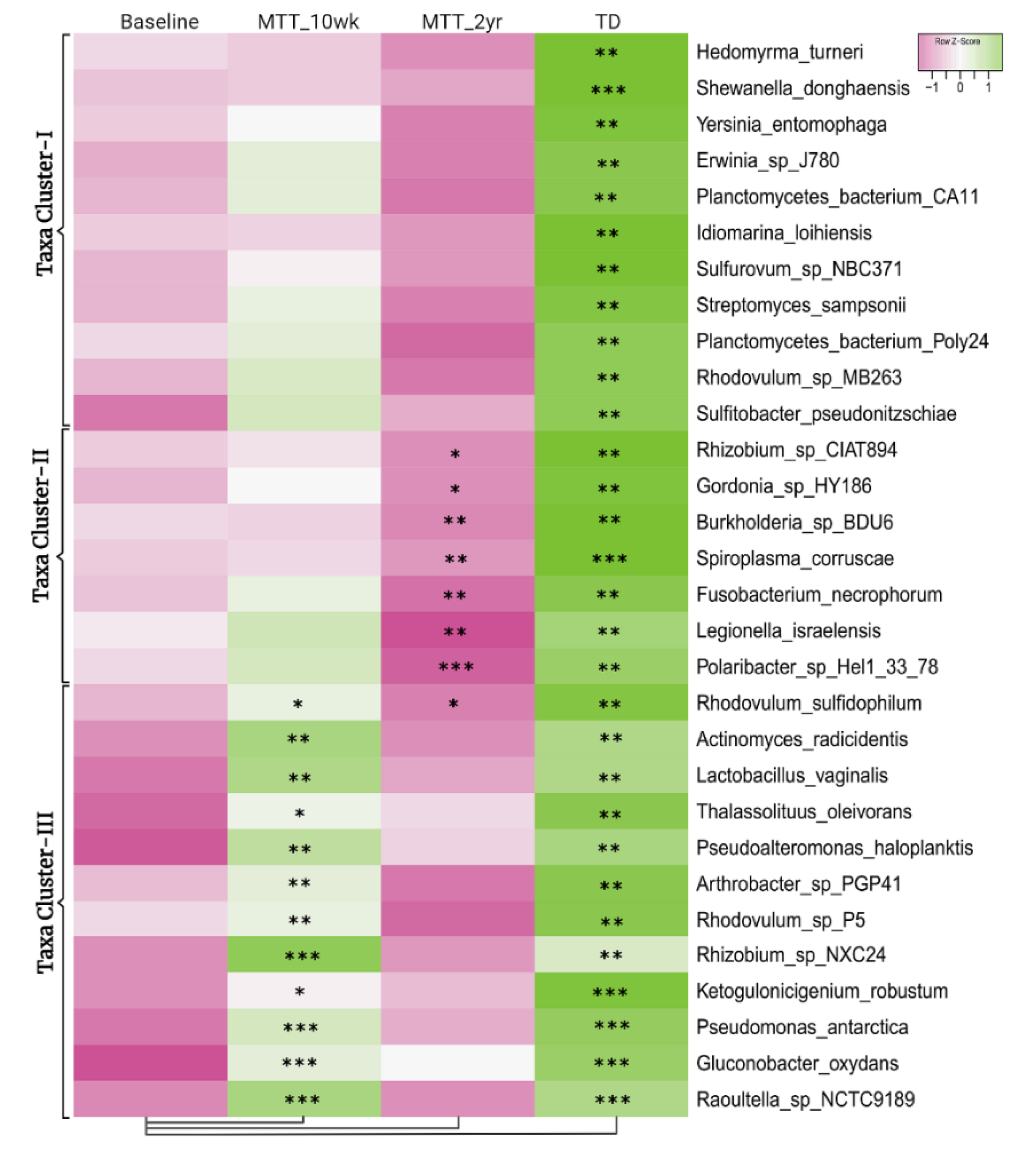
2.1. Many Bacterial Species Shifted after MTT, Mostly after 2 Years
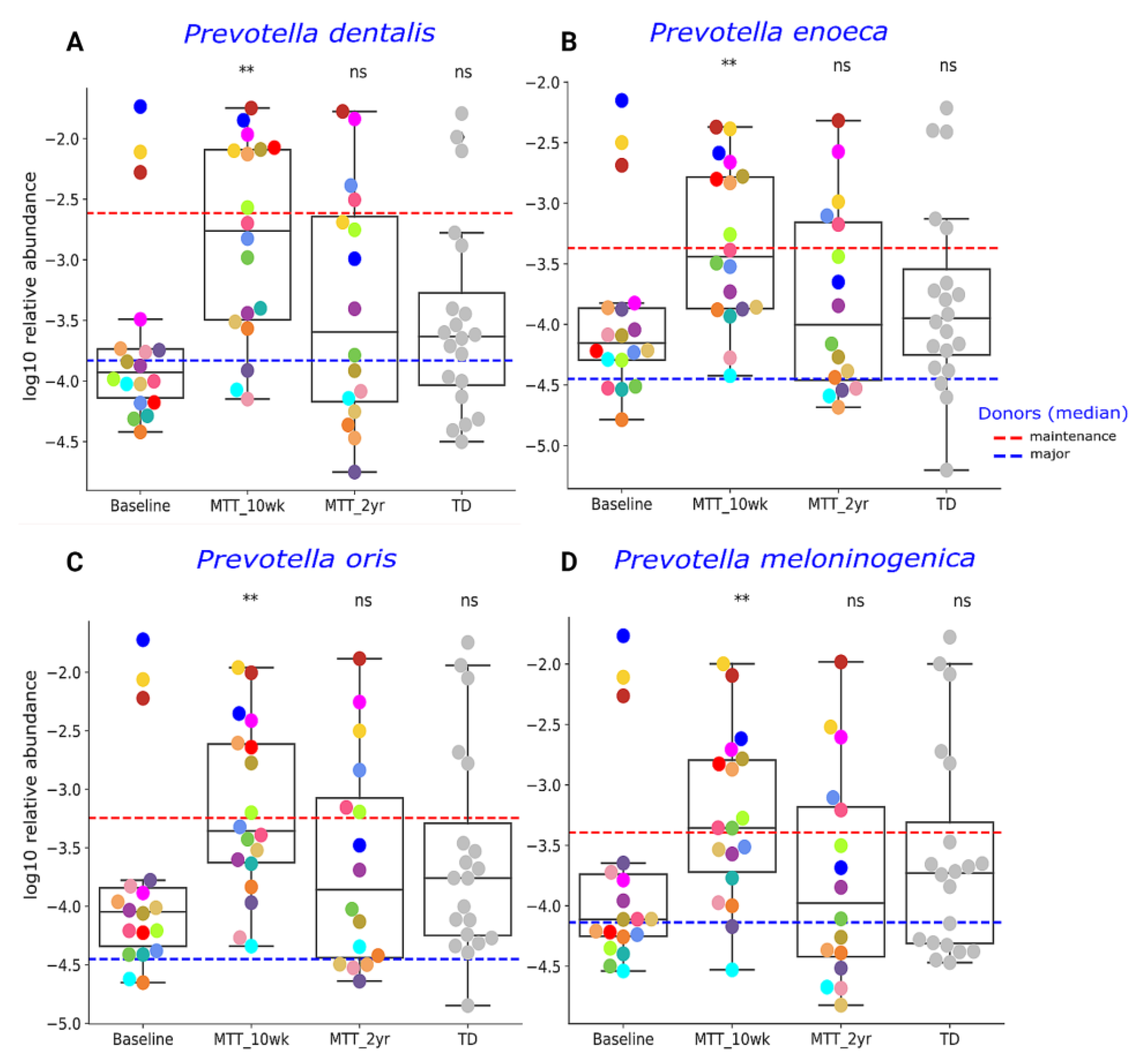
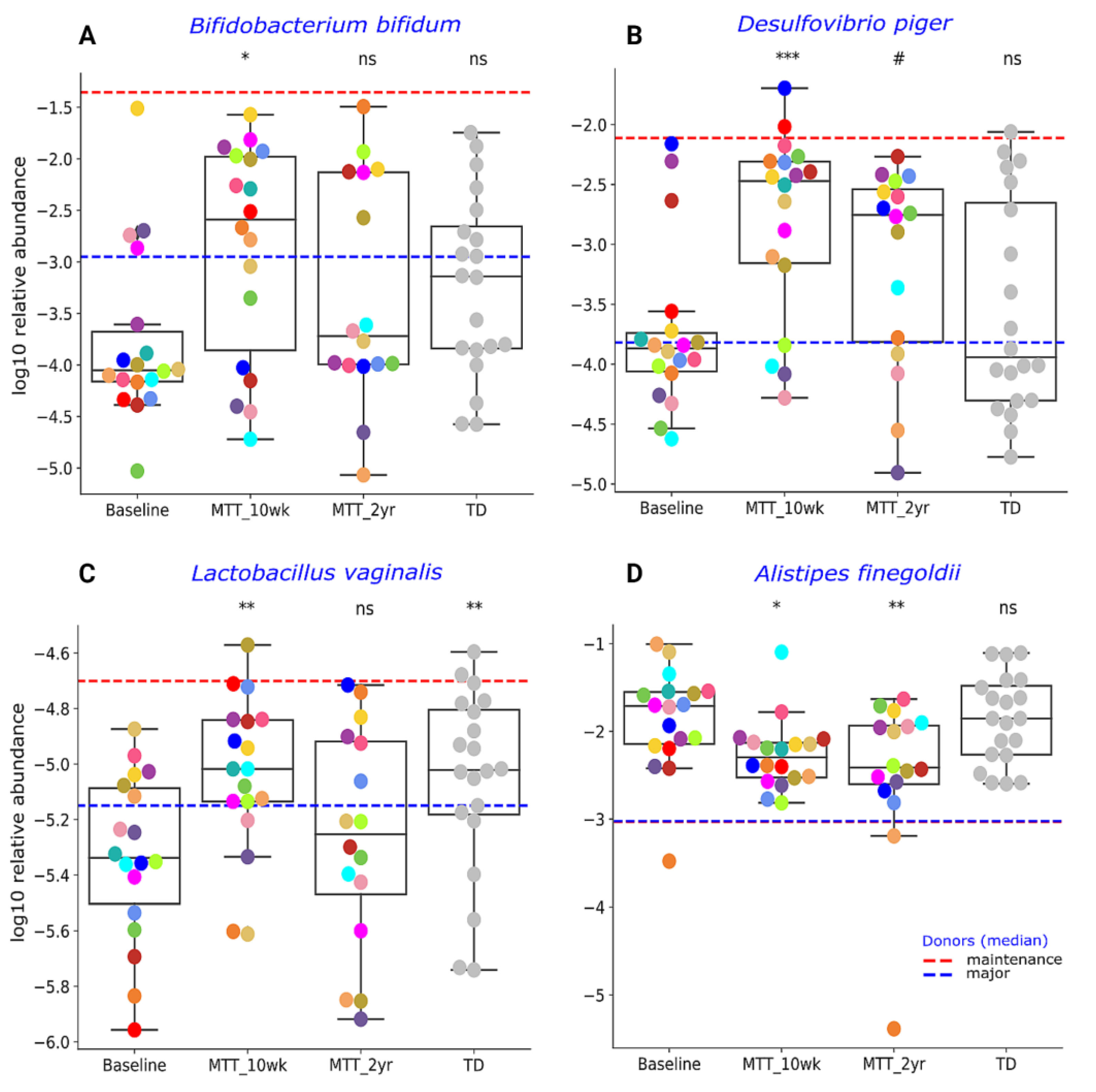
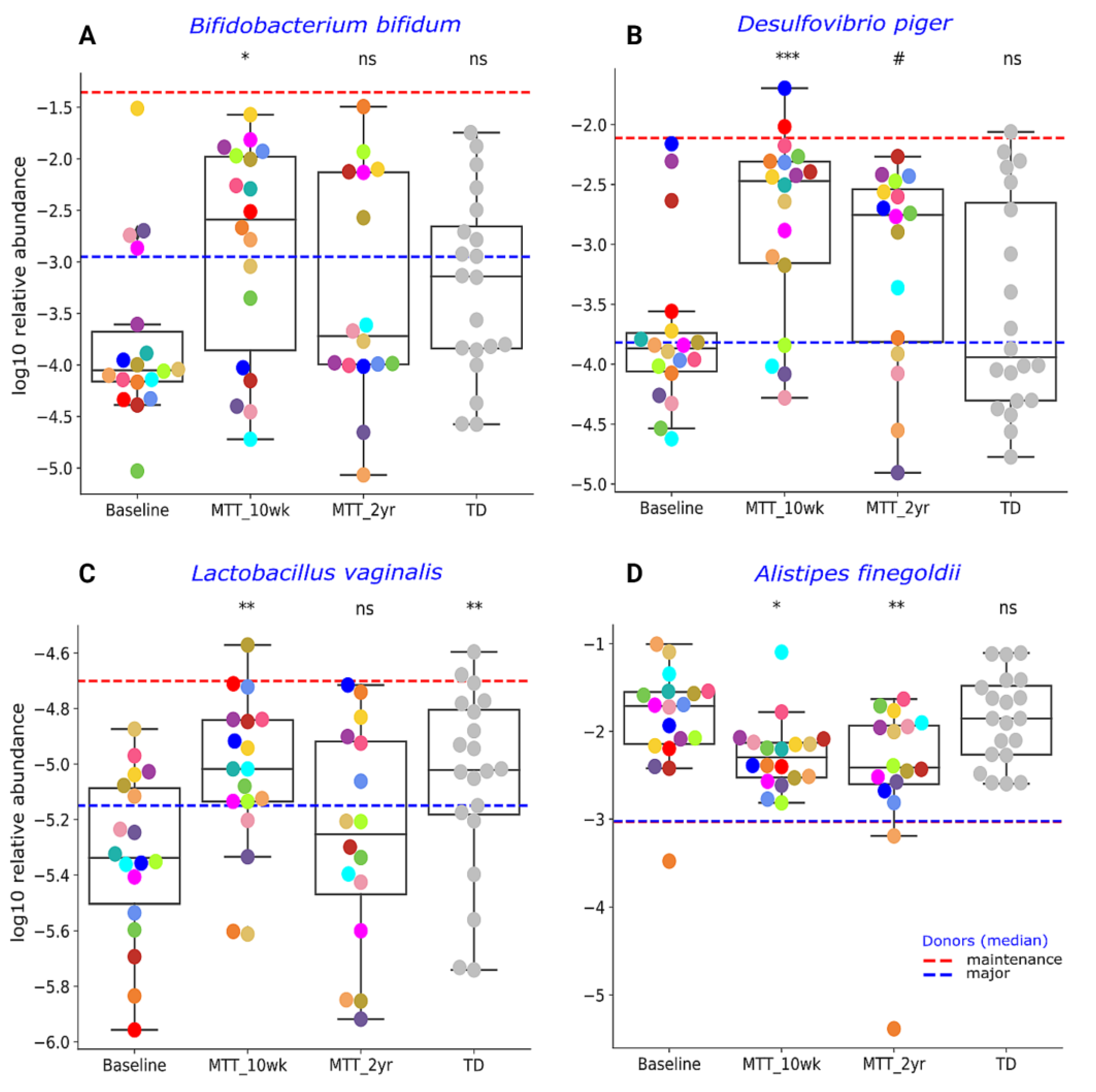
2.2. MTT Changed Specific Bacterial Species including Fiber-Consuming, Probiotic and Sulfur-Reducing Bacteria
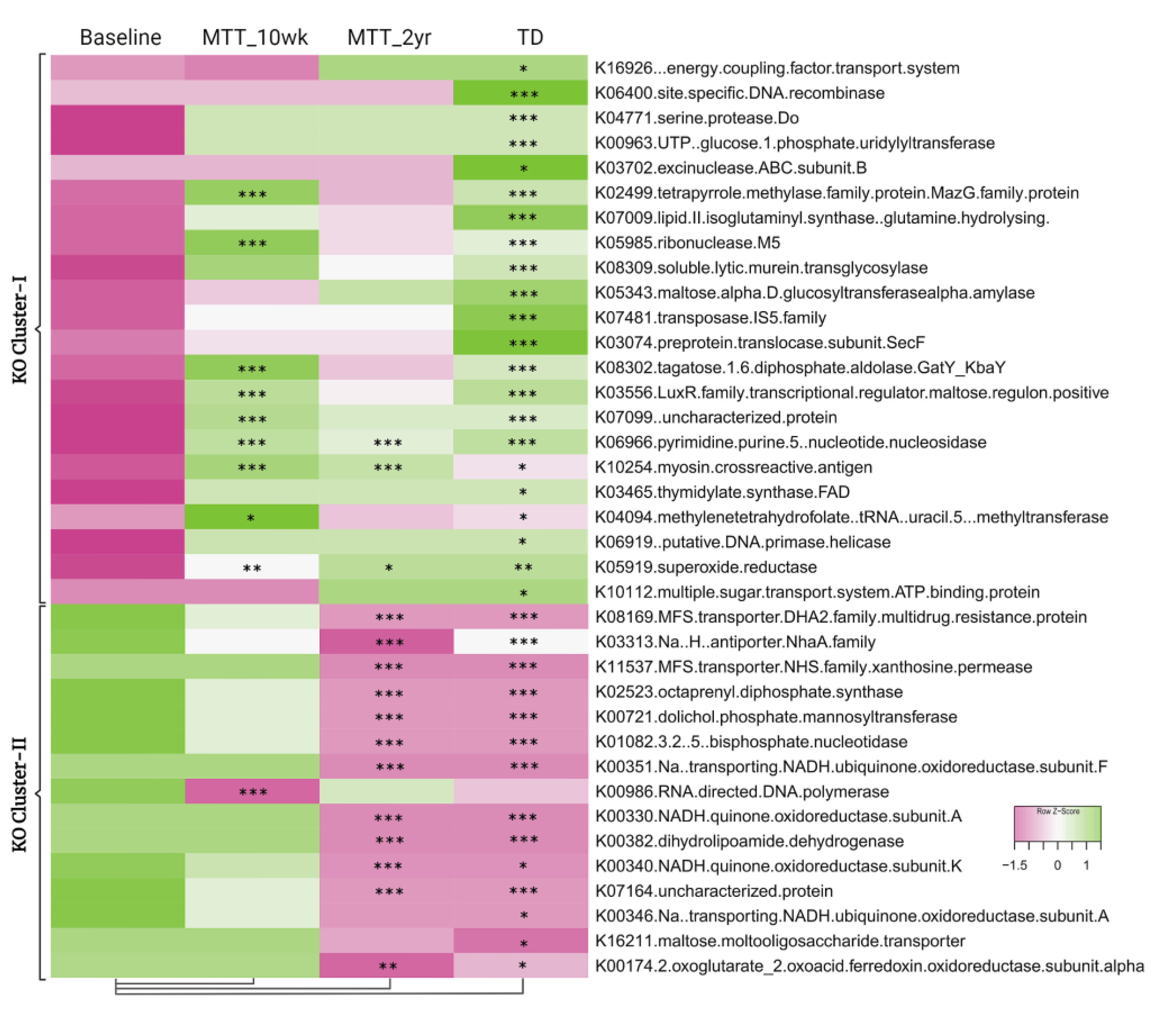
2.3. Microbial Functional Genes Shifted with MTT
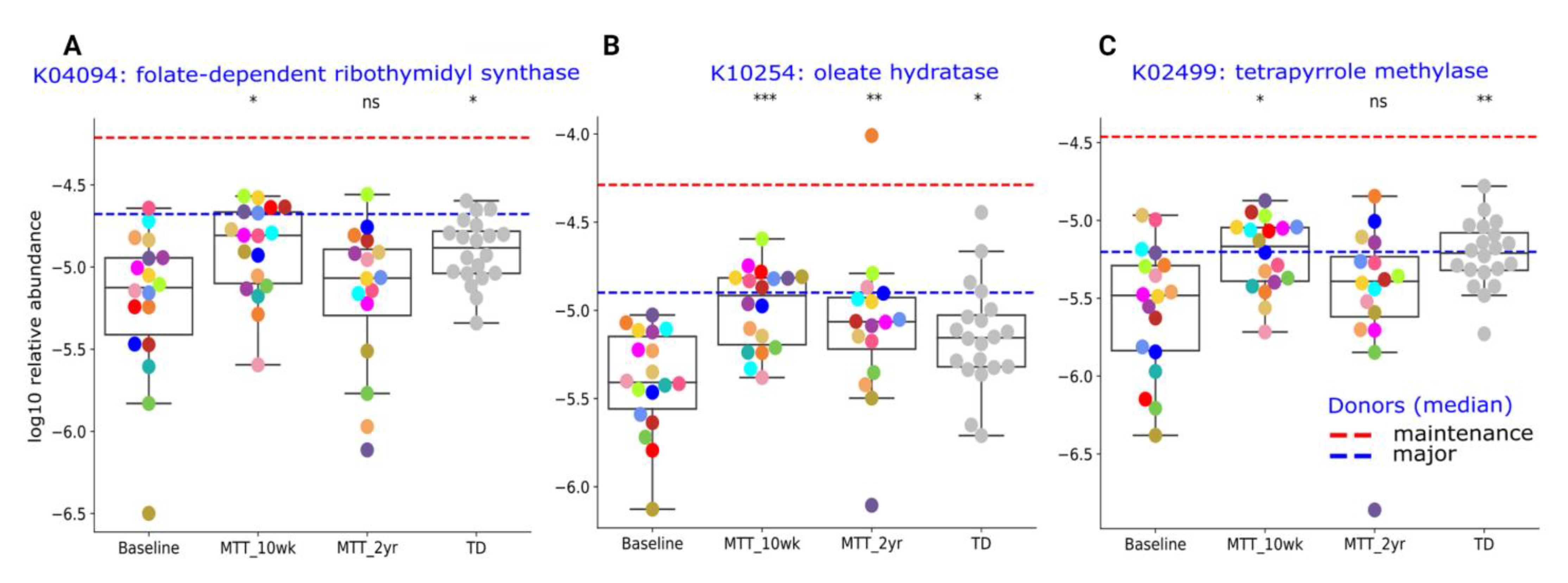
2.4. Relative Abundance of Important Metabolic Genes Changed after MTT
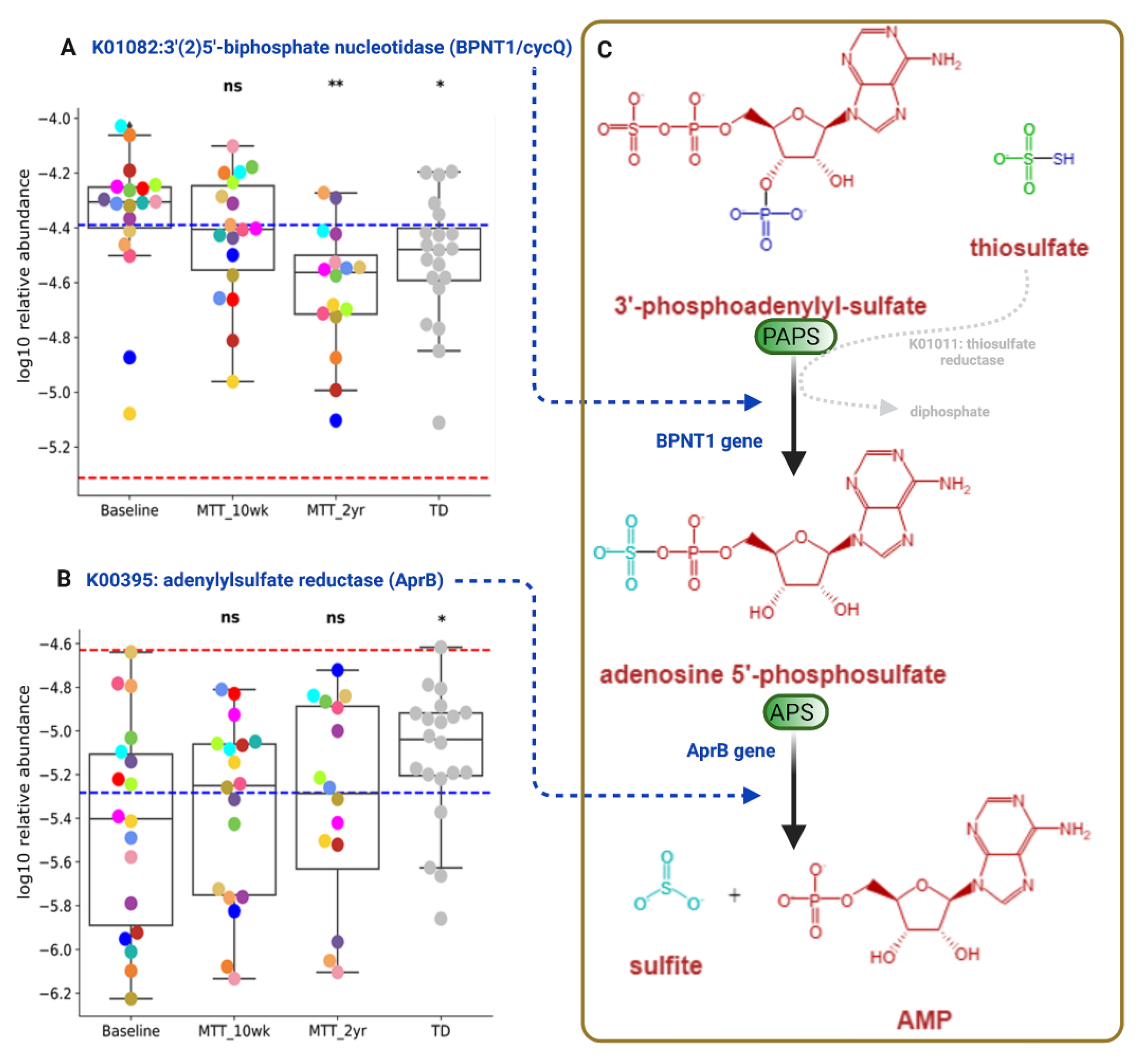
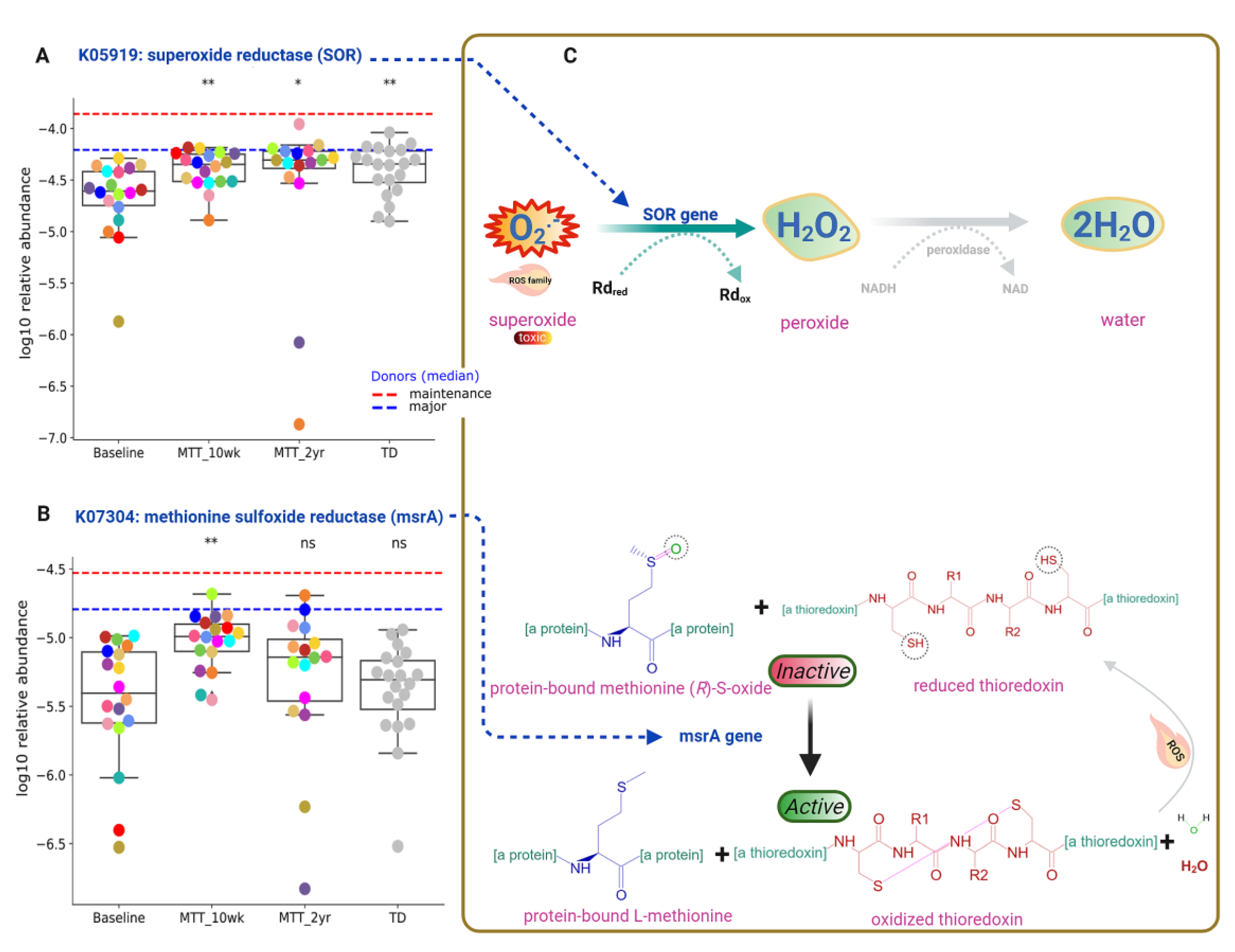
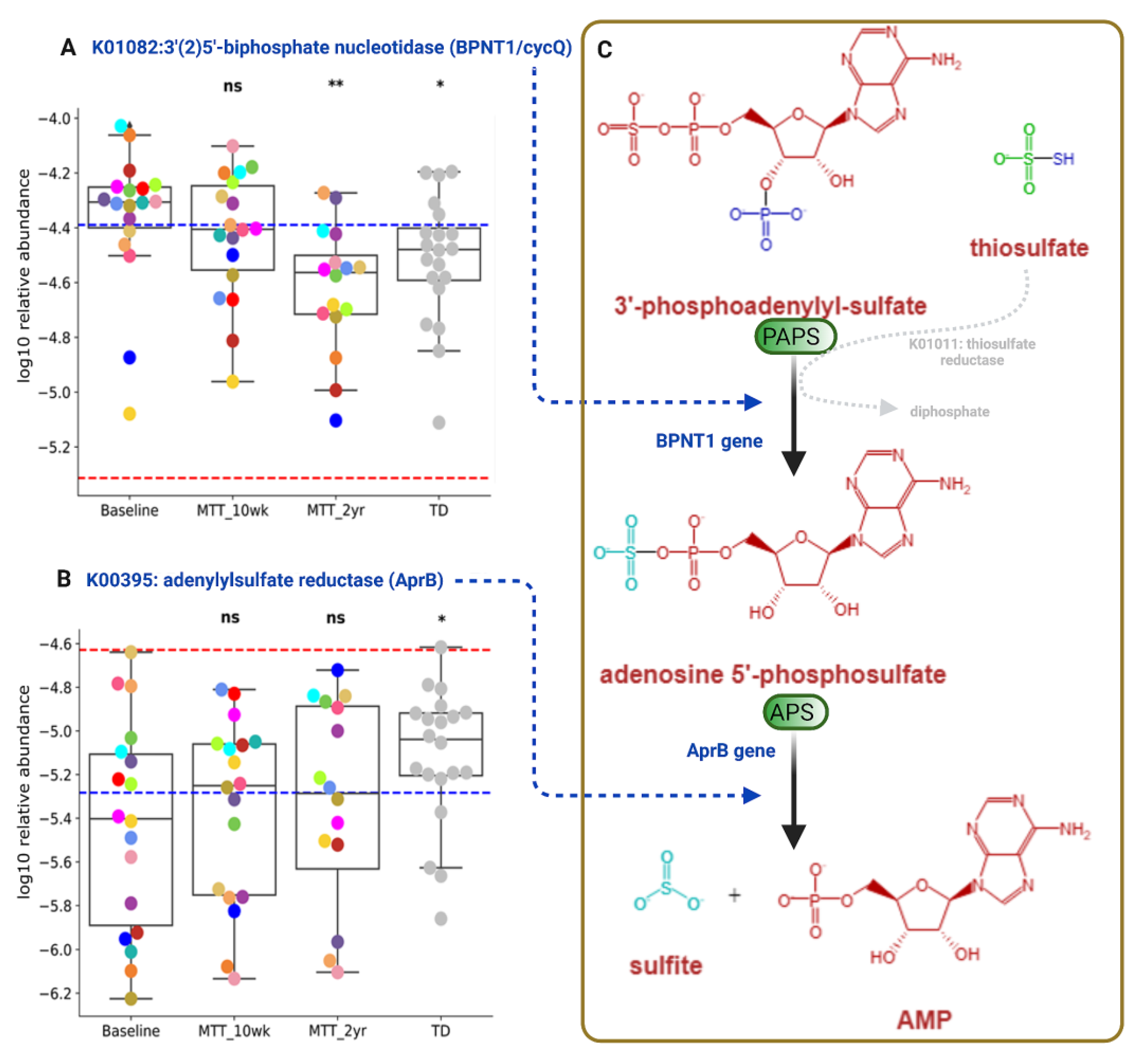
2.5. Abundance of Genes Encoding for Oxidative Stress Protection and Sulfur Metabolism Changed after MTT
2.6. Correlation Analysis Shows Links between Omics and GSRS
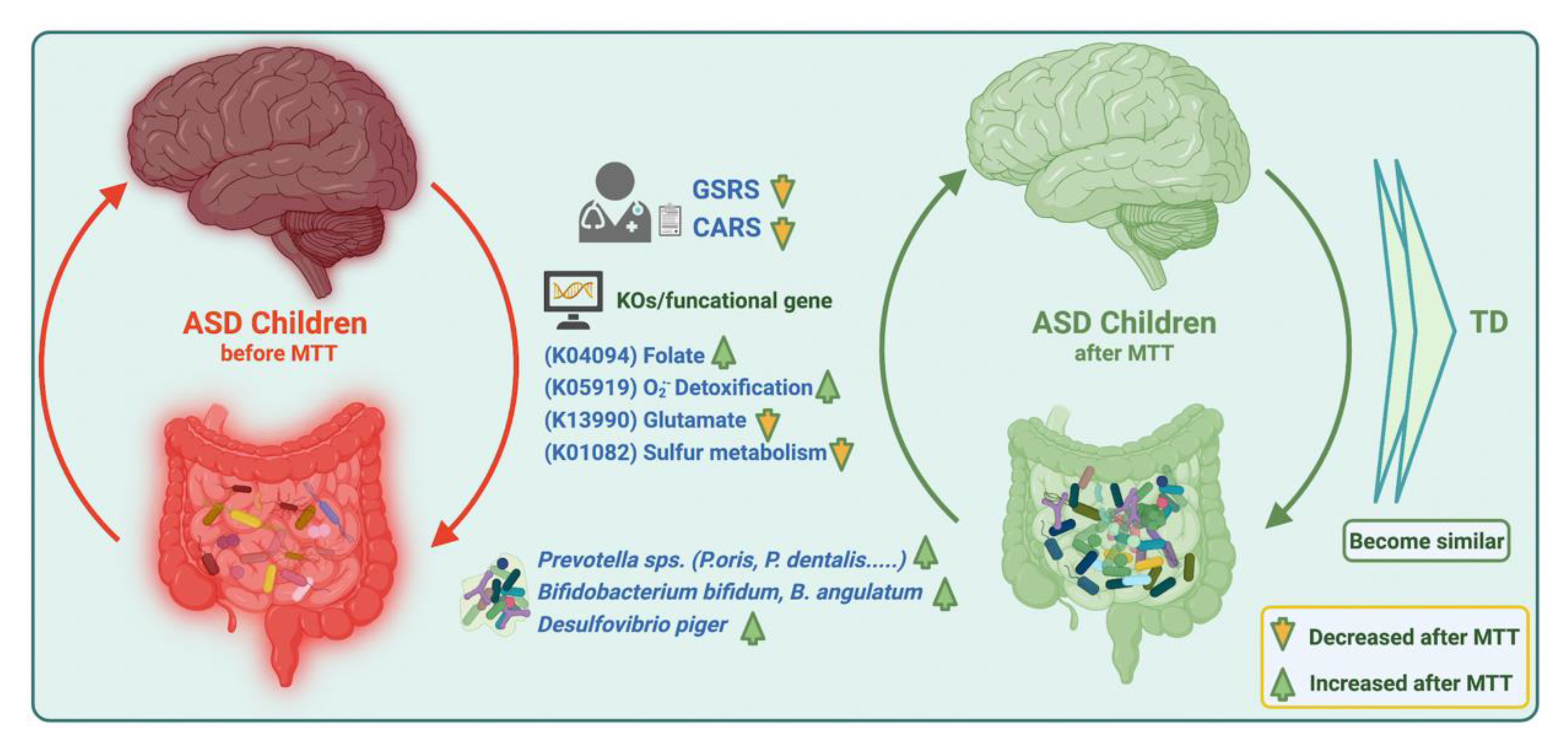
3. Discussion
4. Materials and Methods
4.1. Outline of the Trial
4.2. Metagenomics Sequencing
4.3. Sequencing Analysis
4.4. Fecal and Plasma Metabolomics
4.5. GI and ASD Symptom Assessment
4.6. Diversity Index Calculation
4.7. Multi-Omics Correlation Network
4.8. Statistical Analysis and Plots
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- What Is Autism Spectrum Disorder?|CDC. Available online: https://www.cdc.gov/ncbddd/autism/facts.html (accessed on 3 May 2022).
- Data & Statistics on Autism Spectrum Disorder|CDC. Available online: https://www.cdc.gov/ncbddd/autism/data.html (accessed on 3 May 2022).
- Hsiao, E.Y. Gastrointestinal Issues in Autism Spectrum Disorder. Harv. Rev. Psychiatry 2014, 22, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Restrepo, B.; Angkustsiri, K.; Taylor, S.L.; Rogers, S.J.; Cabral, J.; Heath, B.; Hechtman, A.; Solomon, M.; Ashwood, P.; Amaral, D.G.; et al. Developmental-Behavioral Profiles in Children with Autism Spectrum Disorder and Co-Occurring Gastrointestinal Symptoms. Autism Res. 2020, 13, 1778–1789. [Google Scholar] [CrossRef] [PubMed]
- Vargason, T.; McGuinness, D.L.; Hahn, J. Gastrointestinal Symptoms and Oral Antibiotic Use in Children with Autism Spectrum Disorder: Retrospective Analysis of a Privately Insured U.S. Population. J. Autism Dev. Disord. 2019, 49, 647–659. [Google Scholar] [CrossRef] [PubMed]
- McElhanon, B.O.; McCracken, C.; Karpen, S.; Sharp, W.G. Gastrointestinal Symptoms in Autism Spectrum Disorder: A Meta-Analysis. Pediatrics 2014, 133, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, D.W.; Adams, J.B.; Gregory, A.C.; Borody, T.; Chittick, L.; Fasano, A.; Khoruts, A.; Geis, E.; Maldonado, J.; McDonough-Means, S.; et al. Microbiota Transfer Therapy Alters Gut Ecosystem and Improves Gastrointestinal and Autism Symptoms: An Open-Label Study. Microbiome 2017, 5, 10. [Google Scholar] [CrossRef]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal Flora and Gastrointestinal Status in Children with Autism—Comparisons to Typical Children and Correlation with Autism Severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef] [Green Version]
- Krajmalnik-Brown, R.; Lozupone, C.; Kang, D.-W.; Adams, J.B. Gut Bacteria in Children with Autism Spectrum Disorders: Challenges and Promise of Studying How a Complex Community Influences a Complex Disease. Microb. Ecol. Health Dis. 2015, 26, 26914. [Google Scholar] [CrossRef]
- Garcia-Gutierrez, E.; Narbad, A.; Rodríguez, J.M. Autism Spectrum Disorder Associated With Gut Microbiota at Immune, Metabolomic, and Neuroactive Level. Front. Neurosci. 2020, 14, 578666. [Google Scholar] [CrossRef]
- Kang, D.W.; Adams, J.B.; Coleman, D.M.; Pollard, E.L.; Maldonado, J.; McDonough-Means, S.; Caporaso, J.G.; Krajmalnik-Brown, R. Long-Term Benefit of Microbiota Transfer Therapy on Autism Symptoms and Gut Microbiota. Sci. Rep. 2019, 9, 5821. [Google Scholar] [CrossRef] [Green Version]
- Ruggiero, M. Fecal Microbiota Transplantation and the Brain Microbiota in Neurological Diseases. Clin. Endosc. 2016, 49, 579. [Google Scholar] [CrossRef]
- Collins, S.M.; Surette, M.; Bercik, P. The Interplay between the Intestinal Microbiota and the Brain. Nat. Rev. Microbiol. 2012, 10, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A.; Knight, R.; Mazmanian, S.K.; Cryan, J.F.; Tillisch, K. Gut Microbes and the Brain: Paradigm Shift in Neuroscience. J. Neurosci. 2014, 34, 15490–15496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, Y.; Zuo, T.; Xu, Z.; Zhang, F.; Zhan, H.; Chan, D.; Leung, T.F.; Yeoh, Y.K.; Chan, F.K.L.; Chan, R.; et al. Underdevelopment of the Gut Microbiota and Bacteria Species as Non-Invasive Markers of Prediction in Children with Autism Spectrum Disorder. Gut 2022, 71, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Averina, O.; Kovtun, A.S.; Polyakova, S.I.; Savilova, A.M.; Rebrikov, D.; Danilenko, V.N. The Bacterial Neurometabolic Signature of the Gut Microbiota of Young Children with Autism Spectrum Disorders. J. Med. Microbiol. 2020, 69, 558–571. [Google Scholar] [CrossRef]
- Dan, Z.; Mao, X.; Liu, Q.; Guo, M.; Zhuang, Y.; Liu, Z.; Chen, K.; Chen, J.; Xu, R.; Tang, J.; et al. Altered Gut Microbial Profile Is Associated with Abnormal Metabolism Activity of Autism Spectrum Disorder. Gut Microbes 2020, 11, 1246–1267. [Google Scholar] [CrossRef]
- Wang, M.; Wan, J.; Rong, H.; He, F.; Wang, H.; Zhou, J.; Cai, C.; Wang, Y.; Xu, R.; Yin, Z.; et al. Alterations in Gut Glutamate Metabolism Associated with Changes in Gut Microbiota Composition in Children with Autism Spectrum Disorder. mSystems 2019, 4, e00321-18. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Chu, Y.; Meng, Q.; Ding, R.; Shi, X.; Wang, Z.; He, Y.; Zhang, J.; Liu, J.; Zhang, J.; et al. A Quasi-Paired Cohort Strategy Reveals the Impaired Detoxifying Function of Microbes in the Gut of Autistic Children. Sci. Adv. 2020, 6, eaba3760. [Google Scholar] [CrossRef]
- Kang, D.-W.; Adams, J.B.; Vargason, T.; Santiago, M.; Hahn, J.; Krajmalnik-Brown, R. Distinct Fecal and Plasma Metabolites in Children with Autism Spectrum Disorders and Their Modulation after Microbiota Transfer Therapy. mSphere 2020, 5, e00314-20. [Google Scholar] [CrossRef]
- Sharon, G.; Cruz, N.J.; Kang, D.W.; Gandal, M.J.; Wang, B.; Kim, Y.M.; Zink, E.M.; Casey, C.P.; Taylor, B.C.; Lane, C.J.; et al. Human Gut Microbiota from Autism Spectrum Disorder Promote Behavioral Symptoms in Mice. Cell 2019, 177, 1600–1618.e17. [Google Scholar] [CrossRef] [Green Version]
- Naseribafrouei, A.; Hestad, K.; Avershina, E.; Sekelja, M.; Linløkken, A.; Wilson, R.; Rudi, K. Correlation between the Human Fecal Microbiota and Depression. Neurogastroenterol. Motil. 2014, 26, 1155–1162. [Google Scholar] [CrossRef]
- de Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; de Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal Microbiota and Metabolome of Children with Autism and Pervasive Developmental Disorder Not Otherwise Specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef] [Green Version]
- Indika, N.L.R.; Deutz, N.E.P.; Engelen, M.P.K.J.; Peiris, H.; Wijetunge, S.; Perera, R. Sulfur Amino Acid Metabolism and Related Metabotypes of Autism Spectrum Disorder: A Review of Biochemical Evidence for a Hypothesis. Biochimie 2021, 184, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, K.; Hausmann, B.; Jungbluth, S.P.; Kantor, R.S.; Lavy, A.; Warren, L.A.; Rappé, M.S.; Pester, M.; Loy, A.; Thomas, B.C.; et al. Expanded Diversity of Microbial Groups That Shape the Dissimilatory Sulfur Cycle. ISME J. 2018, 12, 1715–1728. [Google Scholar] [CrossRef] [Green Version]
- Allegretti, J.R.; Kassam, Z.; Mullish, B.H.; Chiang, A.; Carrellas, M.; Hurtado, J.; Marchesi, J.R.; McDonald, J.A.K.; Pechlivanis, A.; Barker, G.F.; et al. Effects of Fecal Microbiota Transplantation With Oral Capsules in Obese Patients. Clin. Gastroenterol. Hepatol. 2020, 18, 855–863.e2. [Google Scholar] [CrossRef] [PubMed]
- Kazemian, N.; Ramezankhani, M.; Sehgal, A.; Khalid, F.M.; Kalkhoran, A.H.Z.; Narayan, A.; Wong, G.K.S.; Kao, D.; Pakpour, S. The Trans-Kingdom Battle between Donor and Recipient Gut Microbiome Influences Fecal Microbiota Transplantation Outcome. Sci. Rep. 2020, 10, 18349. [Google Scholar] [CrossRef]
- Kushak, R.I.; Winter, H.S.; Buie, T.M.; Cox, S.B.; Phillips, C.D.; Ward, N.L. Analysis of the Duodenal Microbiome in Autistic Individuals: Association with Carbohydrate Digestion. J. Pediatr. Gastroenterol. Nutr. 2017, 64, e110–e116. [Google Scholar] [CrossRef]
- Kang, D.W.; Park, J.G.; Ilhan, Z.E.; Wallstrom, G.; LaBaer, J.; Adams, J.B.; Krajmalnik-Brown, R. Reduced Incidence of Prevotella and Other Fermenters in Intestinal Microflora of Autistic Children. PLoS ONE 2013, 8, e68322. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Y.; Wu, M.; Feng, Y.; Zhou, Z.; Chen, L.; Chen, F. Alterations of Oral Microbiota Distinguish Children with Autism Spectrum Disorders from Healthy Controls. Sci. Rep. 2018, 8, 1597. [Google Scholar] [CrossRef] [Green Version]
- Kovatcheva-Datchary, P.; Nilsson, A.; Akrami, R.; Lee, Y.S.; de Vadder, F.; Arora, T.; Hallen, A.; Martens, E.; Björck, I.; Bäckhed, F. Dietary Fiber-Induced Improvement in Glucose Metabolism Is Associated with Increased Abundance of Prevotella. Cell Metab 2015, 22, 971–982. [Google Scholar] [CrossRef] [Green Version]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet Rapidly and Reproducibly Alters the Human Gut Microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Vangay, P.; Johnson, A.J.; Ward, T.L.; Al-Ghalith, G.A.; Shields-Cutler, R.R.; Hillmann, B.M.; Lucas, S.K.; Beura, L.K.; Thompson, E.A.; Till, L.M.; et al. US Immigration Westernizes the Human Gut Microbiome. Cell 2018, 175, 962–972.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Xu, X.; Li, J.; Li, F. Association Between Gut Microbiota and Autism Spectrum Disorder: A Systematic Review and Meta-Analysis. Front Psychiatry 2019, 10, 473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coretti, L.; Paparo, L.; Riccio, M.P.; Amato, F.; Cuomo, M.; Natale, A.; Borrelli, L.; Corrado, G.; Comegna, M.; Buommino, E.; et al. Gut Microbiota Features in Young Children with Autism Spectrum Disorders. Front. Microbiol. 2018, 9, 3146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinan, T.G.; Stanton, C.; Cryan, J.F. Psychobiotics: A Novel Class of Psychotropic. Biol. Psychiatry 2013, 74, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, A.; Lehto, S.M.; Harty, S.; Dinan, T.G.; Cryan, J.F.; Burnet, P.W.J. Psychobiotics and the Manipulation of Bacteria-Gut-Brain Signals. Trends Neurosci. 2016, 39, 763–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosuge, A.; Kunisawa, K.; Arai, S.; Sugawara, Y.; Shinohara, K.; Iida, T.; Wulaer, B.; Kawai, T.; Fujigaki, H.; Yamamoto, Y.; et al. Heat-Sterilized Bifidobacterium Breve Prevents Depression-like Behavior and Interleukin-1β Expression in Mice Exposed to Chronic Social Defeat Stress. Brain Behav. Immun. 2021, 96, 200–211. [Google Scholar] [CrossRef]
- Tian, P.; Zou, R.; Song, L.; Zhang, X.; Jiang, B.; Wang, G.; Lee, Y.K.; Zhao, J.; Zhang, H.; Chen, W. Ingestion of Bifidobacterium Longum Subspecies Infantis Strain CCFM687 Regulated Emotional Behavior and the Central BDNF Pathway in Chronic Stress-Induced Depressive Mice through Reshaping the Gut Microbiota. Food Funct. 2019, 10, 7588–7598. [Google Scholar] [CrossRef]
- Luk, B.; Veeraragavan, S.; Engevik, M.; Balderas, M.; Major, A.; Runge, J.; Luna, R.A.; Versalovic, J. Postnatal Colonization with Human “Infant-Type” Bifidobacterium Species Alters Behavior of Adult Gnotobiotic Mice. PLoS ONE 2018, 13, e0196510. [Google Scholar] [CrossRef]
- Dordević, D.; Jančíková, S.; Vítězová, M.; Kushkevych, I. Hydrogen Sulfide Toxicity in the Gut Environment: Meta-Analysis of Sulfate-Reducing and Lactic Acid Bacteria in Inflammatory Processes. J. Adv. Res. 2020, 27, 55–69. [Google Scholar] [CrossRef]
- Derrien, M.; van Passel, M.W.J.; van de Bovenkamp, J.H.B.; Schipper, R.G.; de Vos, W.M.; Dekker, J. Mucin-Bacterial Interactions in the Human Oral Cavity and Digestive Tract. Gut Microbes 2010, 1, 254–268. [Google Scholar] [CrossRef]
- Tomova, A.; Husarova, V.; Lakatosova, S.; Bakos, J.; Vlkova, B.; Babinska, K.; Ostatnikova, D. Gastrointestinal Microbiota in Children with Autism in Slovakia. Physiol. Behav. 2015, 138, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M. Desulfovibrio Species Are Potentially Important in Regressive Autism. Med. Hypotheses 2011, 77, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Dowd, S.E.; Gontcharova, V.; Liu, C.; Henley, K.E.; Wolcott, R.D.; Youn, E.; Summanen, P.H.; Granpeesheh, D.; Dixon, D.; et al. Pyrosequencing Study of Fecal Microflora of Autistic and Control Children. Anaerobe 2010, 16, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Senok, A.C.; Verstraelen, H.; Temmerman, M.; Botta, G.A. Probiotics for the Treatment of Bacterial Vaginosis. Cochrane Database Syst. Rev. 2009, 4, CD006289. [Google Scholar] [CrossRef] [PubMed]
- Walter, J. Ecological Role of Lactobacilli in the Gastrointestinal Tract: Implications for Fundamental and Biomedical Research. Appl. Environ. Microbiol. 2008, 74, 4985–4996. [Google Scholar] [CrossRef] [Green Version]
- MacFabe, D.F.; Cain, N.E.; Boon, F.; Ossenkopp, K.P.; Cain, D.P. Effects of the Enteric Bacterial Metabolic Product Propionic Acid on Object-Directed Behavior, Social Behavior, Cognition, and Neuroinflammation in Adolescent Rats: Relevance to Autism Spectrum Disorder. Behav. Brain Res. 2011, 217, 47–54. [Google Scholar] [CrossRef]
- Frye, R.E.; Rossignol, D.A.; Scahill, L.; McDougle, C.J.; Huberman, H.; Quadros, E. Treatment of Folate Metabolism Abnormalities in Autism Spectrum Disorder. Semin Pediatr. Neurol. 2020, 35, 100835. [Google Scholar] [CrossRef]
- Bjørklund, G.; Meguid, N.A.; El-Bana, M.A.; Tinkov, A.A.; Saad, K.; Dadar, M.; Hemimi, M.; Skalny, A.V.; Hosnedlová, B.; Kizek, R.; et al. Oxidative Stress in Autism Spectrum Disorder. Mol. Neurobiol. 2020, 57, 2314–2332. [Google Scholar] [CrossRef]
- Frye, R.E.; Vassall, S.; Kaur, G.; Lewis, C.; Karim, M.; Rossignol, D. Emerging Biomarkers in Autism Spectrum Disorder: A Systematic Review. Ann. Transl. Med. 2019, 7, 792. [Google Scholar] [CrossRef]
- Frye, R.E.; Delatorre, R.; Taylor, H.; Slattery, J.; Melnyk, S.; Chowdhury, N.; James, S.J. Redox Metabolism Abnormalities in Autistic Children Associated with Mitochondrial Disease. Transl. Psychiatry 2013, 3, e273. [Google Scholar] [CrossRef]
- El-Ansary, A.; Al-Ayadhi, L. GABAergic/Glutamatergic Imbalance Relative to Excessive Neuroinflammation in Autism Spectrum Disorders. J. Neuroinflammation 2014, 11, 189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, A.; Aja, E.; Fletcher, H.M. Role of Superoxide Reductase FA796 in Oxidative Stress Resistance in Filifactor Alocis. Sci. Rep. 2020, 10, 9178. [Google Scholar] [CrossRef] [PubMed]
- Jenney, F.E.; Verhagen, M.F.J.M.; Cui, X.; Adams, M.W.W. Anaerobic Microbes: Oxygen Detoxification without Superoxide Dismutase. Science 1999, 286, 306–309. [Google Scholar] [CrossRef] [PubMed]
- Dolla, A.; Fournier, M.; Dermoun, Z. Oxygen Defense in Sulfate-Reducing Bacteria. J. Biotechnol. 2006, 126, 87–100. [Google Scholar] [CrossRef]
- Lombard, M.; Touati, D.; Fontecave, M.; Nivière, V. Superoxide Reductase as a Unique Defense System against Superoxide Stress in the Microaerophile Treponema Pallidum. J. Biol. Chem. 2000, 275, 27021–27026. [Google Scholar] [CrossRef]
- Williams, R.J. Sulfate Deficiency as a Risk Factor for Autism. J. Autism Dev. Disord. 2020, 50, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Waring, R.H.; Klovrza, L. Sulphur Metabolism in Autism. J. Nutr. Environ. Med. 2009, 10, 25–32. [Google Scholar] [CrossRef]
- Nunes-Costa, D.; Magalhães, J.D.; G-Fernandes, M.; Cardoso, S.M.; Empadinhas, N. Microbial BMAA and the Pathway for Parkinson’s Disease Neurodegeneration. Front. Aging Neurosci. 2020, 12, 26. [Google Scholar] [CrossRef] [Green Version]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Breitwieser, F.P.; Thielen, P.; Salzberg, S.L. Bracken: Estimating Species Abundance in Metagenomics Data. PeerJ Comput. Sci. 2017, 2017, e104. [Google Scholar] [CrossRef] [Green Version]
- Franzosa, E.A.; McIver, L.J.; Rahnavard, G.; Thompson, L.R.; Schirmer, M.; Weingart, G.; Lipson, K.S.; Knight, R.; Caporaso, J.G.; Segata, N.; et al. Species-Level Functional Profiling of Metagenomes and Metatranscriptomes. Nat. Methods 2018, 15, 962. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, Interactive, Scalable and Extensible Microbiome Data Science Using QIIME 2. Nat. Biotechnol 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; et al. The MetaCyc Database of Metabolic Pathways and Enzymes and the BioCyc Collection of Pathway/Genome Databases. Nucleic Acids Res 2014, 42, D459–D471. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nirmalkar, K.; Qureshi, F.; Kang, D.-W.; Hahn, J.; Adams, J.B.; Krajmalnik-Brown, R. Shotgun Metagenomics Study Suggests Alteration in Sulfur Metabolism and Oxidative Stress in Children with Autism and Improvement after Microbiota Transfer Therapy. Int. J. Mol. Sci. 2022, 23, 13481. https://doi.org/10.3390/ijms232113481
Nirmalkar K, Qureshi F, Kang D-W, Hahn J, Adams JB, Krajmalnik-Brown R. Shotgun Metagenomics Study Suggests Alteration in Sulfur Metabolism and Oxidative Stress in Children with Autism and Improvement after Microbiota Transfer Therapy. International Journal of Molecular Sciences. 2022; 23(21):13481. https://doi.org/10.3390/ijms232113481
Chicago/Turabian StyleNirmalkar, Khemlal, Fatir Qureshi, Dae-Wook Kang, Juergen Hahn, James B. Adams, and Rosa Krajmalnik-Brown. 2022. "Shotgun Metagenomics Study Suggests Alteration in Sulfur Metabolism and Oxidative Stress in Children with Autism and Improvement after Microbiota Transfer Therapy" International Journal of Molecular Sciences 23, no. 21: 13481. https://doi.org/10.3390/ijms232113481
APA StyleNirmalkar, K., Qureshi, F., Kang, D. -W., Hahn, J., Adams, J. B., & Krajmalnik-Brown, R. (2022). Shotgun Metagenomics Study Suggests Alteration in Sulfur Metabolism and Oxidative Stress in Children with Autism and Improvement after Microbiota Transfer Therapy. International Journal of Molecular Sciences, 23(21), 13481. https://doi.org/10.3390/ijms232113481