
Transcriptomic and Metabolomic Analyses Reveal That Fullerol Improves Drought Tolerance in Brassica napus L
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
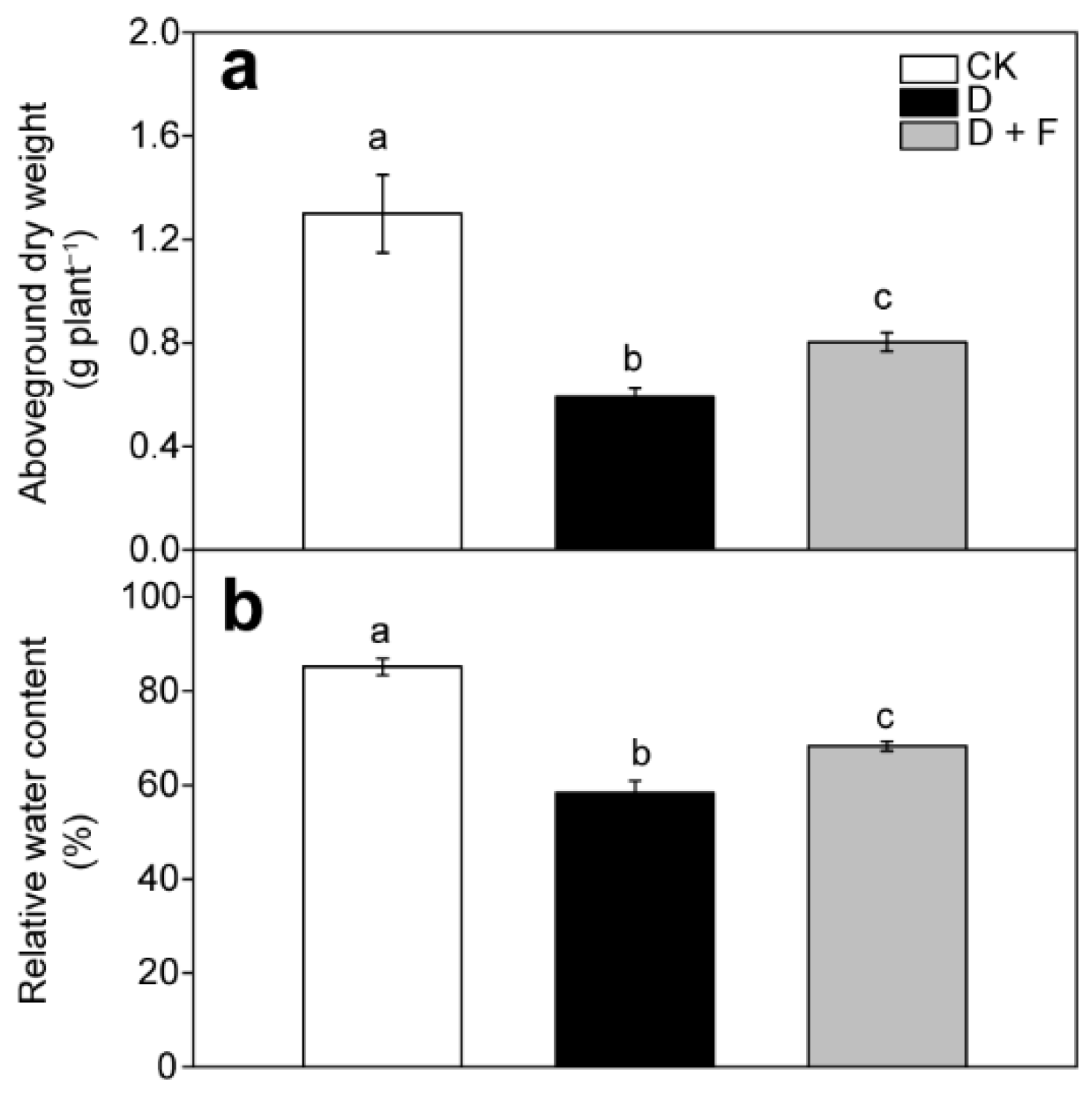
2.1. Aboveground Biomass and Leaf Relative Water Content
2.2. Transcriptomic Analysis
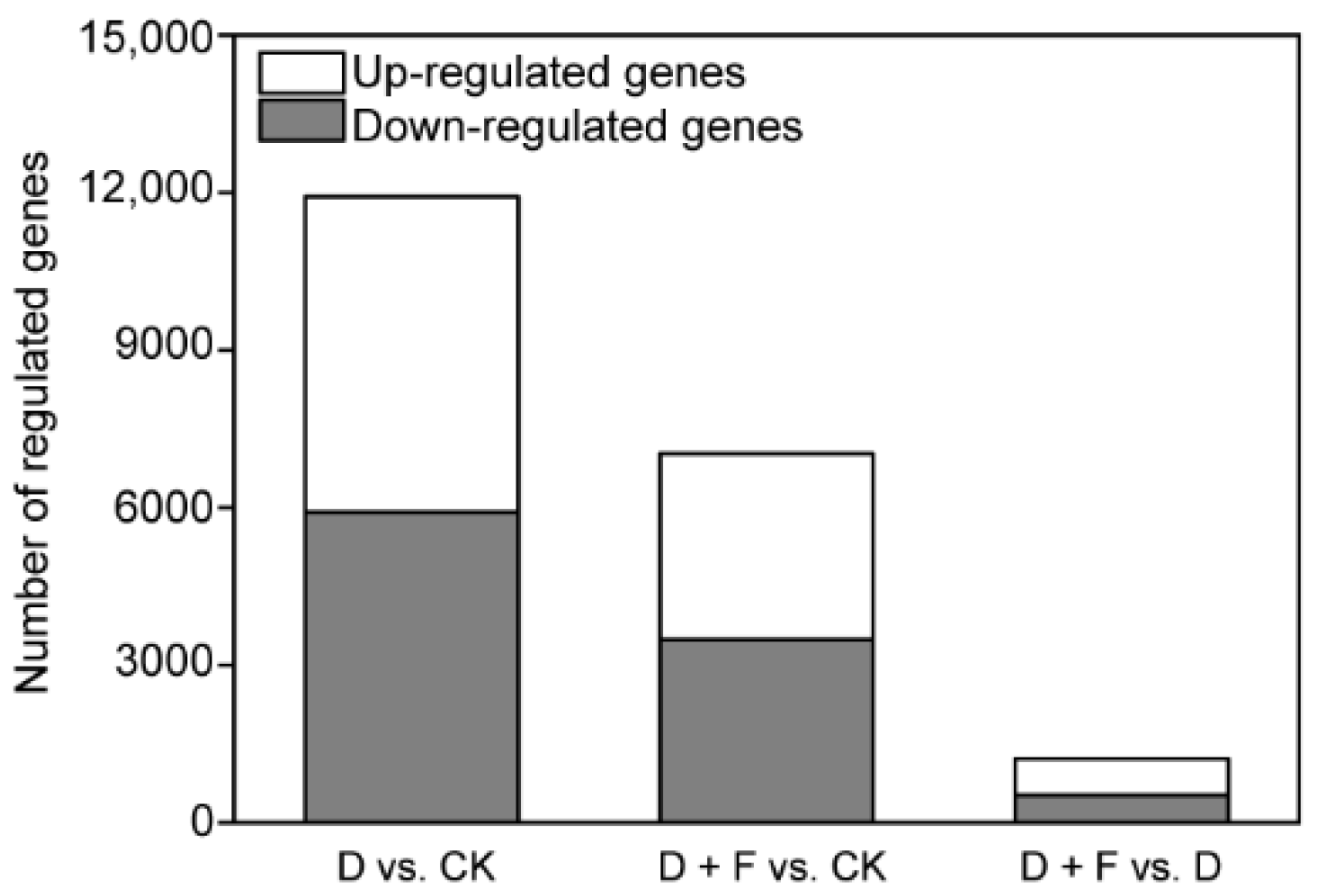
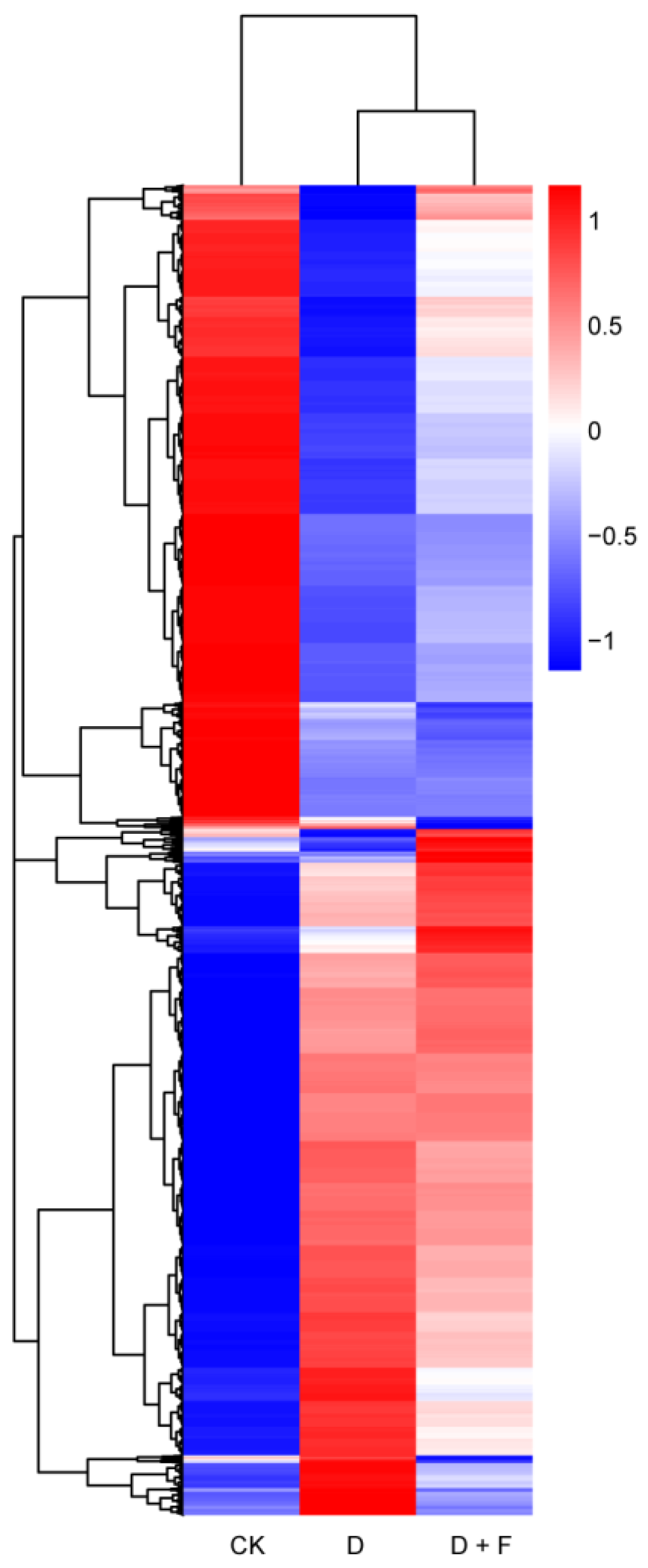
2.2.1. Analysis of Differentially Expressed Genes (DEGs)
2.2.2. Functional Analysis by Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)
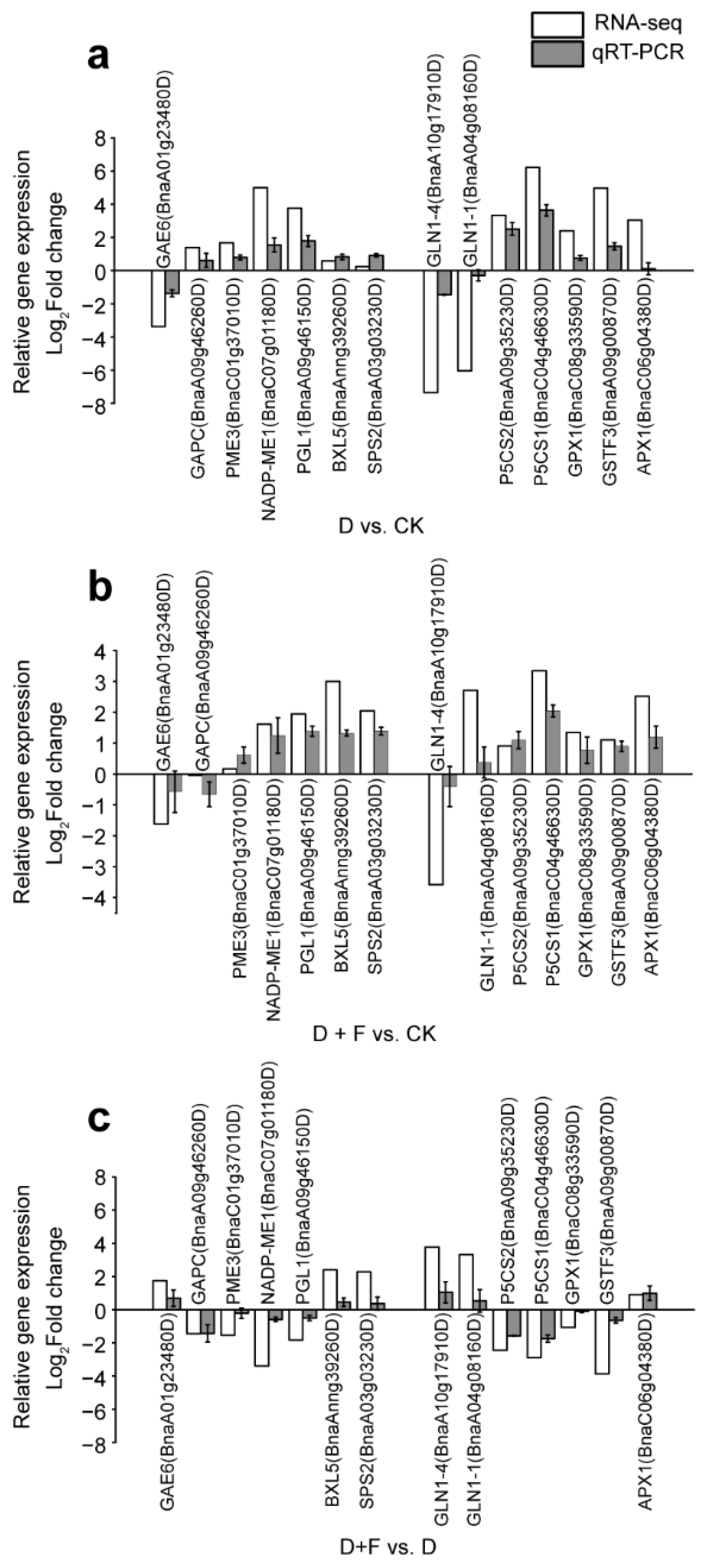
2.2.3. Quantitative Real-Time (qRT)-PCR
2.3. Metabolic Analysis
2.4. Data Integration/Comprehensive Networks of Transcripts and Metabolites
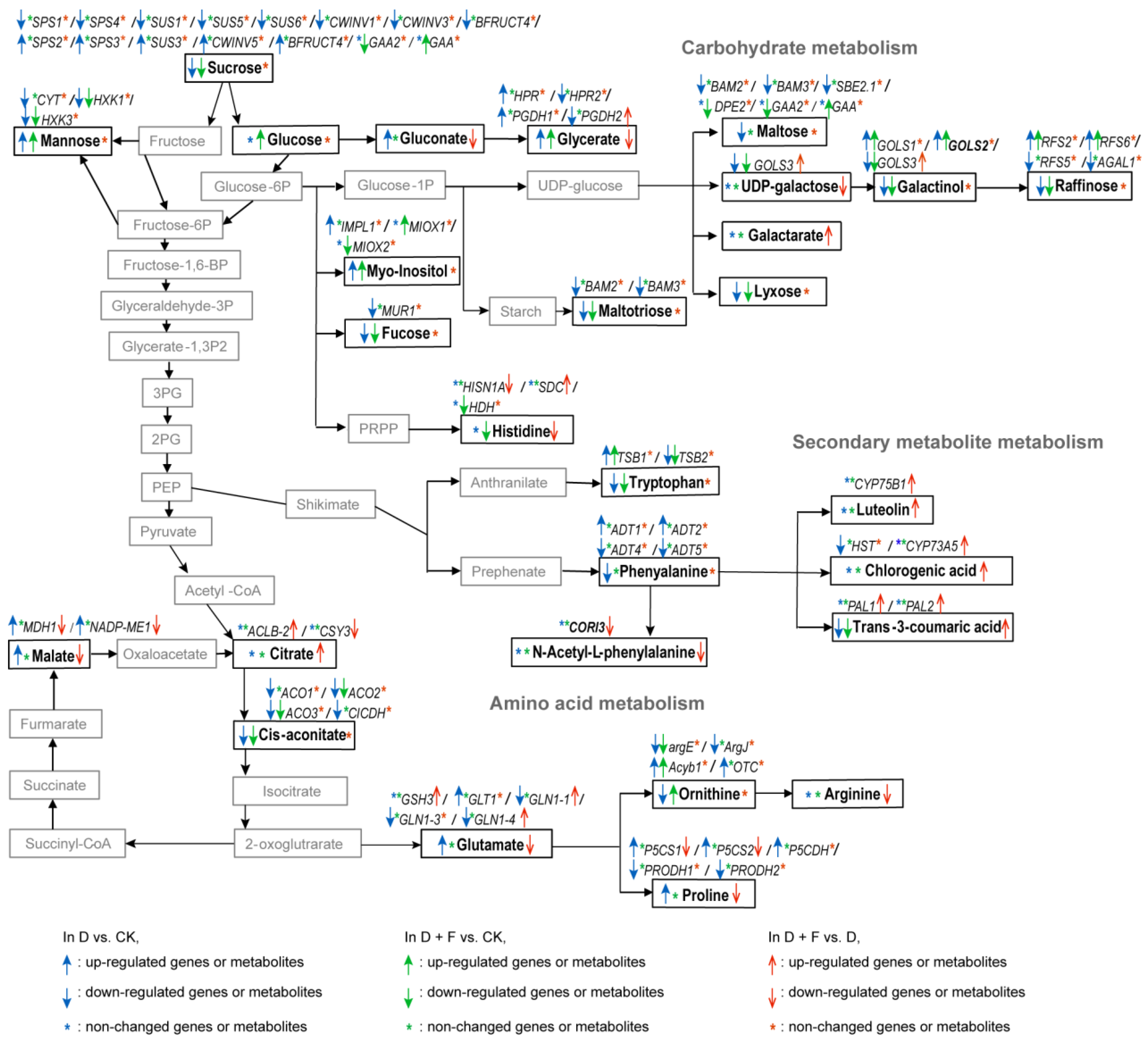
2.4.1. Carbohydrate Metabolism
2.4.2. Amino Acid Metabolism
2.4.3. Secondary Metabolite Metabolism
3. Discussion
3.1. Carbohydrate Metabolism
3.2. Amino Acid Metabolism
3.3. Secondary Metabolite Metabolism
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. RNA-Seq Analysis
4.2.1. RNA Extraction and Quantification
4.2.2. Data Processing
4.2.3. qRT-PCR Analysis
4.3. Metabolomic Analysis
4.3.1. Metabolite Extraction and LC-MS Conditions
4.3.2. Multivariate Data Processing
4.4. Statistical Analyses of Other Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zaytseva, O.; Neumann, G. Carbon nanomaterials: Production, impact on plant development, agricultural and environmental applications. Chem. Biol. Technol. Agric. 2016, 3, 17. [Google Scholar] [CrossRef] [Green Version]
- Duhan, J.S.; Kumar, R.; Kumar, N.; Kaur, P.; Nehra, K.; Duhan, S. Nanotechnology: The new perspective in precision agriculture. Biotechnol. Rep. 2017, 15, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Hatami, M.; Hadian, J.; Ghorbanpour, M. Mechanisms underlying toxicity and stimulatory role of single-walled carbon nanotubes in Hyoscyamus niger during drought stress simulated by polyethylene glycol. J. Hazard. Mater. 2017, 324, 306–320. [Google Scholar] [CrossRef]
- Impa, S.M.; Nadaradjan, S.; Jagadish, S.V.K. Drought stress induced reactive oxygen species and anti-oxidants in plants. In Abiotic Stress Responses in Plants; Ahmad, P., Prasad, M.N.V., Eds.; Springer: New York, NY, USA, 2011; pp. 131–147. [Google Scholar]
- Husen, A.; Siddiqi, K.S. Carbon and fullerene nanomaterials in plant system. J. Nanobiotechnol. 2014, 12, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panova, G.G.; Ktitorova, I.N.; Skobeleva, O.V.; Sinjavina, N.G.; Charykov, N.A.; Semenov, K.N. Impact of polyhydroxy fullerene (fullerol or fullerenol) on growth and biophysical characteristics of barley seedlings in favourable and stressful conditions. Plant Growth Regul. 2016, 79, 309–317. [Google Scholar] [CrossRef]
- Khan, M.N.; Mobin, M.; Abbas, Z.K.; AlMutairi, K.A.; Siddiqui, Z.H. Role of nanomaterials in plants under challenging environments. Plant Physiol. Biochem. 2017, 110, 194–209. [Google Scholar] [CrossRef]
- Borišev, M.; Borišev, I.; Župunski, M.; Arsenov, D.; Pajević, S.; Ćurčić, Ž.; Vasin, J.; Djordjevic, A. Drought impact is alleviated in sugar beets (Beta vulgaris L.) by foliar application of fullerenol nanoparticles. PLoS ONE 2016, 11, e0166248. [Google Scholar] [CrossRef]
- Xiong, J.L.; Li, J.; Wang, H.C.; Zhang, C.L.; Naeem, M.S. Fullerol improves seed germination, biomass accumulation, photosynthesis and antioxidant system in Brassica napus L. under water stress. Plant Physiol. Biochem. 2018, 129, 130–140. [Google Scholar] [CrossRef]
- Wan, J.; Griffiths, R.; Ying, J.; Mccourt, P.; Huang, Y. Development of drought-tolerant canola (Brassica napus L.) through genetic modulation of ABA-mediated stomatal responses. Crop Sci. 2009, 49, 1539–1554. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies–the next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Mibei, E.K.; Owino, W.O.; Ambuko, J.; Giovannoni, J.J.; Onyango, A.N. Metabolomic analyses to evaluate the effect of drought stress on selected African Eggplant accessions. J. Sci. Food Agric. 2018, 98, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Zhu, X.; Wang, Y.; Chen, L.; Duan, Y. Transcriptomic and metabolomic analyses reveal that bacteria promote plant defense during infection of soybean cyst nematode in soybean. BMC Plant Biol. 2018, 18, 86. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Song, S.; Wang, X.; Liu, J.; Dong, S. Transcriptomic and metabolomic analysis of seedling-stage soybean responses to PEG-simulated drought stress. Int. J. Mol. Sci. 2022, 23, 6869. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Huang, L.; Sun, X.; Zhao, L.; Wang, P. Transcriptomic and metabolomic analyses reveal key metabolites, pathways and candidate genes in Sophora davidii (Franch.) Skeels seedlings under drought stress. Front. Plant Sci. 2022, 13, 785702. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Liu, J.; Zhou, Y.; Zhou, S.; Zhang, S.; Tong, H.; Zhao, A. Transcriptome and metabolome profiling unveiled mechanisms of tea (Camellia sinensis) quality improvement by moderate drought on pre-harvest shoots. Phytochemistry 2020, 180, 112515. [Google Scholar] [CrossRef]
- Xiong, J.L.; Dai, L.L.; Ma, N.; Zhang, C.L. Transcriptome and physiological analyses reveal that AM1 as an ABA-mimicking ligand improves drought resistance in Brassica napus. Plant Growth Regul. 2018, 85, 73–90. [Google Scholar] [CrossRef]
- Vital, C.E.; Giordano, A.; de Almeida Soares, E.; Rhys Williams, T.C.; Mesquita, R.O.; Vidigal, P.M.P.; de Santana Lopes, A.; Pacheco, T.G.; Rogalski, M.; de Oliveria Ramos, H.J.; et al. An integrative overview of the molecular and physiological responses of sugarcane under drought conditions. Plant Mol. Biol. 2017, 94, 577–594. [Google Scholar] [CrossRef]
- Rahman, M.A.; Akond, M.; Babar, M.A.; Beecher, C.; Erickson, J.; Thomason, K.; De Jong, F.A.; Mason, R.E. LC-HRMS based non-targeted metabolomic profiling of wheat (Triticum aestivum L.) under post-anthesis drought stress. Am. J. Plant Sci. 2017, 8, 3024–3061. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Jensen, C.R.; Andersen, M.N. Drought stress effect on carbohydrate concentration in soybean leaves and pods during early reproductive development: Its implication in altering pod set. Field Crop Res. 2004, 86, 1–13. [Google Scholar] [CrossRef]
- Mutwakil, M.Z.; Hajrah, N.H.; Atef, A.; Edris, S.; Sabir, M.J.; Al-Ghamdi, A.K.; Sabir, M.J.S.M.; Nelson, C.; Makki, R.M.; Ali, H.M.; et al. Transcriptomic and metabolic responses of Calotropis procera to salt and drought stress. BMC Plant Biol. 2017, 17, 231. [Google Scholar] [CrossRef]
- Rodríguez-Calcerrada, J.; Rodrigues, A.M.; António, C.; Perdiguero, P.; Pita, P.; Collada, C.; Li, M.; Gil, L. Stem metabolism under drought stress–a paradox of increasing respiratory substrates and decreasing respiratory rates. Physiol. Plant. 2021, 172, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Calcerrada, J.; Rodrigues, A.M.; Perdiguero, P.; António, C.; Atkin, O.K.; Li, M.; Collada, C.; Gil, L. A molecular approach to drought-induced reduction in leaf CO2 exchange in drought-resistant Quercus ilex. Physiol. Plant. 2018, 162, 394–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, J.C.; Sheen, J. Sugar sensing in higher plants. Plant Cell 1994, 6, 1665–1679. [Google Scholar] [PubMed] [Green Version]
- Ohto, M.A.; Onai, K.; Furukawa, Y.; Aoki, E.; Araki, T.; Nakamura, K. Effects of sugar on vegetative development and floral transition in Arabidopsis. Plant Physiol. 2001, 127, 252–261. [Google Scholar] [CrossRef] [Green Version]
- Conklin, P.L.; Norris, S.R.; Wheeler, G.L.; Williams, E.H.; Smirnoff, N.; Last, R.L. Genetic evidence for the role of GDP-mannose in plant ascorbic acid (vitamin C) biosynthesis. Proc. Natl. Acad. Sci. USA 1999, 96, 4198–4203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.; Zhou, K.; Li, Y.; Chen, X.; Liu, B.; Li, C.; Gong, X.; Ma, F. Exogenous myo-inositol alleviates salinity-induced stress in Malus hupehensis Rehd. Plant Physiol. Biochem. 2018, 133, 116–126. [Google Scholar] [CrossRef]
- Hatzig, S.; Zaharia, L.I.; Abrams, S.; Hohmann, M.; Legoahec, L.; Bouchereau, A.; Nesi, N.; Snowdon, R.J. Early osmotic adjustment responses in drought-resistant and drought-sensitive oilseed rape. J. Integr. Plant Biol. 2014, 56, 797–809. [Google Scholar] [CrossRef]
- Verbruggen, N.; Hermans, C. Proline accumulation in plants: A review. Amino Acids 2008, 35, 753–759. [Google Scholar] [CrossRef]
- Ben Rejeb, K.; Abdelly, C.; Savouré, A. How reactive oxygen species and proline face stress together. Plant Physiol. Biochem. 2014, 80, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Fàbregas, N.; Fernie, A.R. The metabolic response to drought. J. Exp. Bot. 2019, 70, 1077–1085. [Google Scholar] [CrossRef]
- Tinte, M.M.; Masike, K.; Steenkamp, P.A.; Huyser, J.; van der Hooft, J.J.; Tugizimana, F. Computational metabolomics tools reveal metabolic reconfigurations underlying the effects of biostimulant seaweed extracts on maize plants under drought stress conditions. Metabolites 2022, 12, 487. [Google Scholar] [CrossRef]
- Khan, N.; Bano, A.; Rahman, M.A.; Rathinasabapathi, B.; Babar, M.A. UPLC-HRMS-based untargeted metabolic profiling reveals changes in chickpea (Cicer arietinum) metabolome following long-term drought stress. Plant Cell Environ. 2019, 42, 115–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Wang, P.; Jaini, R.; Dudareva, N.; Chapple, C.; Morgan, J.A. Dynamic modeling of subcellular phenylpropanoid metabolism in Arabidopsis lignifying cells. Metab. Eng. 2018, 49, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Parida, A.K. Metabolomics and network analysis reveal the potential metabolites and biological pathways involved in salinity tolerance of the Halophyte Salvadora Persica. Environ. Exp. Bot. 2018, 148, 85–99. [Google Scholar] [CrossRef]
- Ghorbanpour, M.; Mohammadi, H.; Kariman, K. Nanosilicon-based recovery of barley (Hordeum vulgare) plants subjected to drought stress. Environ. Sci.-Nano. 2020, 7, 443–461. [Google Scholar] [CrossRef]
- Liu, W.C.; Song, R.F.; Zheng, S.Q.; Li, T.T.; Zhang, B.L.; Gao, X.; Lu, Y.T. Coordination of plant growth and abiotic stress responses by tryptophan synthase β subunit 1 through modulation of tryptophan and ABA homeostasis in Arabidopsis. Mol. Plant 2022, 15, 973–990. [Google Scholar] [CrossRef]
- Caillet, S.; Salmiéri, S.; Lacroix, M. Evaluation of free radical-scavenging properties of commercial grape phenol extracts by a fast colorimetric method. Food Chem. 2006, 95, 1–8. [Google Scholar] [CrossRef]
- Weidner, S.; Karamać, M.; Amarowicz, R.; Szypulska, E.; Gołgowska, A. Changes in composition of phenolic compounds and antioxidant properties of Vitis amurensis seeds germinated under osmotic stress. Acta Physiol. Plant. 2007, 29, 283–290. [Google Scholar] [CrossRef]
- Nakabayashi, R.; Mori, T.; Saito, K. Alternation of flavonoid accumulation under drought stress in Arabidopsis thaliana. Plant Signal. Behav. 2014, 9, e29518. [Google Scholar] [CrossRef] [Green Version]
- Yobi, A.; Wone, B.W.; Xu, W.; Alexander, D.C.; Guo, L.; Ryals, J.A.; Oliver, M.J.; Cushman, J.C. Comparative metabolic profiling between desiccation-sensitive and desiccation-tolerant species of Selaginella reveals insights into the resurrection trait. Plant J. 2012, 72, 983–999. [Google Scholar] [CrossRef]
- Wang, W.; Xin, H.; Wang, M.; Ma, Q.; Wang, L.; Kaleri, N.A.; Wang, Y.; Li, X. Transcriptomic analysis reveals the molecular mechanisms of drought-stress-induced decreases in Camellia sinensis leaf quality. Front. Plant Sci. 2016, 7, 385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández, I.; Alegre, L.; Munné-Bosch, S. Drought-induced changes in flavonoids and other low molecular weight antioxidants in Cistus clusii grown under Mediterranean field conditions. Tree Physiol. 2004, 24, 1303–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.L.; Yang, L.; Yang, X.; Zhang, T.; Lan, Y.M.; Zhao, Y.; Han, M.; Yang, L.M. Drought stress induces biosynthesis of flavonoids in leaves and saikosaponins in roots of Bupleurum chinense DC. Phytochemistry 2020, 177, 112434. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Takeuchi, A.; Ozawa, M.; Li, X.H.; Saigo, K.; Kitazawa, K. C60 fullerol formation catalyzed by quatemary ammonium hydroxides. J. Chem. Soc. Chem. Comm. 1993, 23, 460–469. [Google Scholar]
- Cheng, F.; Liu, S.; Wu, J.; Fang, L.; Sun, S.; Liu, B.; Li, P.; Hua, W.; Wang, X. BRAD, the genetics and genomics database for Brassica plants. BMC Plant Biol. 2011, 11, 136. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Zhu, L.P.; Pan, C.; Xu, L.M.; Ke, W.D.; Yang, P.F. Transcriptomic analysis of the regulation of rhizome formation in temperate and tropical lotus (Nelumbo nucifera). Sci. Rep. 2015, 5, 13059. [Google Scholar] [CrossRef] [Green Version]
- D’Oria, A.; Jing, L.; Arkoun, M.; Pluchon, S.; Pateyron, S.; Trouverie, J.; Etienne, P.; Diquélou, S.; Ourry, A. Transcriptomic, metabolomic and ionomic analyses reveal early modulation of leaf mineral content in Brassica napus under mild or severe drought. Int. J. Mol. Sci. 2022, 23, 781. [Google Scholar] [CrossRef]
- Bylesjo, M.; Rantalainen, M.; Cloarec, O.; Nicholson, J.K.; Holmes, E.; Trygg, J. OPLS discriminant analysis: Combining the strengths of PLS-DA and SIMCA classification. J. Chemometr. 2006, 20, 341–351. [Google Scholar] [CrossRef]
- Dill, A.L.; Eberlin, L.S.; Zheng, C.; Costa, A.B.; Ifa, D.R.; Cheng, L.; Masterson, T.A.; Koch, M.O.; Vitek, O.; Cooks, R.G. Multivariate statistical differentiation of renal cell carcinomas based on lipidomic analysis by ambient ionization imaging mass spectrometry. Anal. Bioanal. Chem. 2010, 398, 2969–2978. [Google Scholar] [CrossRef]
- Wu, X.; Zhao, L.L.; Peng, H.B.; She, Y.Q.; Feng, Y.F. Search for potential biomarkers by UPLC/Q-TOF–MS analysis of dynamic changes of glycerophospholipid constituents of RAW264.7 cells treated with NSAID. Chromatographia 2015, 78, 211–220. [Google Scholar] [CrossRef]
- Ratnasekhar, C.; Sonane, M.; Satish, A.; Krishna, M.; Mudiam, K.R. Metabolomics reveals the perturbations in the metabolome of Caenorhabditis elegans exposed to titanium dioxide nanoparticles. Nanotoxicology 2015, 9, 994–1004. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, J.-L.; Ma, N. Transcriptomic and Metabolomic Analyses Reveal That Fullerol Improves Drought Tolerance in Brassica napus L. Int. J. Mol. Sci. 2022, 23, 15304. https://doi.org/10.3390/ijms232315304
Xiong J-L, Ma N. Transcriptomic and Metabolomic Analyses Reveal That Fullerol Improves Drought Tolerance in Brassica napus L. International Journal of Molecular Sciences. 2022; 23(23):15304. https://doi.org/10.3390/ijms232315304
Chicago/Turabian StyleXiong, Jun-Lan, and Ni Ma. 2022. "Transcriptomic and Metabolomic Analyses Reveal That Fullerol Improves Drought Tolerance in Brassica napus L" International Journal of Molecular Sciences 23, no. 23: 15304. https://doi.org/10.3390/ijms232315304