
SCN5A Variants as Genetic Arrhythmias Triggers for Familial Bileaflet Mitral Valve Prolapse
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
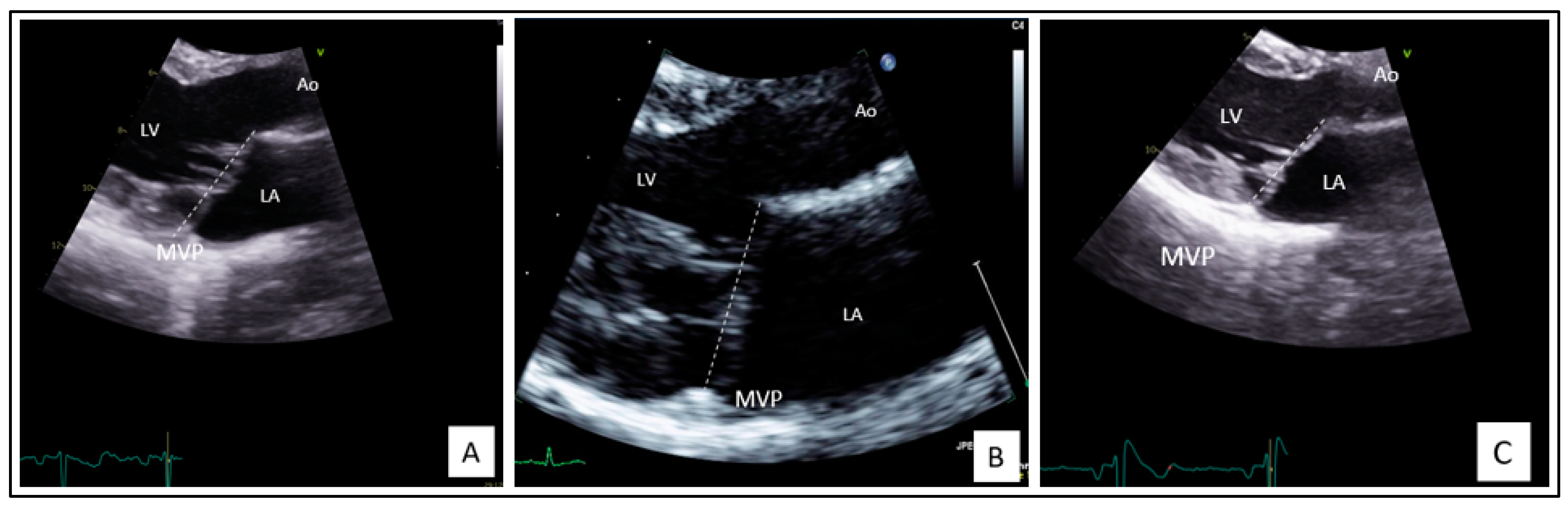
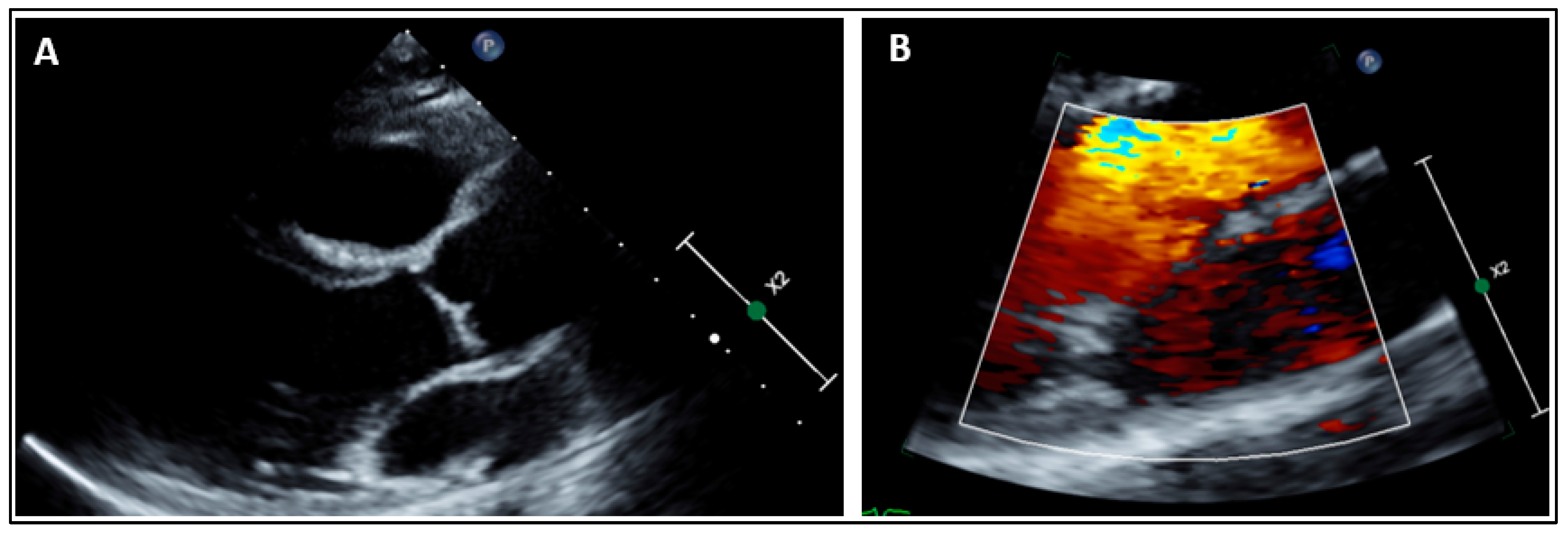
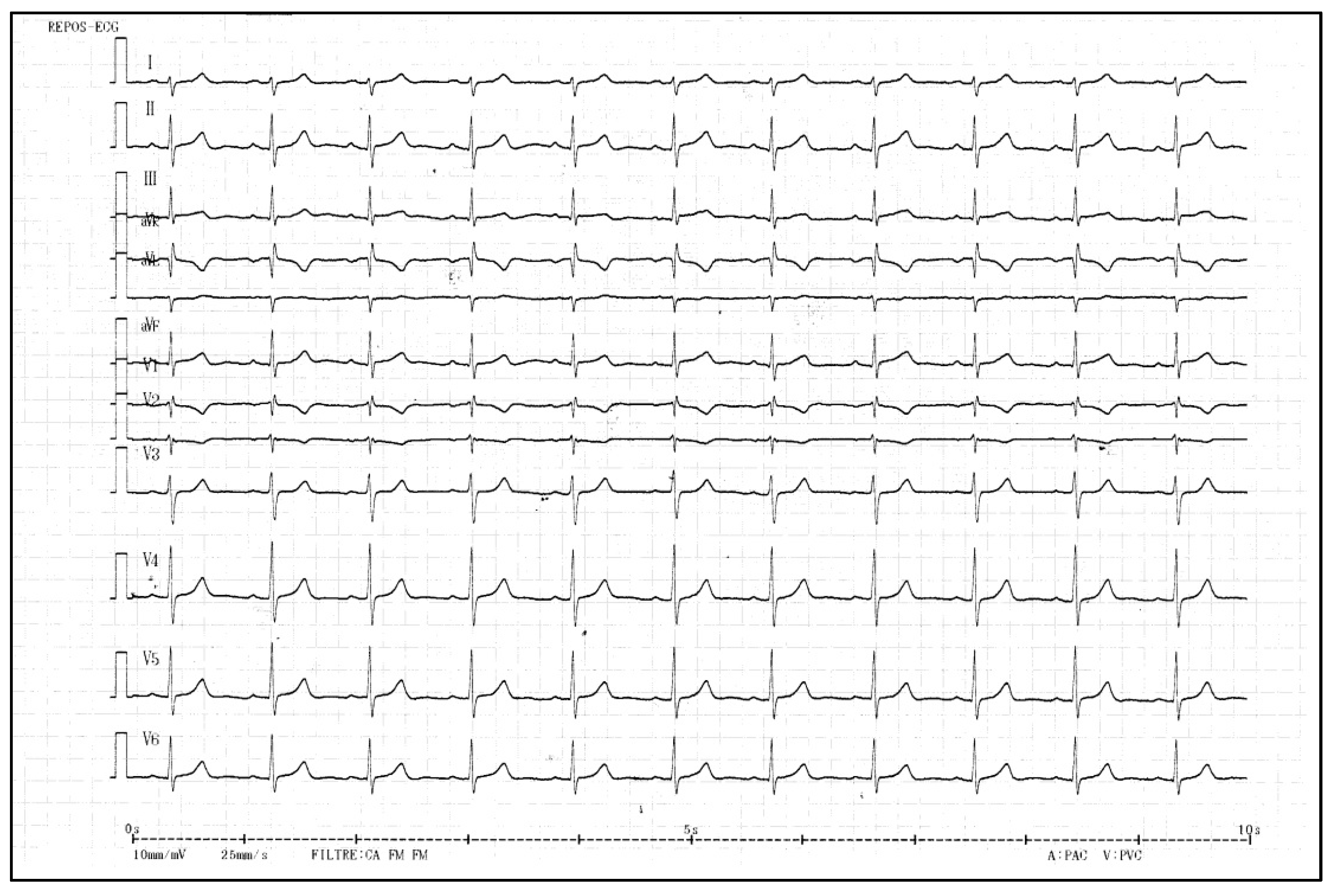
2.1. Clinical Findings
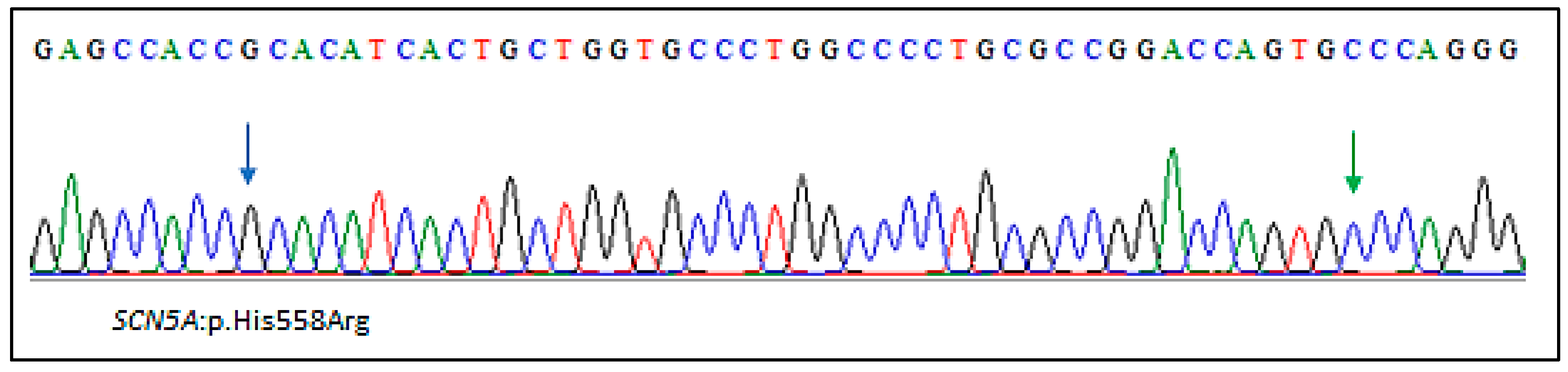
2.2. Genetic Findings
3. Discussion
4. Materials and Methods
4.1. Whole Exome Sequencing (WES)
4.2. In Silico Analysis Tools
Variant Prioritization
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delling, F.N.; Vasan, R.S. Epidemiology and pathophysiology of mitral valve prolapse: New insights into disease progression, genetics, and molecular basis. Circulation 2014, 129, 2158–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.; Roshdy, A.; Sharma, R.; Fletcher, N. Sudden cardiac arrest and coexisting mitral valve prolapse: A case report and literature review. Echo Res. Pract. 2016, 3, D1–D8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukavica, D.; Guglielmo, M.; Baggiano, A.; Muscogiuri, G.; Fusini, L.; Muratori, M.; Tamborini, G.; Mantegazza, V.; Trancuccio, A.; Arnò, C.; et al. Arrhythmic Mitral Valve Prolapse: Introducing an Era of Multimodality Imaging-Based Diagnosis and Risk Stratification. Diagnostics 2021, 11, 467. [Google Scholar] [CrossRef] [PubMed]
- Parwani, P.; Avierinos, J.-F.; Levine, R.A.; Delling, F.N. Mitral Valve Prolapse: Multimodality Imaging and Genetic Insights. Prog. Cardiovasc. Dis. 2017, 60, 361–369. [Google Scholar] [CrossRef]
- Trenkwalder, T.; Krane, M. Mitral valve prolapse: Will genetics finally solve the puzzle? Eur. Heart J. 2022, 43, 1681–1683. [Google Scholar] [CrossRef]
- Althunayyan, A.; Petersen, S.E.; Lloyd, G.; Bhattacharyya, S. Mitral valve prolapse. Expert Rev. Cardiovasc. Ther. 2019, 17, 43–51. [Google Scholar] [CrossRef]
- Coutsoumbas, G.V.; Di Pasquale, G. Mitral valve prolapse with ventricular arrhythmias: Does it carries a worse prognosis? Eur. Heart J. Suppl. 2021, 23, E77–E82. [Google Scholar] [CrossRef]
- Basso, C.; Marra, M.P.; Rizzo, S.; De Lazzari, M.; Giorgi, B.; Cipriani, A.; Frigo, A.C.; Rigato, I.; Migliore, F.; Pilichou, K.; et al. Arrhythmic Mitral Valve Prolapse and Sudden Cardiac Death. Circulation 2015, 132, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Hourdain, J.; Clavel, M.A.; Deharo, J.-C.; Asirvatham, S.; Avierinos, J.F.; Habib, G.; Franceschi, F.; Probst, V.; Sadoul, N.; Martins, R.; et al. Common Phenotype in Patients With Mitral Valve Prolapse Who Experienced Sudden Cardiac Death. Circulation 2018, 138, 1067–1069. [Google Scholar] [CrossRef]
- Sriram, C.S.; Syed, F.F.; Ferguson, M.E.; Johnson, J.N.; Enriquez-Sarano, M.; Cetta, F.; Cannon, B.C.; Asirvatham, S.J.; Ackerman, M.J. Malignant bileaflet mitral valve prolapse syndrome in patients with otherwise idiopathic out-of-hospital cardiac arrest. J. Am. Coll. Cardiol. 2013, 62, 222–230. [Google Scholar] [CrossRef]
- Delling, F.N.; Rong, J.; Larson, M.G.; Lehman, B.; Osypiuk, E.; Stantchev, P.; Slaugenhaupt, S.A.; Benjamin, E.J.; Levine, R.A.; Vasan, R.S. Familial Clustering of Mitral Valve Prolapse in the Community. Circulation 2015, 131, 263–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durst, R.; Sauls, K.; Peal, D.S.; deVlaming, A.; Toomer, K.; Leyne, M.; Salani, M.; Talkowski, M.E.; Brand, H.; Perrocheau, M.; et al. Mutations in DCHS1 cause mitral valve prolapse. Nature 2015, 525, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devereux, R.B.; Brown, W.T.; Kramer-Fox, R.; Sachs, I. Inheritance of mitral valve prolapse: Effect of age and sex on gene expression. Ann. Intern. Med. 1982, 97, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Disse, S.; Abergel, E.; Berrebi, A.; Houot, A.M.; Le Heuzey, J.Y.; Diebold, B.; Guize, L.; Carpentier, A.; Corvol, P.; Jeunemaitre, X. Mapping of a first locus for autosomal dominant myxomatous mitral-valve prolapse to chromosome 16p11.2-p12.1. Am. J. Hum. Genet. 1999, 65, 1242–1251. [Google Scholar] [CrossRef] [Green Version]
- Freed, L.A.; Acierno, J.S.; Dai, D.; Leyne, M.; Marshall, J.E.; Nesta, F.; Levine, R.A.; Slaugenhaupt, S.A. A locus for autosomal dominant mitral valve prolapse on chromosome 11p15.4. Am. J. Hum. Genet. 2003, 72, 1551–1559. [Google Scholar] [CrossRef] [Green Version]
- Nesta, F.; Leyne, M.; Yosefy, C.; Simpson, C.; Dai, D.; Marshall, J.E.; Hung, J.; Slaugenhaupt, S.A.; Levine, R.A. New locus for autosomal dominant mitral valve prolapse on chromosome 13: Clinical insights from genetic studies. Circulation 2005, 112, 2022–2030. [Google Scholar] [CrossRef] [Green Version]
- Toomer, K.A.; Yu, M.; Fulmer, D.; Guo, L.; Moore, K.S.; Moore, R.; Drayton, K.D.; Glover, J.; Peterson, N.; Ramos-Ortiz, S.; et al. Primary cilia defects causing mitral valve prolapse. Sci. Transl. Med. 2019, 11, eaax0290. [Google Scholar] [CrossRef] [Green Version]
- Kyndt, F.; Schott, J.J.; Trochu, J.N.; Baranger, F.; Herbert, O.; Scott, V.; Fressinaud, E.; David, A.; Moisan, J.P.; Bouhour, J.B.; et al. Mapping of X-linked myxomatous valvular dystrophy to chromosome Xq28. Am. J. Hum. Genet. 1998, 62, 627–632. [Google Scholar] [CrossRef] [Green Version]
- Le Tourneau, T.; Mérot, J.; Rimbert, A.; Le Scouarnec, S.; Probst, V.; Le Marec, H.; Levine, R.A.; Schott, J.-J. Genetics of syndromic and non-syndromic mitral valve prolapse. Heart 2018, 104, 978–984. [Google Scholar] [CrossRef]
- Lang, R.M.; Badano, L.P.; Mor-Avi, V.; Afilalo, J.; Armstrong, A.; Ernande, L.; Flachskampf, F.A.; Foster, E.; Goldstein, S.A.; Kuznetsova, T.; et al. Recommendations for cardiac chamber quantification by echocardiography in adults: An update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J. Am. Soc. Echocardiogr. 2015, 28, 1–39.e14. [Google Scholar] [CrossRef]
- Lancellotti, P.; Moura, L.; Pierard, L.A.; Agricola, E.; Popescu, B.A.; Tribouilloy, C.; Hagendorff, A.; Monin, J.-L.; Badano, L.; Zamorano, J.L.; et al. European Association of Echocardiography recommendations for the assessment of valvular regurgitation. Part 2: Mitral and tricuspid regurgitation (native valve disease). Eur. J. Echocardiogr. 2010, 11, 307–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizotte, E.; Junttila, M.J.; Dube, M.P.; Hong, K.; Benito, B.; DE Zutter, M.; Henkens, S.; Sarkozy, A.; Huikuri, H.V.; Towbin, J.; et al. Genetic modulation of brugada syndrome by a common polymorphism. J. Cardiovasc. Electrophysiol. 2009, 20, 1137–1141. [Google Scholar] [CrossRef] [PubMed]
- Poelzing, S.; Forleo, C.; Samodell, M.; Dudash, L.; Sorrentino, S.; Anaclerio, M.; Troccoli, R.; Iacoviello, M.; Romito, R.; Guida, P.; et al. SCN5A polymorphism restores trafficking of a Brugada syndrome mutation on a separate gene. Circulation 2006, 114, 368–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tester, D.J.; Valdivia, C.; Harris-Kerr, C.; Alders, M.; Salisbury, B.A.; Wilde, A.A.M.; Makielski, J.C.; Ackerman, M.J. Epidemiologic, molecular, and functional evidence suggest A572D-SCN5A should not be considered an independent LQT3-susceptibility mutation. Heart Rhythm 2010, 7, 912–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, J.; Wang, T.; Trump, D.; Zimmer, T.; Lei, M. Mutation-specific effects of polymorphism H558R in SCN5A-related sick sinus syndrome. J. Cardiovasc. Electrophysiol. 2010, 21, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yin, L.; Shen, C.; Hu, K.; Ge, J.; Sun, A. SCN5A Variants: Association with Cardiac Disorders. Front. Physiol. 2018, 9, 1372. [Google Scholar] [CrossRef]
- Qureshi, S.F.; Ali, A.; John, P.; Jadhav, A.P.; Venkateshwari, A.; Rao, H.; Jayakrishnan, M.P.; Narasimhan, C.; Shenthar, J.; Thangaraj, K.; et al. Mutational analysis of SCN5A gene in long QT syndrome. Meta Gene 2015, 6, 26–35. [Google Scholar] [CrossRef]
- Mazzaccara, C.; Limongelli, G.; Petretta, M.; Vastarella, R.; Pacileo, G.; Bonaduce, D.; Salvatore, F.; Frisso, G. A common polymorphism in the SCN5A gene is associated with dilated cardiomyopathy. J. Cardiovasc. Med. 2018, 19, 344–350. [Google Scholar] [CrossRef]
- Balla, C.; Mele, D.; Vitali, F.; Andreoli, C.; Tonet, E.; Sanchini, M.; Ferlini, A.; Rapezzi, C.; Gualandi, F.; Bertini, M. Novel SCN5A Variant Shows Multiple Phenotypic Expression in the Same Family. Circ. Genom. Precis. Med. 2021, 14, e003481. [Google Scholar] [CrossRef]
- Missov, E.; Cogswell, R. Sudden cardiac death, mitral valve prolapse, and long QT syndrome. Am. J. Med. 2015, 128, e37–e38. [Google Scholar] [CrossRef]
- Mahajan, A.M.; Itan, Y.; Cerrone, M.; Horowitz, J.; Borneman, L.; Chinitz, L.; Jankelson, L. Sudden Cardiac Arrest in a Patient With Mitral Valve Prolapse and LMNA and SCN5A Mutations. JACC Case Rep. 2021, 3, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Wilde, A.A.M.; Amin, A.S. Clinical Spectrum of SCN5A Mutations: Long QT Syndrome, Brugada Syndrome, and Cardiomyopathy. JACC Clin. Electrophysiol. 2018, 4, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Albert, C.M.; Nam, E.G.; Rimm, E.B.; Jin, H.W.; Hajjar, R.J.; Hunter, D.J.; MacRae, C.A.; Ellinor, P.T. Cardiac sodium channel gene variants and sudden cardiac death in women. Circulation 2008, 117, 16–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spartalis, M.; Tzatzaki, E.; Spartalis, E.; Athanasiou, A.; Moris, D.; Damaskos, C.; Garmpis, N.; Voudris, V. Mitral valve prolapse: An underestimated cause of sudden cardiac death—A current review of the literature. J. Thorac. Dis. 2017, 9, 5390–5398. [Google Scholar] [CrossRef] [Green Version]
- Noseworthy, P.A.; Asirvatham, S.J. The Knot That Binds Mitral Valve Prolapse and Sudden Cardiac Death. Circulation 2015, 132, 551–552. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, K.; Uy-Evanado, A.; Teodorescu, C.; Reinier, K.; Nichols, G.A.; Gunson, K.; Jui, J.; Chugh, S.S. Mitral valve prolapse and sudden cardiac arrest in the community. Heart Rhythm 2016, 13, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Turker, Y.; Ozaydin, M.; Acar, G.; Ozgul, M.; Hoscan, Y.; Varol, E.; Dogan, A.; Erdogan, D.; Yucel, H. Predictors of ventricular arrhythmias in patients with mitral valve prolapse. Int. J. Cardiovasc. Imaging 2010, 26, 139–145. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jaouadi, H.; Théron, A.; Hourdain, J.; Martel, H.; Nguyen, K.; Habachi, R.; Deharo, J.-C.; Collart, F.; Avierinos, J.-F.; Zaffran, S. SCN5A Variants as Genetic Arrhythmias Triggers for Familial Bileaflet Mitral Valve Prolapse. Int. J. Mol. Sci. 2022, 23, 14447. https://doi.org/10.3390/ijms232214447
Jaouadi H, Théron A, Hourdain J, Martel H, Nguyen K, Habachi R, Deharo J-C, Collart F, Avierinos J-F, Zaffran S. SCN5A Variants as Genetic Arrhythmias Triggers for Familial Bileaflet Mitral Valve Prolapse. International Journal of Molecular Sciences. 2022; 23(22):14447. https://doi.org/10.3390/ijms232214447
Chicago/Turabian StyleJaouadi, Hager, Alexis Théron, Jérôme Hourdain, Hélène Martel, Karine Nguyen, Raja Habachi, Jean-Claude Deharo, Frédéric Collart, Jean-François Avierinos, and Stéphane Zaffran. 2022. "SCN5A Variants as Genetic Arrhythmias Triggers for Familial Bileaflet Mitral Valve Prolapse" International Journal of Molecular Sciences 23, no. 22: 14447. https://doi.org/10.3390/ijms232214447