
Antiandrogenic Effects of a Polyphenol in Carex kobomugi through Inhibition of Androgen Synthetic Pathway and Downregulation of Androgen Receptor in Prostate Cancer Cell Lines
 , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
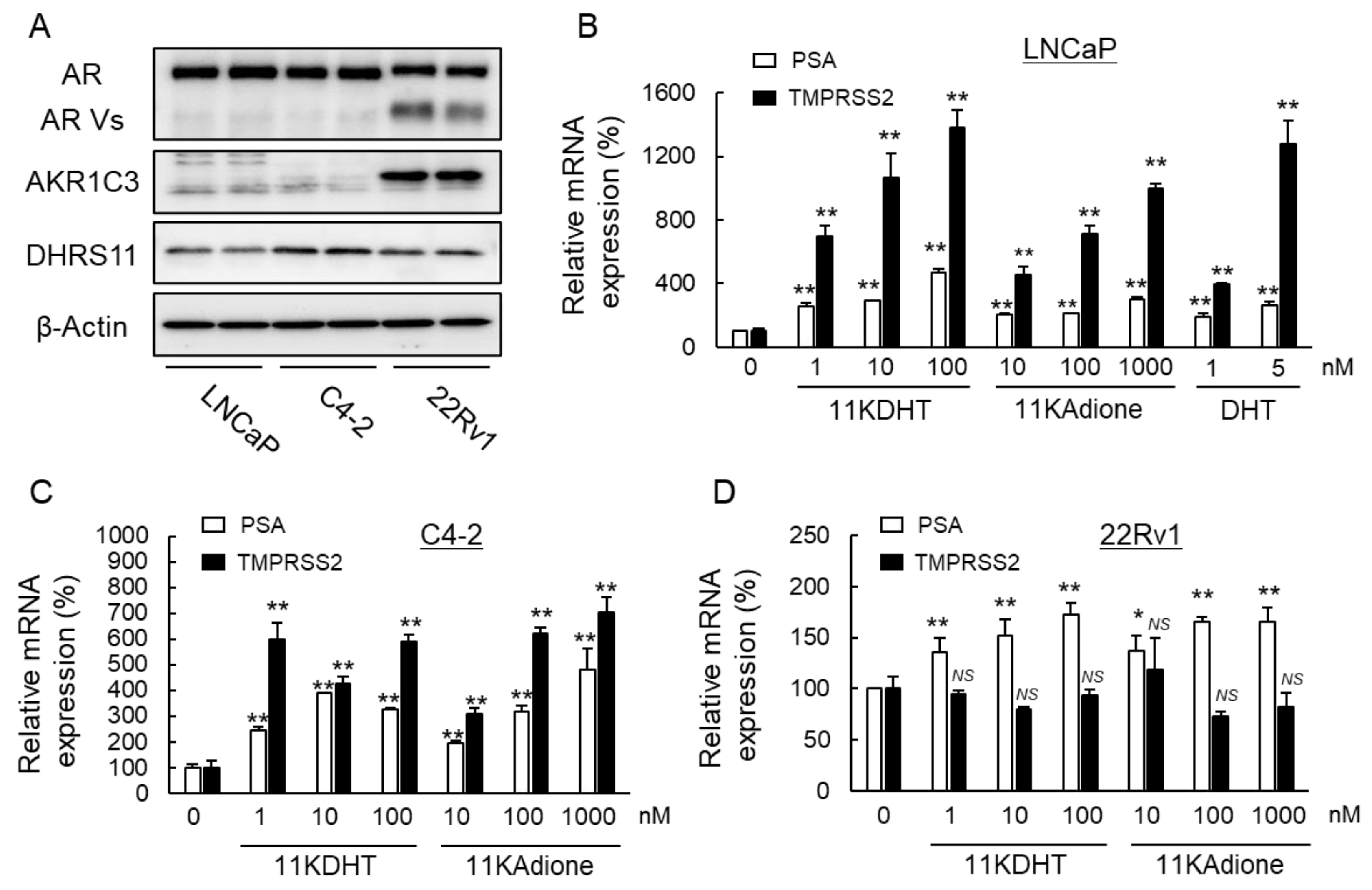
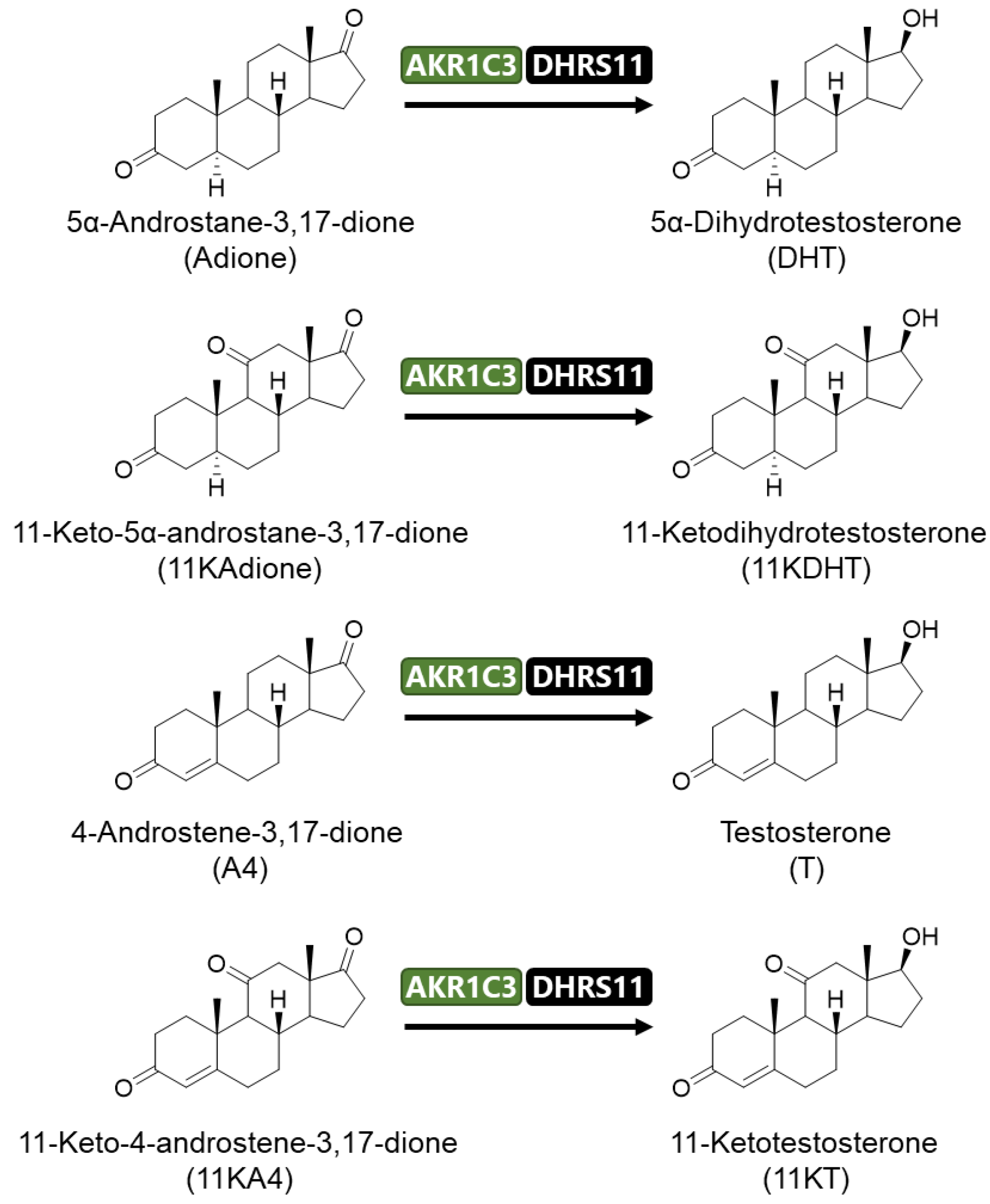
2.1. Activation of Androgen Signaling by 11-Oxygenated Androgens
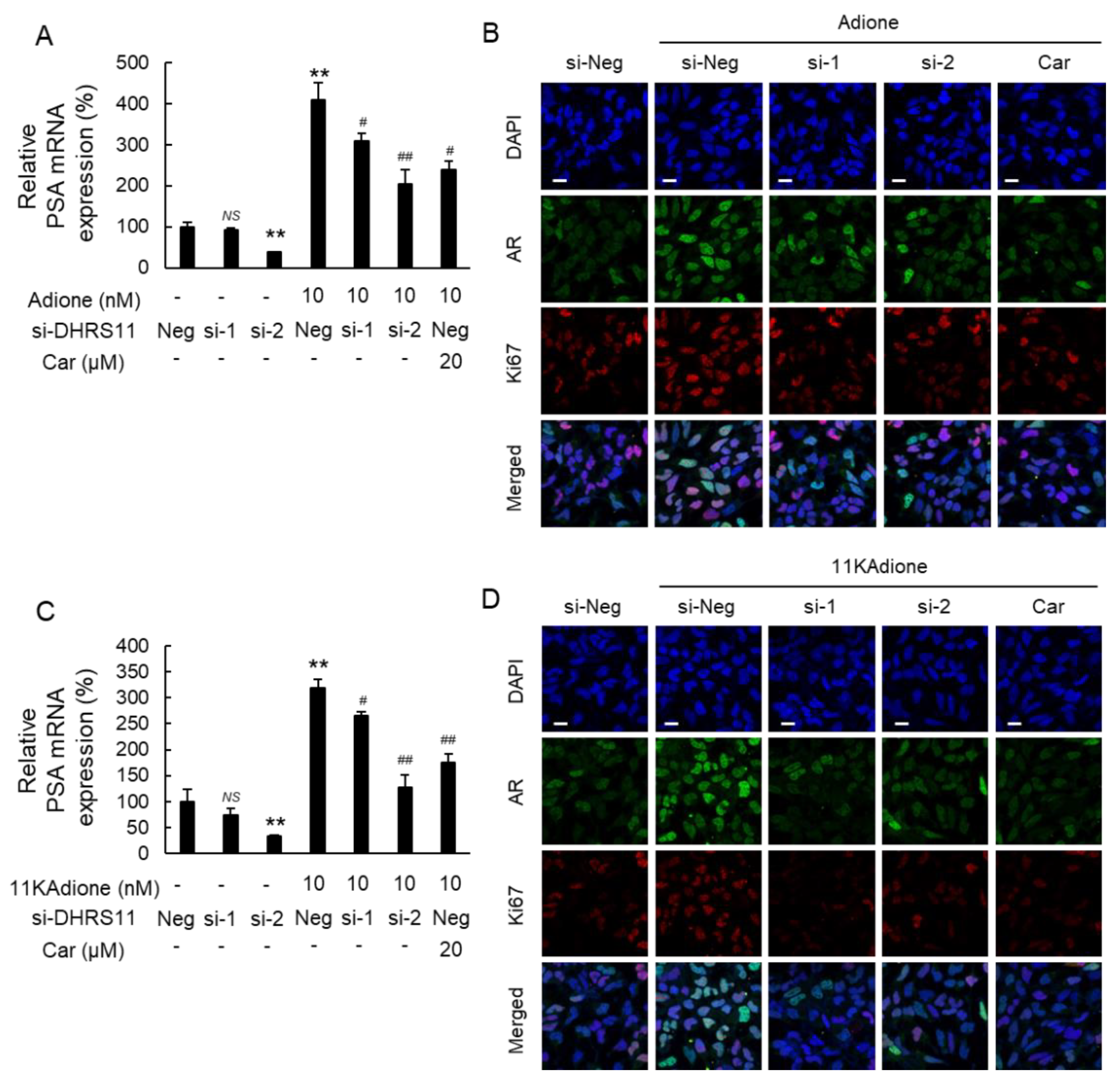
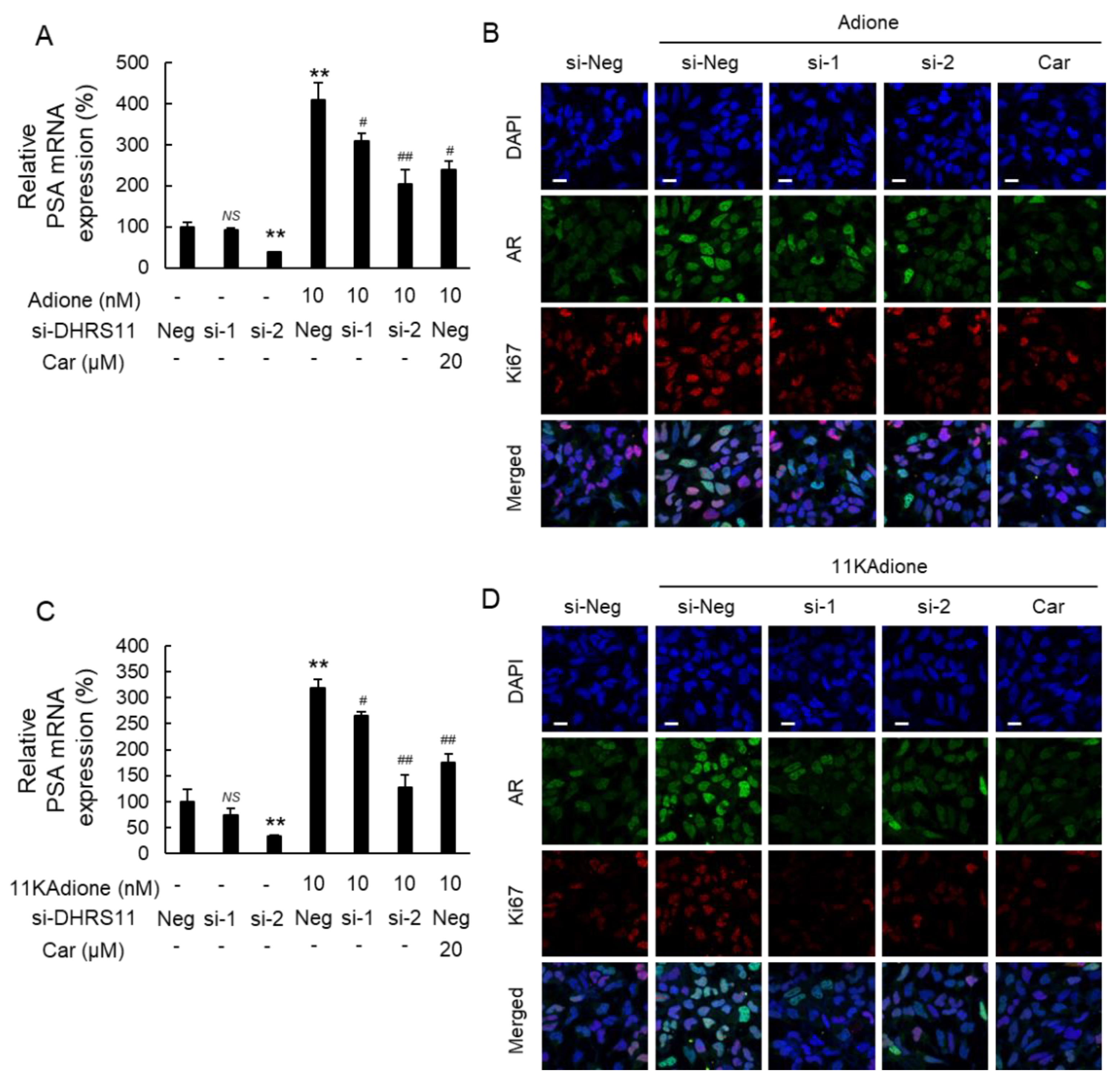
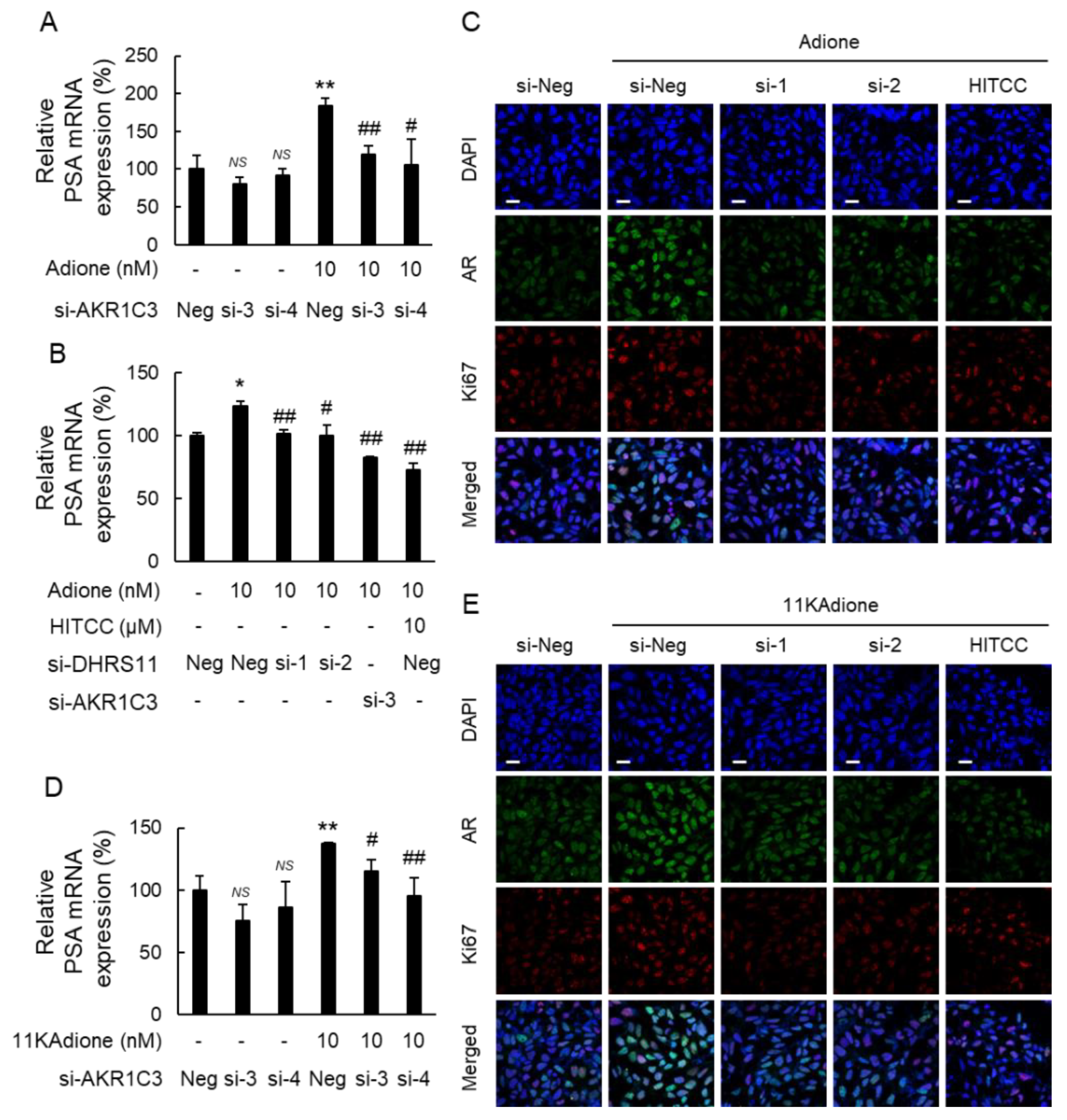
2.2. Effect of DHRS11 Silencing on 11KAdione-Induced Androgen Signaling
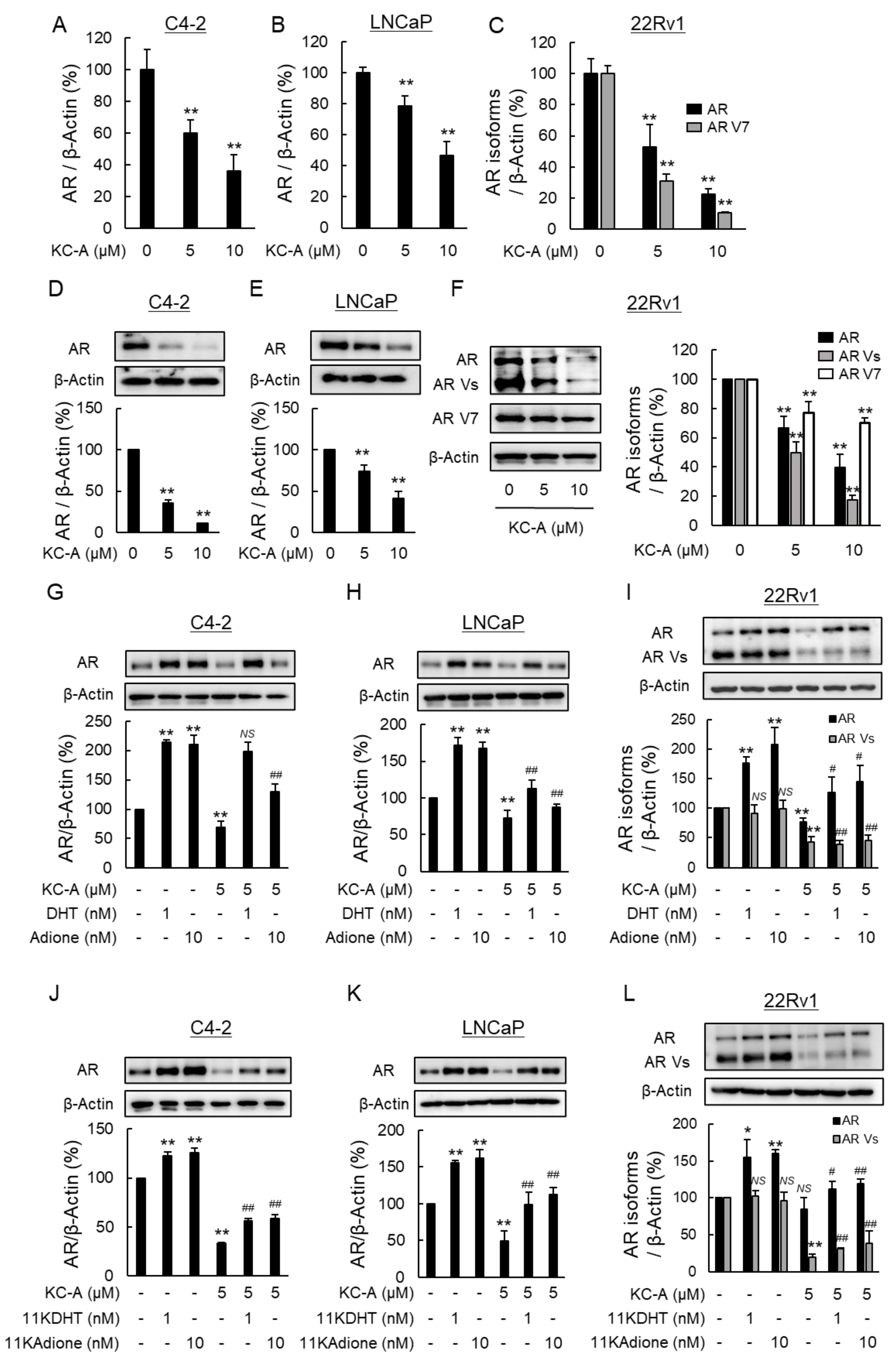
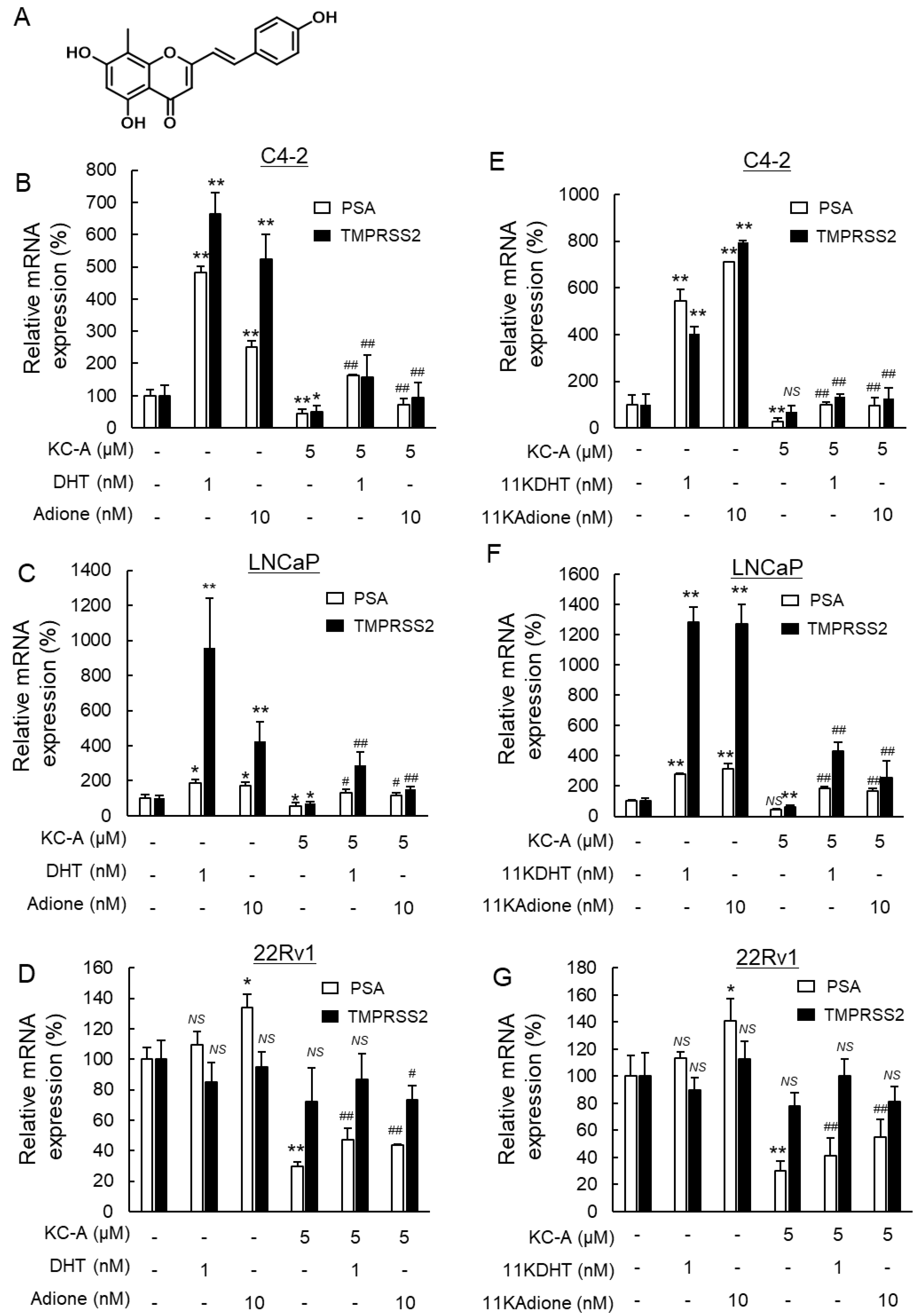
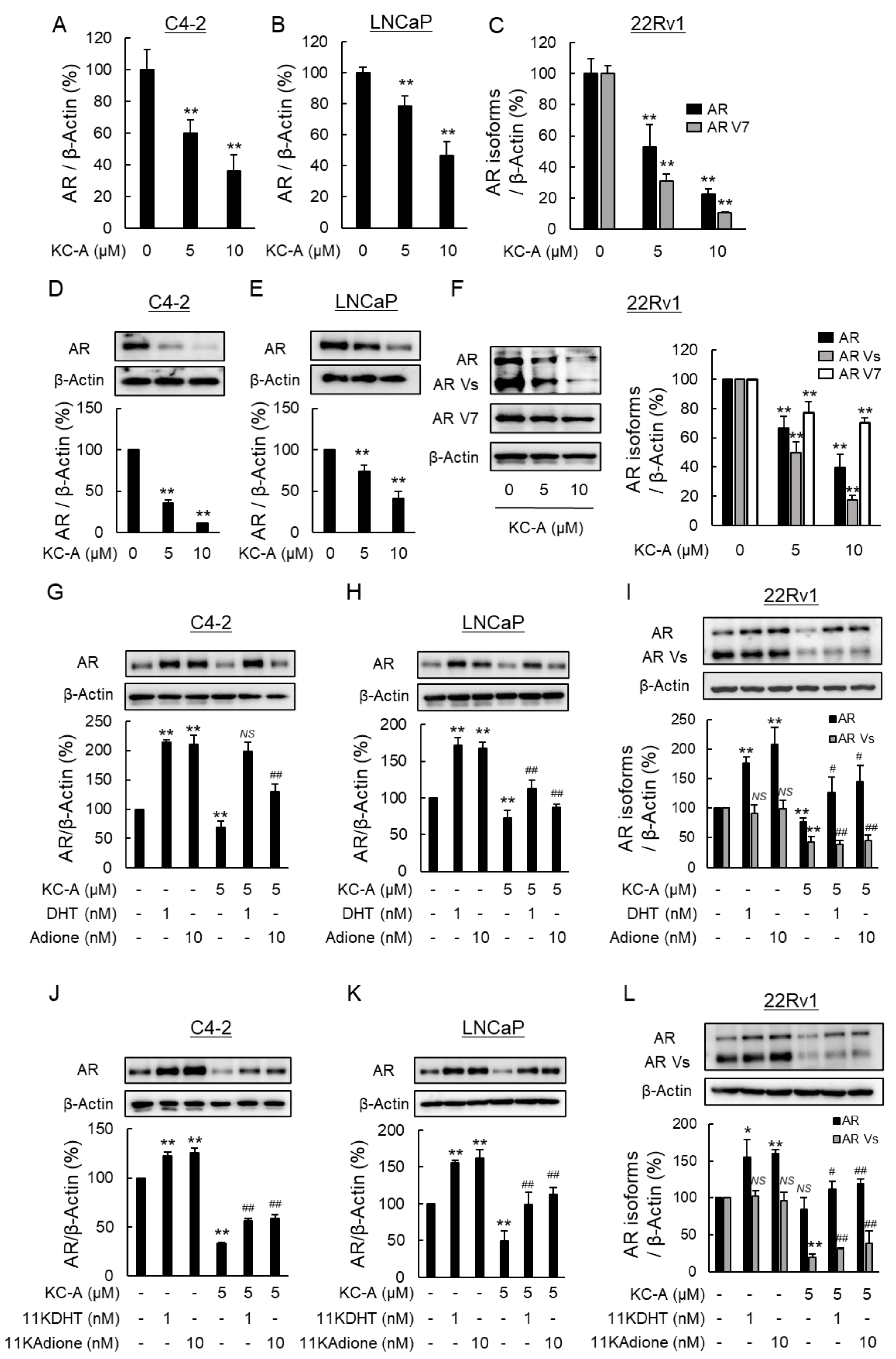
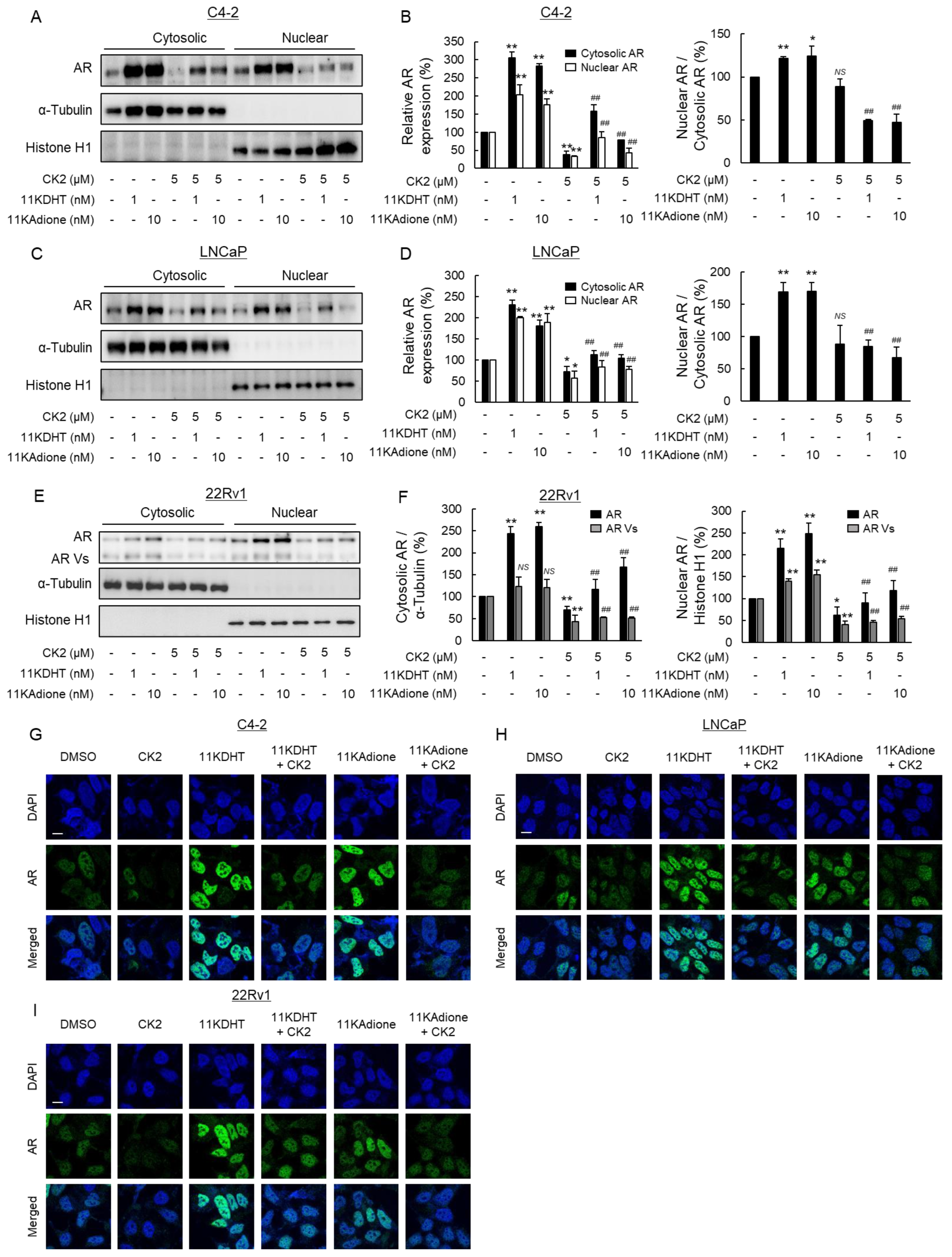
2.3. Suppression of Androgen Signaling Induced by Traditional Androgens and 11-Oxygenated Androgens by Novel Polyphenol KC-A in Carex Kobomugi
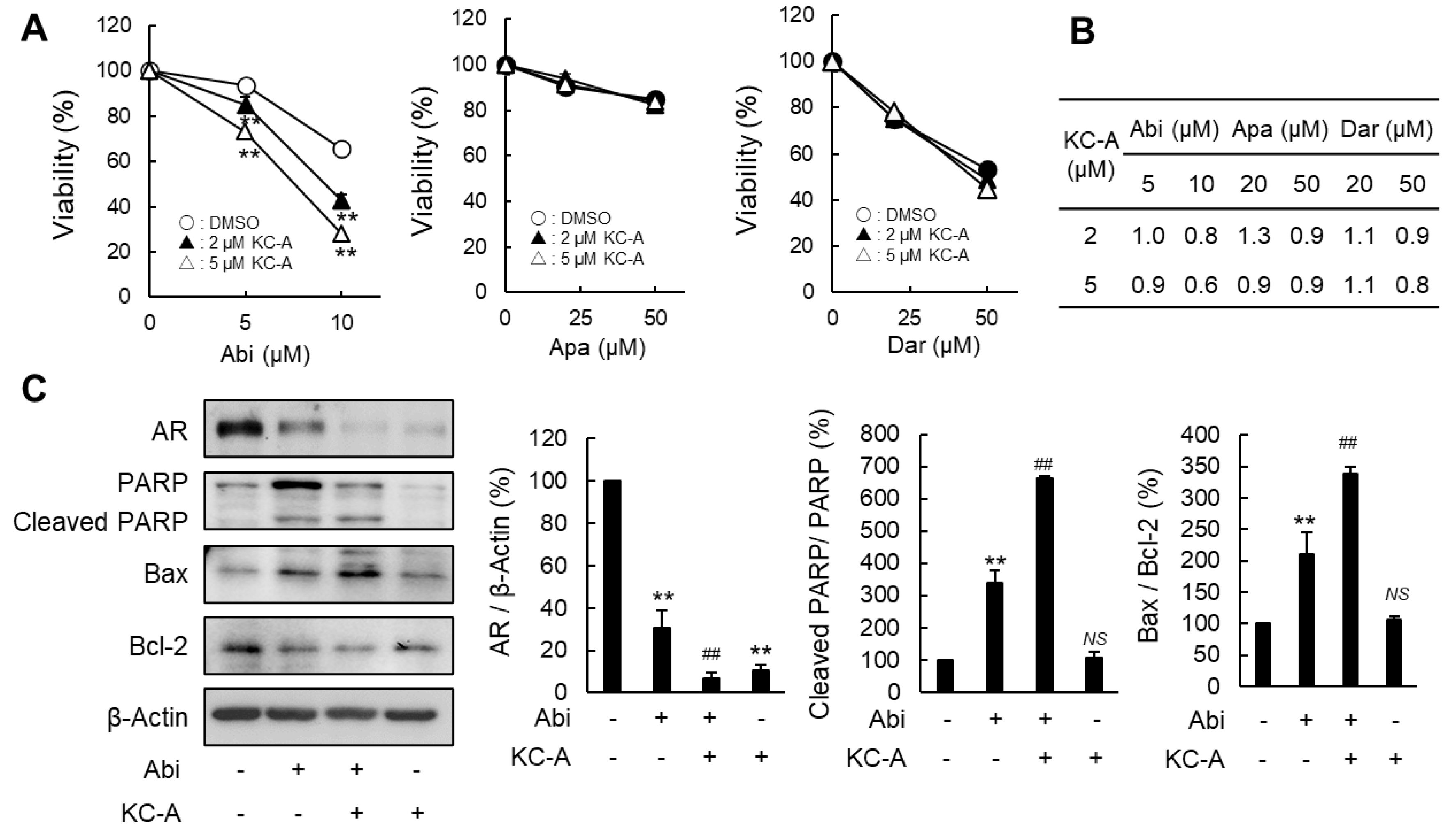
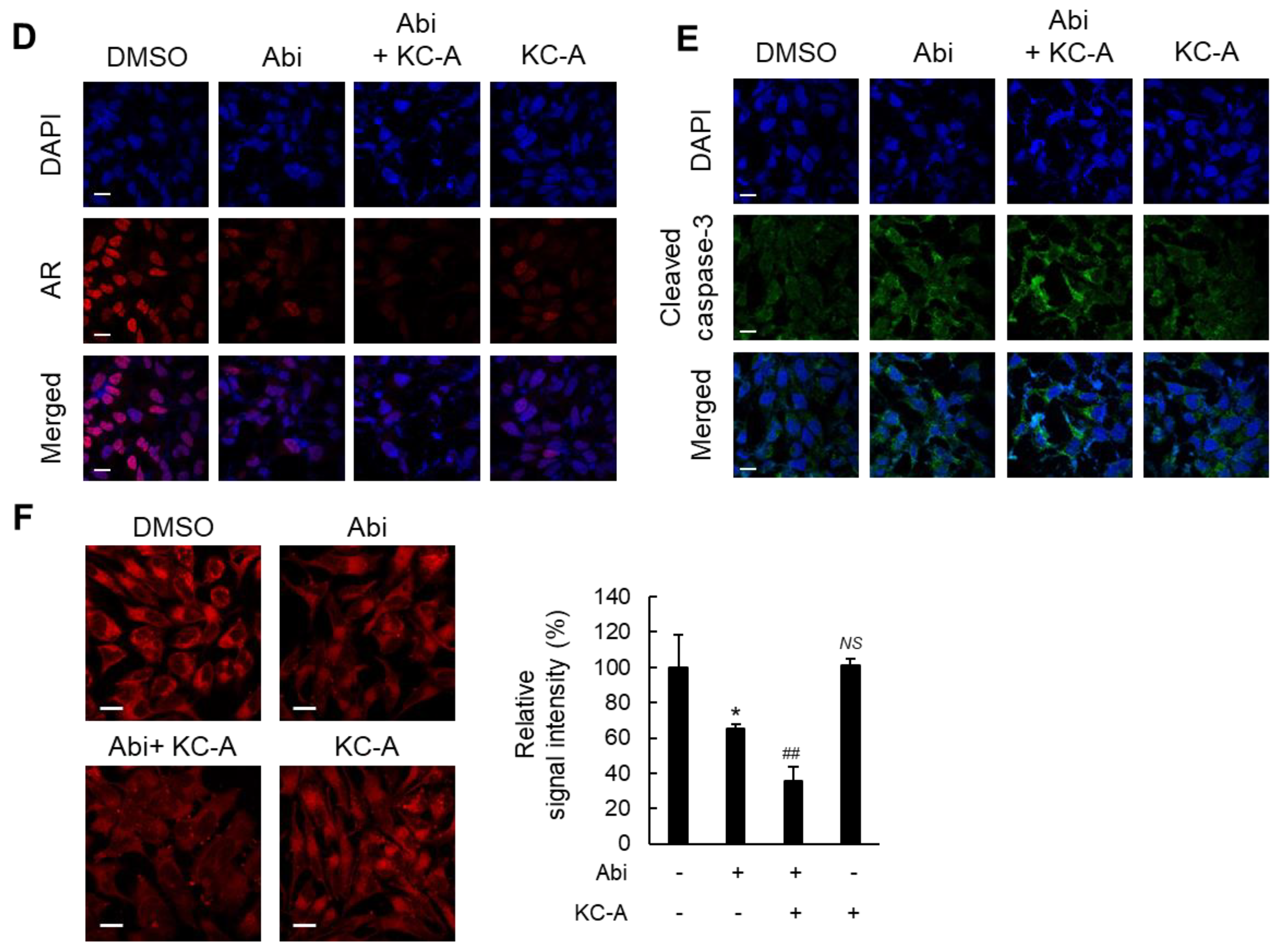
2.4. Increased Sensitivity to Abi by KC-A
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. General Experimental Procedures in Chemistry
3.3. Extraction and Isolation of KC-A
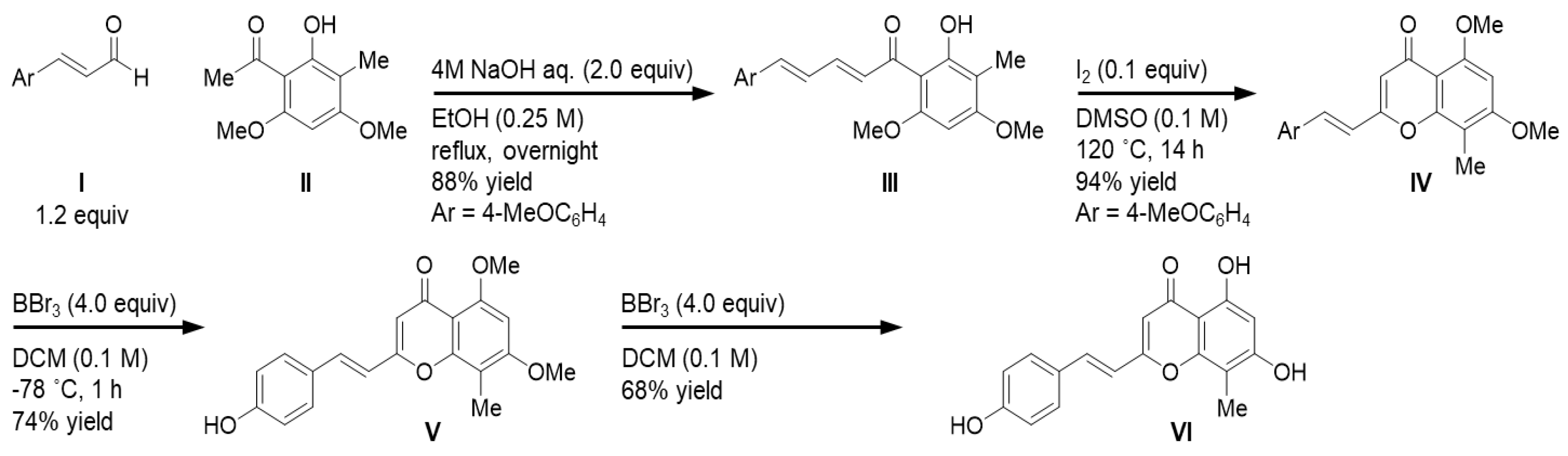
3.4. Synthesis of KC-A
3.5. Enzyme Activity Assay
3.6. Cell Culture and Transfection
3.7. Cell Viability
3.8. RNA Extraction and Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
3.9. Western Blotting
3.10. Immunofluorescence Staining and MT-1 Staining
3.11. AR-EcoScreen Reporter Gene Assay
3.12. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| 11KA4 | 11-keto-4-androstene-3,17-dione |
| 11KAdione | 11-keto-5α-androstane-3,17-dione |
| 11KDHT | 11-keto-5α-dihydrotestosterone |
| 11KT | 11-ketotestosterone |
| 11OHA4 | 11-hydroxy-4-androstene-3,17-dione |
| 17βHSD | 17β-hydroxysteroid dehydrogenase |
| A4 | 4-androstene-3,17-dione |
| Abi | abiraterone acetate |
| Adione | 5α-androstane-3,17-dione |
| AKR | aldo-keto reductase |
| Apa | apalutamide |
| AR | androgen receptor |
| AR Vs | androgen receptor variants |
| CI | combination index |
| CRPC | castration-resistant prostate cancer |
| CS-FBS | charcoal-stripped fetal bovine serum |
| Dar | darolutamide |
| DHEA | dehydroepiandrosterone |
| DHEAS | dehydroepiandrosterone sulfate |
| DHRS11 | dehydrogenase/reductase SDR family member 11 |
| DHT | 5α-dihydrotestosterone |
| DMSO | dimethyl sulfoxide |
| FBS | fetal bovine serum |
| HITCC | 8-hydroxy-2-imino-N-(p-tolyl)-2H-chromene-3-carboxamide |
| PC | prostate cancer |
| PSA | prostate specific antigen |
| RT-qPCR | reverse transcription-quantitative polymerase chain reaction |
| T | testosterone |
| TMPRSS2 | transmembrane protease serine 2 |
References
- Huggins, C.; Hodges, C.V. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J. Clin. 1972, 22, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; Yen, A.E.; Weigel, N.L. Androgen receptors in hormone-dependent and castration-resistant prostate cancer. Pharmacol. Ther. 2013, 140, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, N.; Gulley, J.L.; Dahut, W.L. Androgen deprivation therapy for prostate cancer. JAMA 2005, 294, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef]
- El-Amm, J.; Aragon-Ching, J.B. The Current Landscape of Treatment in Non-Metastatic Castration-Resistant Prostate Cancer. Clin. Med. Insights Oncol. 2019, 13, 1179554919833927. [Google Scholar] [CrossRef]
- Ross, R.W.; Xie, W.; Regan, M.M.; Pomerantz, M.; Nakabayashi, M.; Daskivich, T.J.; Sartor, O.; Taplin, M.E.; Kantoff, P.W.; Oh, W.K. Efficacy of androgen deprivation therapy (ADT) in patients with advanced prostate cancer: Association between Gleason score, prostate-specific antigen level, and prior ADT exposure with duration of ADT effect. Cancer 2008, 112, 1247–1253. [Google Scholar] [CrossRef]
- Mohler, J.L.; Gregory, C.W.; Ford, O.H., 3rd; Kim, D.; Weaver, C.M.; Petrusz, P.; Wilson, E.M.; French, F.S. The androgen axis in recurrent prostate cancer. Clin. Cancer Res. 2004, 10, 440–448. [Google Scholar] [CrossRef] [Green Version]
- Mizokami, A.; Namiki, M. Reconsideration of progression to CRPC during androgen deprivation therapy. J. Steroid Biochem. Mol. Biol. 2015, 145, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Kumagai, J.; Hofland, J.; Erkens-Schulze, S.; Dits, N.F.; Steenbergen, J.; Jenster, G.; Homma, Y.; de Jong, F.H.; van Weerden, W.M. Intratumoral conversion of adrenal androgen precursors drives androgen receptor-activated cell growth in prostate cancer more potently than de novo steroidogenesis. Prostate 2013, 73, 1636–1650. [Google Scholar] [CrossRef]
- Narimoto, K.; Mizokami, A.; Izumi, K.; Mihara, S.; Sawada, K.; Sugata, T.; Shimamura, M.; Miyazaki, K.; Nishino, A.; Namiki, M. Adrenal androgen levels as predictors of outcome in castration-resistant prostate cancer patients treated with combined androgen blockade using flutamide as a second-line anti-androgen. Int. J. Urol. 2010, 17, 337–345. [Google Scholar] [CrossRef]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocr. Relat. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef] [PubMed]
- Ciccarese, C.; Massari, F.; Iacovelli, R.; Fiorentino, M.; Montironi, R.; Di Nunno, V.; Giunchi, F.; Brunelli, M.; Tortora, G. Prostate cancer heterogeneity: Discovering novel molecular targets for therapy. Cancer Treat. Rev. 2017, 54, 68–73. [Google Scholar] [CrossRef]
- Labrie, F. All sex steroids are made intracellularly in peripheral tissues by the mechanisms of intracrinology after menopause. J. Steroid Biochem. Mol. Biol. 2015, 145, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Aka, J.A.; Mazumdar, M.; Lin, S.X. Reductive 17beta-hydroxysteroid dehydrogenases in the sulfatase pathway: Critical in the cell proliferation of breast cancer. Mol. Cell. Endocrinol. 2009, 301, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, E.; Arlt, W.; Storbeck, K.H. A new dawn for androgens: Novel lessons from 11-oxygenated C19 steroids. Mol. Cell. Endocrinol. 2017, 441, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, L.; Arlt, W.; Storbeck, K.H. Intracrine androgen biosynthesis, metabolism and action revisited. Mol. Cell. Endocrinol. 2018, 465, 4–26. [Google Scholar] [CrossRef]
- Pretorius, E.; Africander, D.J.; Vlok, M.; Perkins, M.S.; Quanson, J.; Storbeck, K.H. 11-Ketotestosterone and 11-Ketodihydrotestosterone in Castration Resistant Prostate Cancer: Potent Androgens Which Can No Longer Be Ignored. PLoS ONE 2016, 11, e0159867. [Google Scholar] [CrossRef] [Green Version]
- Turcu, A.F.; Rege, J.; Auchus, R.J.; Rainey, W.E. 11-Oxygenated androgens in health and disease. Nat. Rev. Endocrinol. 2020, 16, 284–296. [Google Scholar] [CrossRef]
- Storbeck, K.H.; Bloem, L.M.; Africander, D.; Schloms, L.; Swart, P.; Swart, A.C. 11β-Hydroxydihydrotestosterone and 11-ketodihydrotestosterone, novel C19 steroids with androgenic activity: A putative role in castration resistant prostate cancer? Mol. Cell. Endocrinol. 2013, 377, 135–146. [Google Scholar] [CrossRef]
- Barnard, M.; Quanson, J.L.; Mostaghel, E.; Pretorius, E.; Snoep, J.L.; Storbeck, K.H. 11-Oxygenated androgen precursors are the preferred substrates for aldo-keto reductase 1C3 (AKR1C3): Implications for castration resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2018, 183, 192–201. [Google Scholar] [CrossRef]
- Snaterse, G.; Visser, J.A.; Arlt, W.; Hofland, J. Circulating steroid hormone variations throughout different stages of prostate cancer. Endocr. Relat. Cancer 2017, 24, R403–R420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnard, M.; Mostaghel, E.A.; Auchus, R.J.; Storbeck, K.H. The role of adrenal derived androgens in castration resistant prostate cancer. J. Steroid Biochem. Mol. Biol. 2020, 197, 105506. [Google Scholar] [CrossRef] [PubMed]
- Wright, C.; O’Day, P.; Alyamani, M.; Sharifi, N.; Auchus, R.J. Abiraterone acetate treatment lowers 11-oxygenated androgens. Eur. J. Endocrinol. 2020, 182, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Endo, S.; Morikawa, Y.; Kudo, Y.; Suenami, K.; Matsunaga, T.; Ikari, A.; Hara, A. Human dehydrogenase/reductase SDR family member 11 (DHRS11) and aldo-keto reductase 1C isoforms in comparison: Substrate and reaction specificity in the reduction of 11-keto-C(19)-steroids. J. Steroid Biochem. Mol. Biol. 2020, 199, 105586. [Google Scholar] [CrossRef]
- Penning, T.M.; Byrns, M.C. Steroid hormone transforming aldo-keto reductases and cancer. Ann. N. Y. Acad. Sci. 2009, 1155, 33–42. [Google Scholar] [CrossRef] [Green Version]
- Endo, S.; Miyagi, N.; Matsunaga, T.; Hara, A.; Ikari, A. Human dehydrogenase/reductase (SDR family) member 11 is a novel type of 17β-hydroxysteroid dehydrogenase. Biochem. Biophys. Res. Commun. 2016, 472, 231–236. [Google Scholar] [CrossRef]
- Liu, L.; Dong, X. Complex impacts of PI3K/AKT inhibitors to androgen receptor gene expression in prostate cancer cells. PLoS ONE 2014, 9, e108780. [Google Scholar] [CrossRef] [Green Version]
- Haendler, B.; Schuttke, I.; Schleuning, W.D. Androgen receptor signalling: Comparative analysis of androgen response elements and implication of heat-shock protein 90 and 14-3-3eta. Mol. Cell. Endocrinol. 2001, 173, 63–73. [Google Scholar] [CrossRef]
- Tararova, N.D.; Narizhneva, N.; Krivokrisenko, V.; Gudkov, A.V.; Gurova, K.V. Prostate cancer cells tolerate a narrow range of androgen receptor expression and activity. Prostate 2007, 67, 1801–1815. [Google Scholar] [CrossRef] [Green Version]
- Endo, S.; Matsunaga, T.; Kanamori, A.; Otsuji, Y.; Nagai, H.; Sundaram, K.; El-Kabbani, O.; Toyooka, N.; Ohta, S.; Hara, A. Selective inhibition of human type-5 17beta-hydroxysteroid dehydrogenase (AKR1C3) by baccharin, a component of Brazilian propolis. J. Nat. Prod. 2012, 75, 716–721. [Google Scholar] [CrossRef]
- Endo, S.; Hu, D.; Matsunaga, T.; Otsuji, Y.; El-Kabbani, O.; Kandeel, M.; Ikari, A.; Hara, A.; Kitade, Y.; Toyooka, N. Synthesis of non-prenyl analogues of baccharin as selective and potent inhibitors for aldo-keto reductase 1C3. Bioorg. Med. Chem. 2014, 22, 5220–5233. [Google Scholar] [CrossRef] [PubMed]
- Endo, S.; Miyagi, N.; Matsunaga, T.; Ikari, A. Rabbit dehydrogenase/reductase SDR family member 11 (DHRS11): Its identity with acetohexamide reductase with broad substrate specificity and inhibitor sensitivity, different from human DHRS11. Chem Biol Interact 2019, 305, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Zwart, N.; Andringa, D.; de Leeuw, W.J.; Kojima, H.; Iida, M.; Houtman, C.J.; de Boer, J.; Kool, J.; Lamoree, M.H.; Hamers, T. Improved androgen specificity of AR-EcoScreen by CRISPR based glucocorticoid receptor knockout. Toxicol. In Vitro 2017, 45, 1–9. [Google Scholar] [CrossRef]
- Kanayama, M.; Lu, C.; Luo, J.; Antonarakis, E.S. AR Splicing Variants and Resistance to AR Targeting Agents. Cancers 2021, 13, 2563. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [Green Version]
- Krongrad, A.; Wilson, C.M.; Wilson, J.D.; Allman, D.R.; McPhaul, M.J. Androgen increases androgen receptor protein while decreasing receptor mRNA in LNCaP cells. Mol. Cell. Endocrinol. 1991, 76, 79–88. [Google Scholar] [CrossRef]
- Vija, L.; Boukari, K.; Loosfelt, H.; Meduri, G.; Viengchareun, S.; Binart, N.; Young, J.; Lombes, M. Ligand-dependent stabilization of androgen receptor in a novel mouse ST38c Sertoli cell line. Mol. Cell. Endocrinol. 2014, 384, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Saleem, M.; Kweon, M.H.; Yun, J.M.; Adhami, V.M.; Khan, N.; Syed, D.N.; Mukhtar, H. A novel dietary triterpene Lupeol induces fas-mediated apoptotic death of androgen-sensitive prostate cancer cells and inhibits tumor growth in a xenograft model. Cancer Res. 2005, 65, 11203–11213. [Google Scholar] [CrossRef] [Green Version]
- Montazeri-Najafabady, N.; Chatrabnous, N.; Arabnezhad, M.R.; Azarpira, N. Anti-androgenic effect of astaxanthin in LNCaP cells is mediated through the aryl hydrocarbon-androgen receptors cross talk. J. Food Biochem. 2021, 45, e13702. [Google Scholar] [CrossRef]
- Liu, S.; Yamauchi, H. Hinokitiol, a metal chelator derived from natural plants, suppresses cell growth and disrupts androgen receptor signaling in prostate carcinoma cell lines. Biochem. Biophys. Res. Commun. 2006, 351, 26–32. [Google Scholar] [CrossRef]
- Chiu, F.L.; Lin, J.K. Downregulation of androgen receptor expression by luteolin causes inhibition of cell proliferation and induction of apoptosis in human prostate cancer cells and xenografts. Prostate 2008, 68, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Naiki-Ito, A.; Naiki, T.; Kato, H.; Iida, K.; Etani, T.; Nagayasu, Y.; Suzuki, S.; Yamashita, Y.; Inaguma, S.; Onishi, M.; et al. Recruitment of miR-8080 by luteolin inhibits androgen receptor splice variant 7 expression in castration-resistant prostate cancer. Carcinogenesis 2020, 41, 1145–1157. [Google Scholar] [CrossRef] [Green Version]
- Ideyama, Y.; Kudoh, M.; Tanimoto, K.; Susaki, Y.; Nanya, T.; Nakahara, T.; Ishikawa, H.; Yoden, T.; Okada, M.; Fujikura, T.; et al. Novel nonsteroidal inhibitor of cytochrome P45017α (17α-hydroxylase/C17–20 lyase), YM116, decreased prostatic weights by reducing serum concentrations of testosterone and adrenal androgens in rats. Prostate 1998, 37, 10–18. [Google Scholar] [CrossRef]
- Haidar, S.; Ehmer, P.B.; Barassin, S.; Batzl-Hartmann, C.; Hartmann, R.W. Effects of novel 17α-hydroxylase/C17, 20-lyase (P450 17, CYP 17) inhibitors on androgen biosynthesis in vitro and in vivo. J. Steroid Biochem. Mol. Biol. 2003, 84, 555–562. [Google Scholar] [CrossRef]
- Han, J.; Zhang, J.; Zhang, W.; Zhang, D.; Li, Y.; Zhang, J.; Zhang, Y.; Diao, T.; Cui, L.; Li, W.; et al. Abiraterone and MDV3100 inhibits the proliferation and promotes the apoptosis of prostate cancer cells through mitophagy. Cancer Cell Int. 2019, 19, 332. [Google Scholar] [CrossRef] [PubMed]
- Soifer, H.S.; Souleimanian, N.; Wu, S.; Voskresenskiy, A.M.; Collak, F.K.; Cinar, B.; Stein, C.A. Direct regulation of androgen receptor activity by potent CYP17 inhibitors in prostate cancer cells. J. Biol. Chem. 2012, 287, 3777–3787. [Google Scholar] [CrossRef] [Green Version]
- Ashrafi, G.; Wu, Z.; Farrell, R.J.; Ryan, T.A. GLUT4 Mobilization Supports Energetic Demands of Active Synapses. Neuron 2017, 93, 606–615.e603. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Endo, S.; Kawai, M.; Hoshi, M.; Segawa, J.; Fujita, M.; Matsukawa, T.; Fujimoto, N.; Matsunaga, T.; Ikari, A. Targeting Nrf2-antioxidant signaling reverses acquired cabazitaxel resistance in prostate cancer cells. J. Biochem. 2021, 170, 89–96. [Google Scholar] [CrossRef]
- Endo, S.; Oguri, H.; Segawa, J.; Kawai, M.; Hu, D.; Xia, S.; Okada, T.; Irie, K.; Fujii, S.; Gouda, H.; et al. Development of Novel AKR1C3 Inhibitors as New Potential Treatment for Castration-Resistant Prostate Cancer. J. Med. Chem. 2020, 63, 10396–10411. [Google Scholar] [CrossRef]
- Endo, S.; Matsuoka, T.; Nishiyama, T.; Arai, Y.; Kashiwagi, H.; Abe, N.; Oyama, M.; Matsunaga, T.; Ikari, A. Flavonol glycosides of Rosa multiflora regulates intestinal barrier function through inhibiting claudin expression in differentiated Caco-2 cells. Nutr Res 2019, 72, 92–104. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kudo, Y.; Endo, S.; Tanio, M.; Saka, T.; Himura, R.; Abe, N.; Takeda, M.; Yamaguchi, E.; Yoshino, Y.; Arai, Y.; et al. Antiandrogenic Effects of a Polyphenol in Carex kobomugi through Inhibition of Androgen Synthetic Pathway and Downregulation of Androgen Receptor in Prostate Cancer Cell Lines. Int. J. Mol. Sci. 2022, 23, 14356. https://doi.org/10.3390/ijms232214356
Kudo Y, Endo S, Tanio M, Saka T, Himura R, Abe N, Takeda M, Yamaguchi E, Yoshino Y, Arai Y, et al. Antiandrogenic Effects of a Polyphenol in Carex kobomugi through Inhibition of Androgen Synthetic Pathway and Downregulation of Androgen Receptor in Prostate Cancer Cell Lines. International Journal of Molecular Sciences. 2022; 23(22):14356. https://doi.org/10.3390/ijms232214356
Chicago/Turabian StyleKudo, Yudai, Satoshi Endo, Masatoshi Tanio, Tomofumi Saka, Rin Himura, Naohito Abe, Mitsumi Takeda, Eiji Yamaguchi, Yuta Yoshino, Yuki Arai, and et al. 2022. "Antiandrogenic Effects of a Polyphenol in Carex kobomugi through Inhibition of Androgen Synthetic Pathway and Downregulation of Androgen Receptor in Prostate Cancer Cell Lines" International Journal of Molecular Sciences 23, no. 22: 14356. https://doi.org/10.3390/ijms232214356
APA StyleKudo, Y., Endo, S., Tanio, M., Saka, T., Himura, R., Abe, N., Takeda, M., Yamaguchi, E., Yoshino, Y., Arai, Y., Kashiwagi, H., Oyama, M., Itoh, A., Shiota, M., Fujimoto, N., & Ikari, A. (2022). Antiandrogenic Effects of a Polyphenol in Carex kobomugi through Inhibition of Androgen Synthetic Pathway and Downregulation of Androgen Receptor in Prostate Cancer Cell Lines. International Journal of Molecular Sciences, 23(22), 14356. https://doi.org/10.3390/ijms232214356