
Occurrence and Variability of the Efflux Pump Gene norA across the Staphylococcus Genus †



Abstract
:1. Introduction
2. Results and Discussion
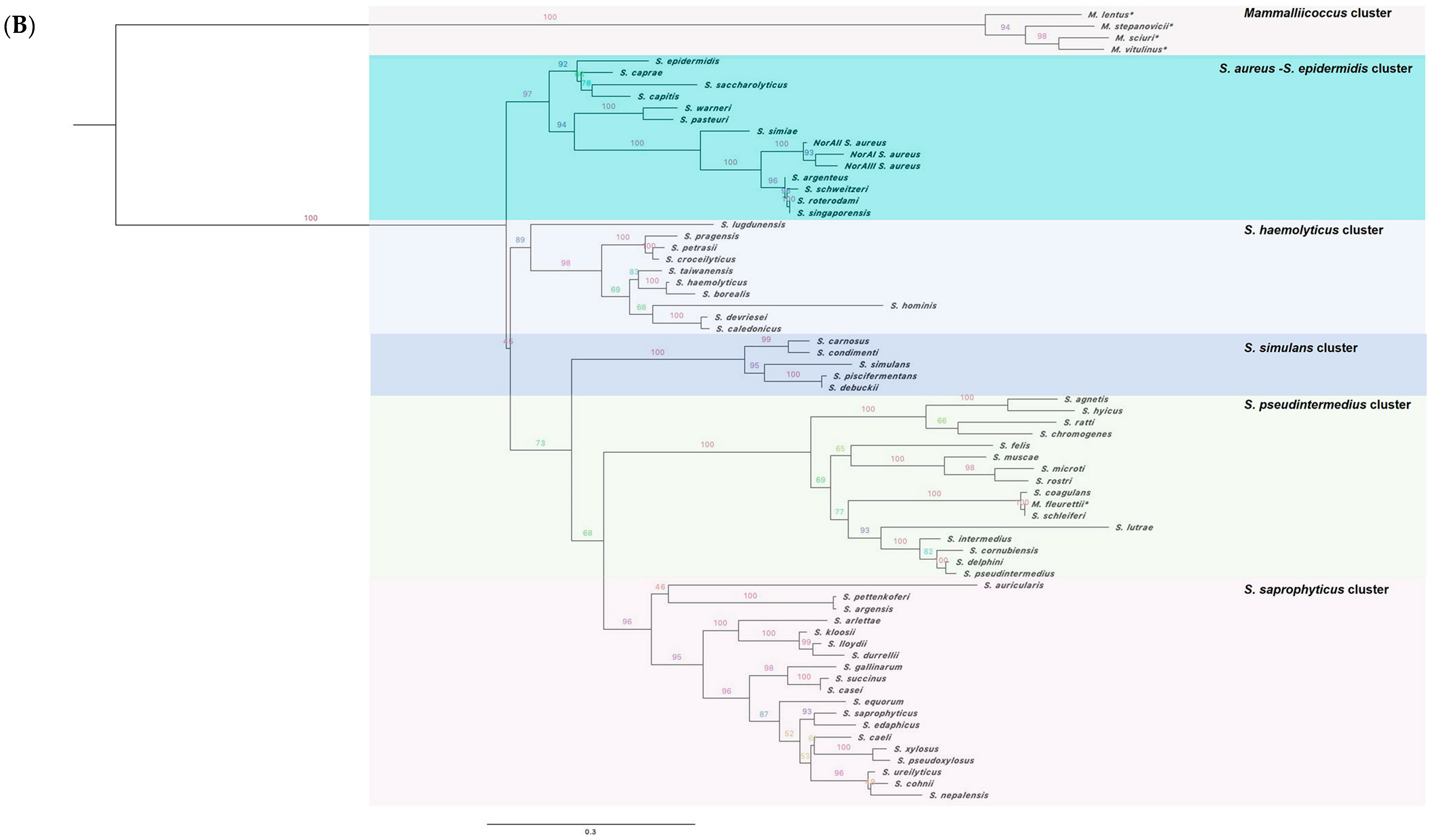
2.1. The norA Gene Is Ubiquitous across the Staphylococcus Genus
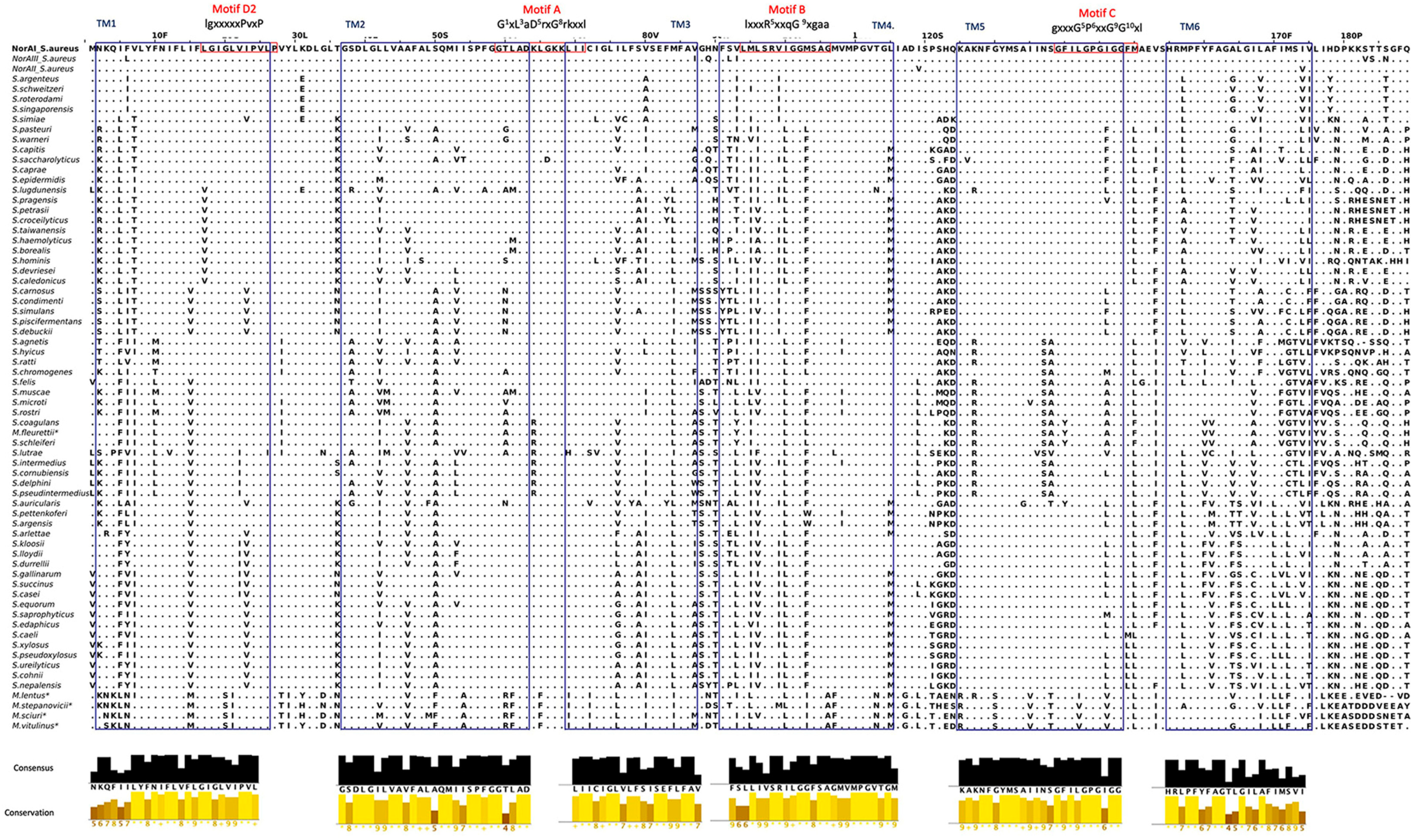
2.2. Characterization of the NorA Predicted Polypeptide Sequences across Staphylococci


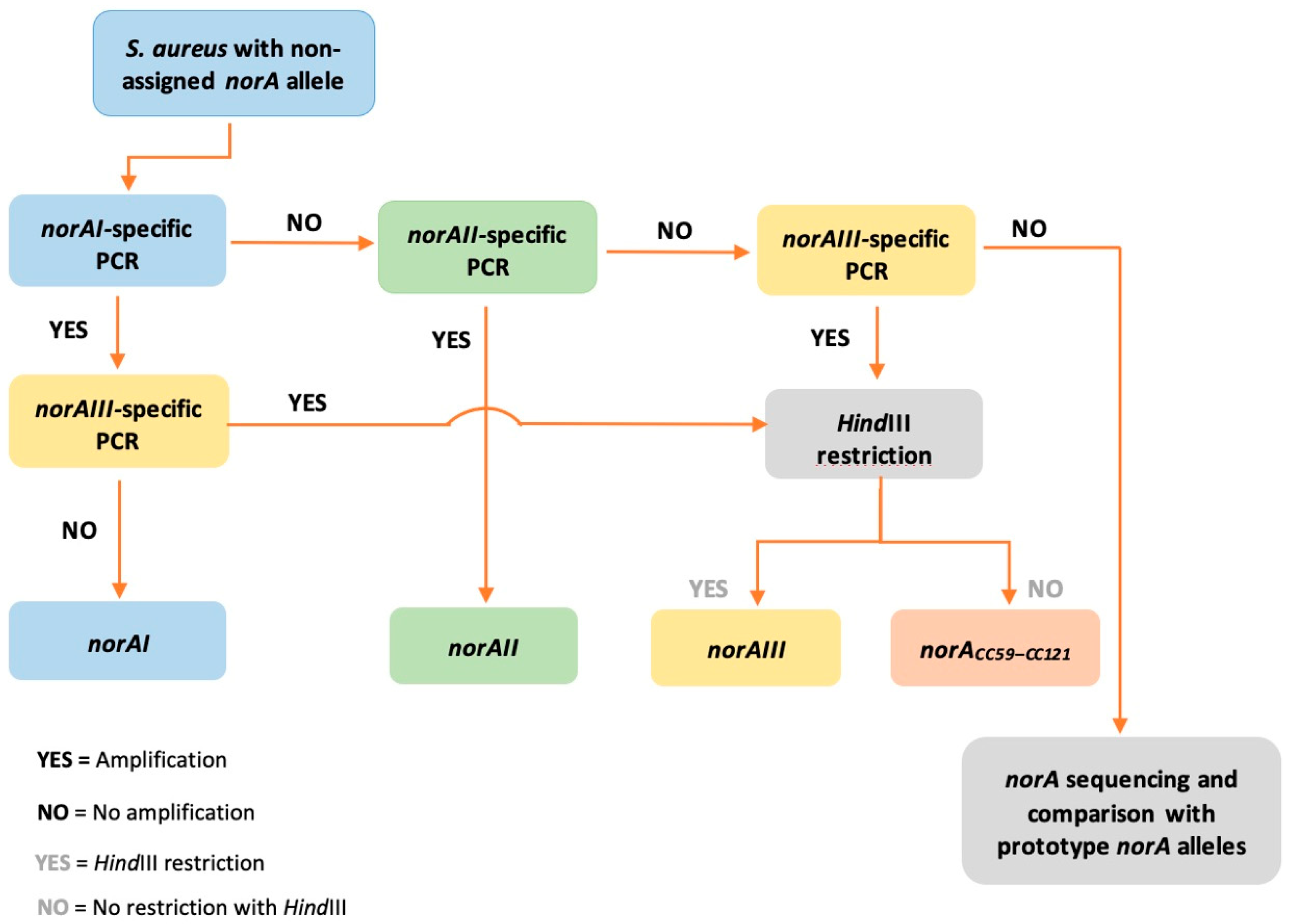
2.3. Applicability of a Molecular Approach for Rapid norA Allele Screening in S. aureus
2.4. Prevalence of S. aureus norA Alleles and Their Association with Specific Clonal Lineages
2.5. Implications for Future Work
3. Materials and Methods
3.1. Screening of norA Alleles among Contemporary S. aureus Strains
3.1.1. Bacterial Strains
3.1.2. Screening of norA Alleles
3.1.3. norA Sequencing
3.1.4. Relation between norA and S. aureus Clonal Lineages
3.2. Survey of the Gene Coding for the NorA Efflux Pump among the Staphylococcus Genus
3.3. Characterization of NorA Polypeptide Sequences across Staphylococci
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ubukata, K.; Itoh-Yamashita, N.; Konno, M. Cloning and expression of the norA gene for fluoroquinolone resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 1989, 33, 1535–1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, H.; Bogaki, M.; Nakamura, S.; Ubukata, K.; Konno, M. Nucleotide sequence and characterization of the Staphylococcus aureus norA gene, which confers resistance to quinolones. J. Bacteriol. 1990, 172, 6942–6949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, K. Multidrug resistance pumps in bacteria: Variations on a theme. Trends Biochem. Sci. 1994, 19, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Neyfakh, A.A.; Borsch, C.M.; Kaatz, G.W. Fluoroquinolone resistance protein NorA of Staphylococcus aureus is a multidrug efflux transporter. Antimicrob. Agents Chemother. 1993, 37, 128–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, X.; Ji, Q.; Liang, H.; Missiakas, D.; Lan, L.; He, C. Expression of multidrug resistance efflux pump gene norA is iron responsive in Staphylococcus aureus. J. Bacteriol. 2012, 194, 1753–1762. [Google Scholar] [CrossRef] [Green Version]
- Marchi, E.; Furi, L.; Arioli, S.; Morrissey, I.; Di Lorenzo, V.; Mora, D.; Giovannetti, L.; Oggioni, M.R.; Viti, C. Novel insight into antimicrobial resistance and sensitivity phenotypes associated to qac and norA genotypes in Staphylococcus aureus. Microbiol. Res. 2015, 170, 184–194. [Google Scholar] [CrossRef]
- DeMarco, C.E.; Cushing, L.A.; Frempong-Manso, E.; Seo, S.M.; Jaravaza, T.A.A.; Kaatz, G.W. Efflux-related resistance to norfloxacin, dyes, and biocides in bloodstream isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 2007, 51, 3235–3239. [Google Scholar] [CrossRef] [Green Version]
- Huet, A.A.; Raygada, J.L.; Mendiratta, K.; Seo, S.M.; Kaatz, G.W. Multidrug efflux pump overexpression in Staphylococcus aureus after single and multiple in vitro exposures to biocides and dyes. Microbiology 2008, 154, 3144–3153. [Google Scholar] [CrossRef] [Green Version]
- Kosmidis, C.; DeMarco, C.E.; Frempong-Manso, E.; Seo, S.M.; Kaatz, G.W. In silico genetic correlations of multidrug efflux pump gene expression in Staphylococcus aureus. Int. J. Antimicrob. Agents 2010, 36, 222–229. [Google Scholar] [CrossRef]
- Costa, S.S.; Falcão, C.; Viveiros, M.; Machado, D.; Martins, M.; Melo-Cristino, J.; Amaral, L.; Couto, I. Exploring the contribution of efflux on the resistance to fluoroquinolones in clinical isolates of Staphylococcus aureus. BMC Microbiol. 2011, 11, e241. [Google Scholar] [CrossRef]
- Furi, L.; Ciusa, M.L.; Knight, S.; Di Lorenzo, V.; Tocci, N.; Cirasola, D.; Aragones, L.; Coelho, J.R.; Freitas, A.T.; Marchi, E.; et al. Evaluation of reduced susceptibility to quaternary ammonium compounds and bisbiguanidines in clinical isolates and laboratory-generated mutants of Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 3488–3497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, S.S.; Viveiros, M.; Rosato, A.E.; Melo-Cristino, J.; Couto, I. Impact of efflux in the development of multidrug resistance phenotypes in Staphylococcus aureus. BMC Microbiol. 2015, 15, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papkou, A.; Hedge, J.; Kapel, N.; Young, B.; MacLean, R.C. Efflux pump activity potentiates the evolution of antibiotic resistance across S. aureus isolates. Nat. Commun. 2020, 11, 3970. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.S.; Viveiros, M.; Pomba, C.; Couto, I. Active antimicrobial efflux in Staphylococcus epidermidis: Building up of resistance to fluoroquinolones and biocides in a major opportunistic pathogen. J. Antimicrob. Chemother. 2018, 73, 320–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marco, L.; Liliana, G.; Anna, B.; Annarita, M. Intrinsic role of coagulase negative staphylococci norA-like efflux system in fluoroquinolones resistance. AIMS Microbiol. 2017, 3, 908–914. [Google Scholar] [CrossRef]
- Rampacci, E.; Felicetti, T.; Pietrella, D.; Sabatini, S.; Passamonti, F. Drug efflux transporters in Staphylococcus pseudintermedius: In silico prediction and characterization of resistance. J. Antimicrob. Chemother. 2022, 77, 3283–3290. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, N.; Okada, H.; Narui, K.; Sasatsu, M. Comparison of the nucleotide sequence and expression of norA genes and microbial susceptibility in 21 strains of Staphylococcus aureus. Microb. Drug Resist. 2004, 10, 197–203. [Google Scholar] [CrossRef]
- Kaatz, G.W.; Seo, S.M.; Ruble, C.A. Efflux-mediated fluoroquinolone resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 1993, 37, 1086–1094. [Google Scholar] [CrossRef] [Green Version]
- Costa, S.S.; Sobkowiak, B.; Parreira, R.; Edgeworth, J.D.; Viveiros, M.; Clark, T.G.; Couto, I. Genetic diversity of norA, coding for a main efflux pump of Staphylococcus aureus. Front. Genet. 2019, 9, 710. [Google Scholar] [CrossRef]
- Madhaiyan, M.; Wirth, J.S.; Saravanan, V.S. Phylogenomic analyses of the Staphylococcaceae family suggest the reclassification of five species within the genus Staphylococcus as heterotypic synonyms, the promotion of five subspecies to novel species, the taxonomic reassignment of five Staphylococcus species to Mammalliicoccus gen. nov., and the formal assignment of Nosocomiicoccus to the family Staphylococcaceae. Int. J. Syst. Evol. Microbiol. 2020, 70, 5926–5936. [Google Scholar] [CrossRef]
- Becker, K.; Schaumburg, F.; Kearns, A.; Larsen, A.R.; Lindsay, J.A.; Skov, R.L.; Westh, H. Implications of identifying the recently defined members of the Staphylococcus aureus complex S. argenteus and S. schweitzeri: A position paper of members of the ESCMID Study Group for Staphylococci and Staphylococcal Diseases (ESGS). Clin. Microbiol. Infect. 2019, 25, 1064–1070. [Google Scholar] [CrossRef]
- Schutte, A.H.J.; Strepis, N.; Zandijk, W.H.A.; Bexkens, M.L.; Bode, L.G.M.; Klaassen, C.H.W. Characterization of Staphylococcus roterodami sp. Nov., a new species within the Staphylococcus aureus complex isolated from a human foot infection. Int. J. Syst. Evol. Microbiol. 2021, 71, 4996. [Google Scholar] [CrossRef] [PubMed]
- Chew, K.L.; Octavia, S.; Lai, D.; Lin, R.T.P.; Teo, J.W.P. Staphylococcus singaporensis sp. nov., a new member of the Staphylococcus aureus complex, isolated from human clinical specimens. Int. J. Syst. Evol. Microbiol. 2021, 71, 005067. [Google Scholar] [CrossRef] [PubMed]
- Kosecka-Strojek, M.; Sabat, A.J.; Akkerboom, V.; Becker, K.; van Zanten, E.; Wisselink, G.; Miedzobrodzki, J.; Kooistra-Smid, A.M.D.M.; Friedrich, A.W. Development and validation of a reference data set for assigning Staphylococcus species based on Next-Generation Sequencing of the 16S-23S rRNA region. Front. Cell Infect. Microbiol. 2019, 7, 278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, I.T.; Brown, M.H.; Skurray, R.A. Proton-dependent multidrug efflux systems. Microbiol. Rev. 1996, 60, 575–608. [Google Scholar] [CrossRef] [PubMed]
- Ginn, S.L.; Brown, M.H.; Skurray, R.A. The TetA(K) tetracycline/H+ antiporter from Staphylococcus aureus: Mutagenesis and functional analysis of motif C. J. Bacteriol. 2000, 182, 1492–1498. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.C.; Zhao, Y.; Heng, J.; Jiang, D. Energy coupling mechanisms of MFS transporters. Protein Sci. 2015, 24, 1560–1579. [Google Scholar] [CrossRef] [Green Version]
- Shang, Y.; Lv, P.; Li, S.; Wang, W.; Liu, Y.; Yang, C. Allele-based analysis revealed the critical functions of region 277-297 in the NorA efflux pump of Staphylococcus aureus. J. Antimicrob. Chemother. 2021, 76, 1420–1427. [Google Scholar] [CrossRef]
- Brawley, D.N.; Sauer, D.B.; Li, J.; Zheng, X.; Koide, A.; Jedhe, G.S.; Suwatthee, T.; Song, J.; Liu, Z.; Arora, P.S.; et al. Structural basis for inhibition of the drug efflux pump NorA from Staphylococcus aureus. Nat. Chem. Biol. 2022, 18, 706–712. [Google Scholar] [CrossRef]
- Ferreira, C.; Costa, S.S.; Serrano, M.; Oliveira, K.; Trigueiro, G.; Pomba, C.; Couto, I. Clonal lineages, antimicrobial resistance, and PVL carriage of Staphylococcus aureus associated to skin and soft-tissue infections from ambulatory patients in Portugal. Antibiotics 2021, 10, 345. [Google Scholar] [CrossRef]
- Costa, S.S.; Ribeiro, R.; Serrano, M.; Oliveira, K.; Ferreira, C.; Leal, M.; Pomba, C.; Couto, I. Staphylococcus aureus causing skin and soft tissue infections in companion animals: Antimicrobial resistance profiles and clonal lineages. Antibiotics 2022, 11, 599. [Google Scholar] [CrossRef] [PubMed]
- Sierra, J.M.; Ruiz, J.; de Anta, M.T.J.; Vila, J. Prevalence of two different genes encoding NorA in 23 clinical strains of Staphylococcus aureus. J. Antimicrob. Chemother. 2000, 46, 145–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, F.J.; Hertel, B.; Hofmann, B.; Scheuring, S.; Verhoef, J.; Fluit, A.C.; Heinz, H.P.; Köhrer, K.; Jones, M.E. Relationship between mutations in the coding and promoter regions of the norA genes in 42 unrelated clinical isolates of Staphylococcus aureus and the MICs of norfloxacin for these strains. J. Antimicrob. Chemother. 1998, 42, 561–563. [Google Scholar] [CrossRef] [Green Version]
- Kosmidis, C.; Schindler, B.D.; Jacinto, P.L.; Patel, D.; Bains, K.; Seo, S.M.; Kaatz, G.W. Expression of multidrug resistance efflux pump genes in clinical and environmental isolates of Staphylococcus aureus. Int. J. Antimicrob. Agents 2012, 40, 204–209. [Google Scholar] [CrossRef]
- Brooks, L.E.; Ul-Hasan, S.; Chan, B.K.; and Sistrom, M.J. Quantifying the evolutionary conservation of genes encoding multidrug efflux pumps in the ESKAPE pathogens to identify antimicrobial drug targets. mSystems 2018, 3, e00024-18. [Google Scholar] [CrossRef] [Green Version]
- Kaatz, G.W.; Seo, S.M.; Ruble, C.A. Mechanisms of fluoroquinolone resistance in Staphylococcus aureus. J. Infect. Dis. 1991, 163, 1080–1086. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Francisco, A.P.; Bugalho, M.; Ramirez, M.; Carriço, J.A. Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinform. 2009, 10, 152. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro-Gonçalves, B.; Francisco, A.P.; Vaz, C.; Ramirez, M.; Carriço, J.A. PHYLOViZ Online: Web-based tool for visualization, phylogenetic inference, analysis and sharing of minimum spanning trees. Nucl. Acids Res. 2016, 44, W246–W251. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucl. Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Trifinopoulos, J.; Nguyen, L.-T.; von Haeseler, A.; Minh, B.Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucl. Acids Res. 2016, 44, W232–W235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, S.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview version 2: A Multiple Sequence Alignment and Analysis Workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Yates, C.M.; Filippis, I.; Kelley, L.A.; Sternberg, M.J. SuSPect: Enhanced prediction of single amino acid variant (SAV) phenotype using network features. J. Mol. Biol. 2014, 426, 2692–2701. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allele | Primers (5′ → 3′) | Amplicon Size (nt) | PCR Conditions | Restriction with HindIII (nt) |
|---|---|---|---|---|
| Forward primers | ||||
| norAI | YonorA (Fw) a: ATATTCAGTTGTTGTCTTAATAT | 230 | 94 °C, 4 min 35 cycles of 94 °C, 30 s; 55 °C, 30 s; 72 °C, 30 s 72 °C, 5 min | No |
| norAII | NorAII (Fw) b: CTGTATTCTTTATATACATCG | 391 | 94 °C, 4 min 35 cycles of 94 °C, 30 s; 57 °C, 30 s; 72 °C, 30 s 72 °C, 5 min | No |
| norAIII | NorAIII (Fw) b: GACCCCTAAAAAAGTTTCGAC | 526 | 94 °C, 3 min 35 cycles of 94 °C, 30 s; 54 °C, 30 s; 72 °C, 30 s 72 °C, 5 min | 166 + 360 |
| norACC59–CC121 | No restriction | |||
| Reverse primer | ||||
| All alleles | NorA2 (Rv) a,c: GCACATCAAATAACGCACCT | |||
| Clonal Complex (CC) /Clonal Lineages (ST) | Origin (No. of Strains) | Confirmed Allele | |||
|---|---|---|---|---|---|
| norAI | norAII | norAIII | norACC59–CC121 | ||
| Expected allele: norAI a | |||||
| CC1 | |||||
| ST188 | Companion animal (n = 1) | + | - | - | - |
| ST6565 | Companion animal (n = 1) | + | - | - | - |
| CC5 | |||||
| ST5 | Companion animal (n = 3); human (n = 2) | + | - | - | - |
| ST105 | Companion animal (n = 2); human (n = 5) | + | - | - | - |
| ST6531 | Human (n = 1) | + | - | - | - |
| ST6535 | Companion animal (n = 1) | + | - | - | - |
| CC8 | |||||
| ST8 | Human (n = 4) | + | - | - | - |
| ST72 | Companion animal (n = 2); human (n = 1) | + | - | - | - |
| ST6566 | Companion animal (n = 1) | + | - | - | - |
| CC15 | |||||
| ST15 | Companion animal (n = 1); human (n = 2) | + | - | - | - |
| Expected allele: norAII a | |||||
| CC22 | |||||
| ST22 | Companion animal (n = 10); human (n = 3) | - | + | - | - |
| CC30 | |||||
| ST30 | Human (n = 1) | - | + | - | - |
| CC152 | |||||
| ST152 | Human (n = 1) | - | + | - | - |
| CC398 | |||||
| ST398 | Companion animal (n = 5) | - | + | - | - |
| Expected allele: norAIII or norACC59–CC121 a | |||||
| CC45 | |||||
| ST278 | Human (n = 1) | - | - | + b | - |
| CC121 | |||||
| ST121 | Companion animal (n = 1) | + | - | - | + c |
| No previous association with norA allele | |||||
| CC7 | |||||
| ST7 | Companion animal (n =1); human (n = 1) | + | - | - | - |
| CC25 | |||||
| ST25 | Human (n = 2) | + | - | - | - |
| CC97 | |||||
| ST97 | Companion animal (n =1); human (n = 1) | + | - | - | - |
| --- | |||||
| ST816 | Companion animal (n = 1) | + | - | - | + c |
| --- | |||||
| ST6564 | Human (n = 1) | - | + | - | - |
| norA Allele | No. Strains (%) | Host | ST (CC) [30,31] |
|---|---|---|---|
| norAI | 32 (61.6%) | Human (n = 18) Companion animal (n = 14) | ST188, ST6565 (CC1); ST5, ST105, ST6531, ST6535 (CC5); ST7 (CC7); ST8, ST72, ST6566 (CC8); ST15 (CC15); ST25 (CC25); ST97 (CC97) |
| norAII | 17 (32.7%) | Human (n = 6) Companion animal (n = 11) | ST22 (CC22); ST30 (CC30); ST152 (CC152); ST398 (CC398); ST6564 |
| norAIII | 1 (1.9%) | Human (n = 1) | ST278 (CC45) |
| norACC59–CC121 | 2 (3.8%) | Companion animal (n = 2) | ST121 (CC121); ST816 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, C.; Abrantes, P.; Costa, S.S.; Viveiros, M.; Couto, I. Occurrence and Variability of the Efflux Pump Gene norA across the Staphylococcus Genus. Int. J. Mol. Sci. 2022, 23, 15306. https://doi.org/10.3390/ijms232315306
Ferreira C, Abrantes P, Costa SS, Viveiros M, Couto I. Occurrence and Variability of the Efflux Pump Gene norA across the Staphylococcus Genus. International Journal of Molecular Sciences. 2022; 23(23):15306. https://doi.org/10.3390/ijms232315306
Chicago/Turabian StyleFerreira, Carolina, Patrícia Abrantes, Sofia Santos Costa, Miguel Viveiros, and Isabel Couto. 2022. "Occurrence and Variability of the Efflux Pump Gene norA across the Staphylococcus Genus" International Journal of Molecular Sciences 23, no. 23: 15306. https://doi.org/10.3390/ijms232315306