
Inflammation Regulates Haematopoietic Stem Cells and Their Niche


Abstract
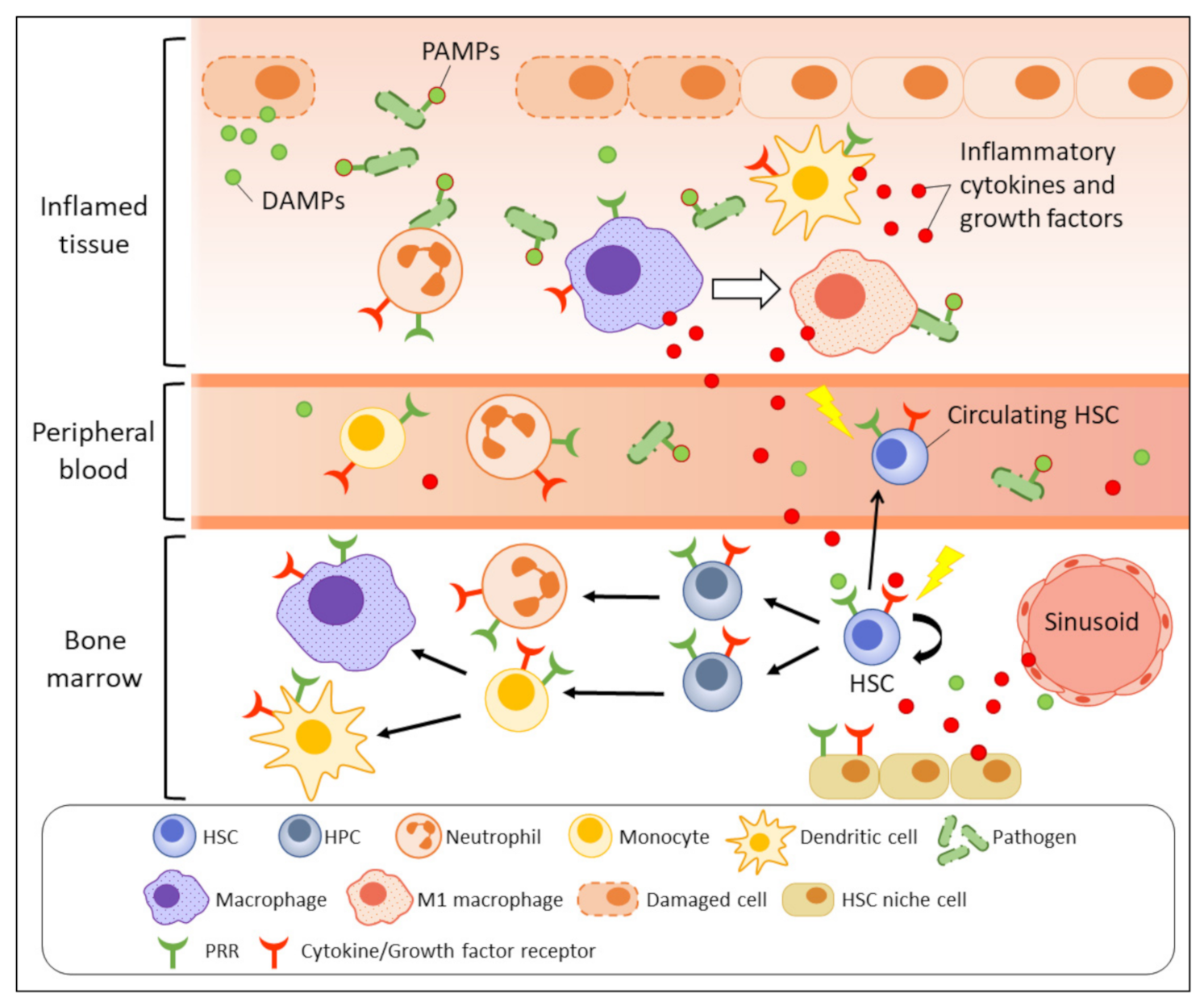
:1. Introduction
2. Inflammatory Processes
3. Acute Inflammation and HSPCs
4. Metabolic Changes upon Inflammation
5. Chronic Inflammation
6. Inflammation and HSC Niche
7. Future Directions and Clinical Implications
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kakiuchi, M.; Hirata, Y.; Robson, S.C.; Fujisaki, J. Paradoxical Regulation of Allogeneic Bone Marrow Engraftment and Immune Privilege by Mesenchymal Cells and Adenosine. Transplant. Cell. Ther. Off. Publ. Am. Soc. Transplant. Cell. Ther. 2021, 27, 92.e1–92.e5. [Google Scholar] [CrossRef]
- Fujisaki, J.; Wu, J.; Carlson, A.L.; Silberstein, L.; Putheti, P.; Larocca, R.; Gao, W.; Saito, T.I.; Lo Celso, C.; Tsuyuzaki, H.; et al. In Vivo Imaging of Treg Cells Providing Immune Privilege to the Haematopoietic Stem-Cell Niche. Nature 2011, 474, 216–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Contreras-Lopez, R.; Elizondo-Vega, R.; Paredes, M.J.; Luque-Campos, N.; Torres, M.J.; Tejedor, G.; Vega-Letter, A.M.; Figueroa-Valdés, A.; Pradenas, C.; Oyarce, K.; et al. HIF1α-Dependent Metabolic Reprogramming Governs Mesenchymal Stem/Stromal Cell Immunoregulatory Functions. FASEB J. 2020, 34, 8250–8264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietras, E.M.; Warr, M.R.; Passegué, E. Cell Cycle Regulation in Hematopoietic Stem Cells. J. Cell Biol. 2011, 195, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Krzyszczyk, P.; Schloss, R.; Palmer, A.; Berthiaume, F. The Role of Macrophages in Acute and Chronic Wound Healing and Interventions to Promote Pro-Wound Healing Phenotypes. Front. Physiol. 2018, 9, 419. [Google Scholar] [CrossRef] [PubMed]
- Kolaczkowska, E.; Kubes, P. Neutrophil Recruitment and Function in Health and Inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Nathan, C. Neutrophils and Immunity: Challenges and Opportunities. Nat. Rev. Immunol. 2006, 6, 173–182. [Google Scholar] [CrossRef]
- Ravindran, M.; Khan, M.A.; Palaniyar, N. Neutrophil Extracellular Trap Formation: Physiology, Pathology, and Pharmacology. Biomolecules 2019, 9, E365. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Sinha, M.; Datta, S.; Abas, M.; Chaffee, S.; Sen, C.K.; Roy, S. Monocyte and Macrophage Plasticity in Tissue Repair and Regeneration. Am. J. Pathol. 2015, 185, 2596–2606. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Gómez, A.; Perretti, M.; Soehnlein, O. Resolution of Inflammation: An Integrated View. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef]
- Novak, N.; Koch, S.; Allam, J.-P.; Bieber, T. Dendritic Cells: Bridging Innate and Adaptive Immunity in Atopic Dermatitis. J. Allergy Clin. Immunol. 2010, 125, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Tai, Y.; Wang, Q.; Korner, H.; Zhang, L.; Wei, W. Molecular Mechanisms of T Cells Activation by Dendritic Cells in Autoimmune Diseases. Front. Pharmacol. 2018, 9, 642. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [Green Version]
- Schultze, J.L.; Mass, E.; Schlitzer, A. Emerging Principles in Myelopoiesis at Homeostasis and during Infection and Inflammation. Immunity 2019, 50, 288–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manz, M.G.; Boettcher, S. Emergency Granulopoiesis. Nat. Rev. Immunol. 2014, 14, 302–314. [Google Scholar] [CrossRef]
- Takizawa, H.; Boettcher, S.; Manz, M.G. Demand-Adapted Regulation of Early Hematopoiesis in Infection and Inflammation. Blood 2012, 119, 2991–3002. [Google Scholar] [CrossRef] [Green Version]
- Granick, J.L.; Simon, S.I.; Borjesson, D.L. Hematopoietic Stem and Progenitor Cells as Effectors in Innate Immunity. Bone Marrow Res. 2012, 2012, 165107. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, H.; Fritsch, K.; Kovtonyuk, L.V.; Saito, Y.; Yakkala, C.; Jacobs, K.; Ahuja, A.K.; Lopes, M.; Hausmann, A.; Hardt, W.-D.; et al. Pathogen-Induced TLR4-TRIF Innate Immune Signaling in Hematopoietic Stem Cells Promotes Proliferation but Reduces Competitive Fitness. Cell Stem Cell 2017, 21, 225–240.e5. [Google Scholar] [CrossRef]
- Allakhverdi, Z.; Comeau, M.R.; Smith, D.E.; Toy, D.; Endam, L.M.; Desrosiers, M.; Liu, Y.-J.; Howie, K.J.; Denburg, J.A.; Gauvreau, G.M.; et al. CD34+ Hemopoietic Progenitor Cells Are Potent Effectors of Allergic Inflammation. J. Allergy Clin. Immunol. 2009, 123, 472–478.e1. [Google Scholar] [CrossRef]
- Allakhverdi, Z.; Delespesse, G. Hematopoietic Progenitor Cells Are Innate Th2 Cytokine-Producing Cells. Allergy 2012, 67, 4–9. [Google Scholar] [CrossRef]
- Zhao, J.L.; Ma, C.; O’Connell, R.M.; Mehta, A.; DiLoreto, R.; Heath, J.R.; Baltimore, D. Conversion of Danger Signals into Cytokine Signals by Hematopoietic Stem and Progenitor Cells for Regulation of Stress-Induced Hematopoiesis. Cell Stem Cell 2014, 14, 445–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeibundGut-Landmann, S.; Weidner, K.; Hilbi, H.; Oxenius, A. Nonhematopoietic Cells Are Key Players in Innate Control of Bacterial Airway Infection. J. Immunol. 2011, 186, 3130–3137. [Google Scholar] [CrossRef]
- Teijaro, J.R.; Walsh, K.B.; Cahalan, S.; Fremgen, D.M.; Roberts, E.; Scott, F.; Martinborough, E.; Peach, R.; Oldstone, M.B.A.; Rosen, H. Endothelial Cells Are Central Orchestrators of Cytokine Amplification during Influenza Virus Infection. Cell 2011, 146, 980–991. [Google Scholar] [CrossRef] [Green Version]
- Mai, J.; Virtue, A.; Shen, J.; Wang, H.; Yang, X.-F. An Evolving New Paradigm: Endothelial Cells – Conditional Innate Immune Cells. J. Hematol. Oncol. 2013, 6, 61. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Pérez, A.; Franco-Trepat, E.; Guillán-Fresco, M.; Jorge-Mora, A.; López, V.; Pino, J.; Gualillo, O.; Gómez, R. Role of Toll-Like Receptor 4 on Osteoblast Metabolism and Function. Front. Physiol. 2018, 9, 504. [Google Scholar] [CrossRef]
- AlQranei, M.S.; Senbanjo, L.T.; Aljohani, H.; Hamza, T.; Chellaiah, M.A. Lipopolysaccharide- TLR-4 Axis Regulates Osteoclastogenesis Independent of RANKL/RANK Signaling. BMC Immunol. 2021, 22, 23. [Google Scholar] [CrossRef] [PubMed]
- Rashedi, I.; Gómez-Aristizábal, A.; Wang, X.-H.; Viswanathan, S.; Keating, A. TLR3 or TLR4 Activation Enhances Mesenchymal Stromal Cell-Mediated Treg Induction via Notch Signaling. Stem Cells 2017, 35, 265–275. [Google Scholar] [CrossRef]
- Siegel, G.; Schäfer, R.; Dazzi, F. The Immunosuppressive Properties of Mesenchymal Stem Cells. Transplantation 2009, 87, S45. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Passegué, E. TNF-α Coordinates Hematopoietic Stem Cell Survival and Myeloid Regeneration. Cell Stem Cell 2019, 25, 357–372.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahandideh, B.; Derakhshani, M.; Abbaszadeh, H.; Akbar Movassaghpour, A.; Mehdizadeh, A.; Talebi, M.; Yousefi, M. The Pro-Inflammatory Cytokines Effects on Mobilization, Self-Renewal and Differentiation of Hematopoietic Stem Cells. Hum. Immunol. 2020, 81, 206–217. [Google Scholar] [CrossRef]
- Florez, M.A.; Matatall, K.A.; Jeong, Y.; Ortinau, L.; Shafer, P.W.; Lynch, A.M.; Jaksik, R.; Kimmel, M.; Park, D.; King, K.Y. Interferon Gamma Mediates Hematopoietic Stem Cell Activation and Niche Relocalization through BST2. Cell Rep. 2020, 33, 108530. [Google Scholar] [CrossRef]
- Morales-Mantilla, D.E.; King, K.Y. The Role of Interferon-Gamma in Hematopoietic Stem Cell Development, Homeostasis, and Disease. Curr. Stem Cell Rep. 2018, 4, 264–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caux, C.; Favre, C.; Saeland, S.; Duvert, V.; Durand, I.; Mannoni, P.; Banchereau, J. Potentiation of Early Hematopoiesis by Tumor Necrosis Factor-Alpha Is Followed by Inhibition of Granulopoietic Differentiation and Proliferation. Blood 1991, 78, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Snoeck, H.W.; Weekx, S.; Moulijn, A.; Lardon, F.; Lenjou, M.; Nys, G.; Van Ranst, P.C.; Van Bockstaele, D.R.; Berneman, Z.N. Tumor Necrosis Factor Alpha Is a Potent Synergistic Factor for the Proliferation of Primitive Human Hematopoietic Progenitor Cells and Induces Resistance to Transforming Growth Factor Beta but Not to Interferon Gamma. J. Exp. Med. 1996, 183, 705–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, Y.; Cain, D.W.; Kuraoka, M.; Kondo, M.; Kelsoe, G. IL-1RI Dependent HSC Proliferation Is Necessary for Inflammatory Granulopoiesis and Reactive Neutrophilia. J. Immunol. 2009, 182, 6477–6484. [Google Scholar] [CrossRef]
- Mossadegh-Keller, N.; Sarrazin, S.; Kandalla, P.K.; Espinosa, L.; Stanley, E.R.; Nutt, S.L.; Moore, J.; Sieweke, M.H. M-CSF Instructs Myeloid Lineage Fate in Single Haematopoietic Stem Cells. Nature 2013, 497, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Pietras, E.M. Inflammation: A Key Regulator of Hematopoietic Stem Cell Fate in Health and Disease. Blood 2017, 130, 1693–1698. [Google Scholar] [CrossRef] [Green Version]
- Takizawa, H.; Regoes, R.R.; Boddupalli, C.S.; Bonhoeffer, S.; Manz, M.G. Dynamic Variation in Cycling of Hematopoietic Stem Cells in Steady State and Inflammation. J. Exp. Med. 2011, 208, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldridge, M.T.; King, K.Y.; Boles, N.C.; Weksberg, D.C.; Goodell, M.A. Quiescent Hematopoietic Stem Cells Are Activated by IFNγ in Response to Chronic Infection. Nature 2010, 465, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Zhang, C. The Regulatory Role of IFN-γ on the Proliferation and Differentiation of Hematopoietic Stem and Progenitor Cells. Stem Cell Rev. Rep. 2017, 13, 705–712. [Google Scholar] [CrossRef]
- de Bruin, A.M.; Demirel, Ö.; Hooibrink, B.; Brandts, C.H.; Nolte, M.A. Interferon-γ Impairs Proliferation of Hematopoietic Stem Cells in Mice. Blood 2013, 121, 3578–3585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Dybedal, I.; Bryder, D.; Nilsson, L.; Sitnicka, E.; Sasaki, Y.; Jacobsen, S.E.W. IFN-γ Negatively Modulates Self-Renewal of Repopulating Human Hemopoietic Stem Cells. J. Immunol. 2005, 174, 752–757. [Google Scholar] [CrossRef]
- Qin, Y.; Fang, K.; Lu, N.; Hu, Y.; Tian, Z.; Zhang, C. Interferon Gamma Inhibits the Differentiation of Mouse Adult Liver and Bone Marrow Hematopoietic Stem Cells by Inhibiting the Activation of Notch Signaling. Stem Cell Res. Ther. 2019, 10, 210. [Google Scholar] [CrossRef] [PubMed]
- Mirantes, C.; Passegué, E.; Pietras, E.M. Pro-Inflammatory Cytokines: Emerging Players Regulating HSC Function in Normal and Diseased Hematopoiesis. Exp. Cell Res. 2014, 329, 248–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platanias, L.C. Mechanisms of Type-I- and Type-II-Interferon-Mediated Signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Rezzoug, F.; Huang, Y.; Tanner, M.K.; Wysoczynski, M.; Schanie, C.L.; Chilton, P.M.; Ratajczak, M.Z.; Fugier-Vivier, I.J.; Ildstad, S.T. TNF-α Is Critical to Facilitate Hemopoietic Stem Cell Engraftment and Function. J. Immunol. 2008, 180, 49–57. [Google Scholar] [CrossRef]
- Chen, J. Senescence and Functional Failure in Hematopoietic Stem Cells. Exp. Hematol. 2004, 32, 1025–1032. [Google Scholar] [CrossRef]
- Sekulovic, S.; Gylfadottir, V.; Vulto, I.; Gasparetto, M.; Even, Y.; Brookes, C.; Smith, C.; Eaves, C.J.; Lansdorp, P.M.; Rossi, F.M.; et al. Prolonged Self-Renewal Activity Unmasks Telomerase Control of Telomere Homeostasis and Function of Mouse Hematopoietic Stem Cells. Blood 2011, 118, 1766–1773. [Google Scholar] [CrossRef] [Green Version]
- Burberry, A.; Zeng, M.Y.; Ding, L.; Wicks, I.; Inohara, N.; Morrison, S.J.; Núñez, G. Infection Mobilizes Hematopoietic Stem Cells through Cooperative NOD-like Receptor and Toll-like Receptor Signaling. Cell Host Microbe 2014, 15, 779–791. [Google Scholar] [CrossRef] [Green Version]
- Ratajczak, M.Z.; Adamiak, M.; Plonka, M.; Abdel-Latif, A.; Ratajczak, J. Mobilization of Hematopoietic Stem Cells as a Result of Innate Immunity-Mediated Sterile Inflammation in the Bone Marrow Microenvironment—The Involvement of Extracellular Nucleotides and Purinergic Signaling. Leukemia 2018, 32, 1116–1123. [Google Scholar] [CrossRef] [Green Version]
- Lemoli, R.M.; D’Addio, A. Hematopoietic Stem Cell Mobilization. Haematologica 2008, 93, 321–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herman, A.; Romine, M.; Monlish, D.; Schuettpelz, L.G. Toll like Receptor 2 Signaling Regulates Hematopoietic Stem Cell Function and Promotes G-CSF Mediated HSC Mobilization. Blood 2014, 124, 4334. [Google Scholar] [CrossRef]
- Xie, M.; Zhang, S.; Dong, F.; Zhang, Q.; Wang, J.; Wang, C.; Zhu, C.; Zhang, S.; Luo, B.; Wu, P.; et al. Granulocyte Colony-Stimulating Factor Directly Acts on Mouse Lymphoid-Biased but Not Myeloid-Biased Hematopoietic Stem Cells. Haematologica 2021, 106, 1647–1658. [Google Scholar] [CrossRef] [Green Version]
- Bernitz, J.M.; Daniel, M.G.; Fstkchyan, Y.S.; Moore, K. Granulocyte Colony-Stimulating Factor Mobilizes Dormant Hematopoietic Stem Cells without Proliferation in Mice. Blood 2017, 129, 1901–1912. [Google Scholar] [CrossRef] [Green Version]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLean, A.L.; Smith, M.A.; Liepe, J.; Sim, A.; Khorshed, R.; Rashidi, N.M.; Scherf, N.; Krinner, A.; Roeder, I.; Lo Celso, C.; et al. Single Cell Phenotyping Reveals Heterogeneity Among Hematopoietic Stem Cells Following Infection. Stem Cells 2017, 35, 2292–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct Measurement of Local Oxygen Concentration in the Bone Marrow of Live Animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Simsek, T.; Kocabas, F.; Zheng, J.; DeBerardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The Distinct Metabolic Profile of Hematopoietic Stem Cells Reflects Their Location in a Hypoxic Niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef] [Green Version]
- Takubo, K.; Goda, N.; Yamada, W.; Iriuchishima, H.; Ikeda, E.; Kubota, Y.; Shima, H.; Johnson, R.S.; Hirao, A.; Suematsu, M.; et al. Regulation of the HIF-1α Level Is Essential for Hematopoietic Stem Cells. Cell Stem Cell 2010, 7, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Miharada, K.; Karlsson, G.; Rehn, M.; Rörby, E.; Siva, K.; Cammenga, J.; Karlsson, S. Cripto Regulates Hematopoietic Stem Cells as a Hypoxic-Niche-Related Factor through Cell Surface Receptor GRP78. Cell Stem Cell 2011, 9, 330–344. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Chen, Y.; Li, Q.; Han, X.; Cheng, C.; Wang, Z.; Danielpour, D.; Dunwoodie, S.L.; Bunting, K.D.; Yang, Y.-C. HIF-1α Deletion Partially Rescues Defects of Hematopoietic Stem Cell Quiescence Caused by Cited2 Deficiency. Blood 2012, 119, 2789–2798. [Google Scholar] [CrossRef] [Green Version]
- Vukovic, M.; Sepulveda, C.; Subramani, C.; Guitart, A.V.; Mohr, J.; Allen, L.; Panagopoulou, T.I.; Paris, J.; Lawson, H.; Villacreces, A.; et al. Adult Hematopoietic Stem Cells Lacking Hif-1α Self-Renew Normally. Blood 2016, 127, 2841–2846. [Google Scholar] [CrossRef] [Green Version]
- Sykes, S.M. The Identity Crisis of Hif-1α in HSC Biology. Blood 2016, 127, 2782–2784. [Google Scholar] [CrossRef] [Green Version]
- Mistry, J.J.; Marlein, C.R.; Moore, J.A.; Hellmich, C.; Wojtowicz, E.E.; Smith, J.G.W.; Macaulay, I.; Sun, Y.; Morfakis, A.; Patterson, A.; et al. ROS-Mediated PI3K Activation Drives Mitochondrial Transfer from Stromal Cells to Hematopoietic Stem Cells in Response to Infection. Proc. Natl. Acad. Sci. USA 2019, 116, 24610–24619. [Google Scholar] [CrossRef] [Green Version]
- Mistry, J.J.; Hellmich, C.; Moore, J.A.; Jibril, A.; Macaulay, I.; Moreno-Gonzalez, M.; Di Palma, F.; Beraza, N.; Bowles, K.M.; Rushworth, S.A. Free Fatty-Acid Transport via CD36 Drives β-Oxidation-Mediated Hematopoietic Stem Cell Response to Infection. Nat. Commun. 2021, 12, 7130. [Google Scholar] [CrossRef]
- Vannini, N.; Girotra, M.; Naveiras, O.; Nikitin, G.; Campos, V.; Giger, S.; Roch, A.; Auwerx, J.; Lutolf, M.P. Specification of Haematopoietic Stem Cell Fate via Modulation of Mitochondrial Activity. Nat. Commun. 2016, 7, 13125. [Google Scholar] [CrossRef] [PubMed]
- Ludin, A.; Gur-Cohen, S.; Golan, K.; Kaufmann, K.B.; Itkin, T.; Medaglia, C.; Lu, X.-J.; Ledergor, G.; Kollet, O.; Lapidot, T. Reactive Oxygen Species Regulate Hematopoietic Stem Cell Self-Renewal, Migration and Development, As Well As Their Bone Marrow Microenvironment. Antioxid. Redox Signal. 2014, 21, 1605–1619. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.; Li, H.; Pazhanisamy, S.K.; Meng, A.; Wang, Y.; Zhou, D. Reactive Oxygen Species and Hematopoietic Stem Cell Senescence. Int. J. Hematol 2011, 94, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Dépond, M.; He, L.; Foudi, A.; Kwarteng, E.O.; Lauret, E.; Plo, I.; Desterke, C.; Dessen, P.; Fujii, N.; et al. CXCR4/CXCL12 Axis Counteracts Hematopoietic Stem Cell Exhaustion through Selective Protection against Oxidative Stress. Sci. Rep. 2016, 6, 37827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Yu, J.; Pan, Q.; Yang, J.; Hao, G.; Wang, Y.; Li, L.; Cao, H. Hypoxia-Inducible Factor-2 Alpha Promotes the Proliferation of Human Placenta-Derived Mesenchymal Stem Cells through the MAPK/ERK Signaling Pathway. Sci. Rep. 2016, 6, 35489. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.S.; Ko, Y.J.; Lee, M.W.; Park, H.J.; Park, Y.J.; Kim, D.-I.; Sung, K.W.; Koo, H.H.; Yoo, K.H. Effect of Low Oxygen Tension on the Biological Characteristics of Human Bone Marrow Mesenchymal Stem Cells. Cell Stress Chaperones 2016, 21, 1089–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Kim, H.J.; Chang, E.-J.; Lee, Z.H.; Hwang, S.J.; Kim, H.-M.; Lee, Y.; Kim, H.-H. IL-17 Stimulates the Proliferation and Differentiation of Human Mesenchymal Stem Cells: Implications for Bone Remodeling. Cell Death Differ. 2009, 16, 1332–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Cássia Noronha, N.; Mizukami, A.; Caliári-Oliveira, C.; Cominal, J.G.; Rocha, J.L.M.; Covas, D.T.; Swiech, K.; Malmegrim, K.C.R. Priming Approaches to Improve the Efficacy of Mesenchymal Stromal Cell-Based Therapies. Stem Cell Res. Ther. 2019, 10, 131. [Google Scholar] [CrossRef] [Green Version]
- Arra, M.; Swarnkar, G.; Ke, K.; Otero, J.E.; Ying, J.; Duan, X.; Maruyama, T.; Rai, M.F.; O’Keefe, R.J.; Mbalaviele, G.; et al. LDHA-Mediated ROS Generation in Chondrocytes Is a Potential Therapeutic Target for Osteoarthritis. Nat. Commun. 2020, 11, 3427. [Google Scholar] [CrossRef] [PubMed]
- Matatall, K.A.; Jeong, M.; Chen, S.; Sun, D.; Chen, F.; Mo, Q.; Kimmel, M.; King, K.Y. Chronic Infection Depletes Hematopoietic Stem Cells Through Stress-Induced Terminal Differentiation. Cell Rep. 2016, 17, 2584–2595. [Google Scholar] [CrossRef] [Green Version]
- Kovtonyuk, L.V.; Fritsch, K.; Feng, X.; Manz, M.G.; Takizawa, H. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Front. Immunol 2016, 7, 502. [Google Scholar] [CrossRef] [Green Version]
- Coppack, S.W. Pro-Inflammatory Cytokines and Adipose Tissue. Proc. Nutr. Soc. 2001, 60, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trottier, M.D.; Naaz, A.; Li, Y.; Fraker, P.J. Enhancement of Hematopoiesis and Lymphopoiesis in Diet-Induced Obese Mice. Proc. Natl. Acad. Sci. USA 2012, 109, 7622–7629. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.S.; Ete Chan, M.; Rubin, J.; Rubin, C.T. Marrow Adiposity and Hematopoiesis in Aging and Obesity: Exercise as an Intervention. Curr. Osteoporos. Rep. 2018, 16, 105–115. [Google Scholar] [CrossRef]
- Ambrosi, T.H.; Scialdone, A.; Graja, A.; Gohlke, S.; Jank, A.-M.; Bocian, C.; Woelk, L.; Fan, H.; Logan, D.W.; Schürmann, A.; et al. Adipocyte Accumulation in the Bone Marrow during Obesity and Aging Impairs Stem Cell-Based Hematopoietic and Bone Regeneration. Cell Stem Cell 2017, 20, 771–784.e6. [Google Scholar] [CrossRef] [Green Version]
- Naveiras, O.; Nardi, V.; Wenzel, P.L.; Fahey, F.; Daley, G.Q. Bone Marrow Adipocytes as Negative Regulators of the Hematopoietic Microenvironment. Nature 2009, 460, 259–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poller, W.C.; Nahrendorf, M.; Swirski, F.K. Hematopoiesis and Cardiovascular Disease. Circ. Res. 2020, 126, 1061–1085. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Pagler, T.; Gautier, E.L.; Avagyan, S.; Siry, R.L.; Han, S.; Welch, C.L.; Wang, N.; Randolph, G.J.; Snoeck, H.W.; et al. ATP-Binding Cassette Transporters and HDL Suppress Hematopoietic Stem Cell Proliferation. Science 2010, 328, 1689–1693. [Google Scholar] [CrossRef] [Green Version]
- Murphy, A.J.; Akhtari, M.; Tolani, S.; Pagler, T.; Bijl, N.; Kuo, C.-L.; Wang, M.; Sanson, M.; Abramowicz, S.; Welch, C.; et al. ApoE Regulates Hematopoietic Stem Cell Proliferation, Monocytosis, and Monocyte Accumulation in Atherosclerotic Lesions in Mice. J. Clin. Invest. 2011, 121, 4138–4149. [Google Scholar] [CrossRef] [Green Version]
- Yvan-Charvet, L.; Wang, N.; Tall, A.R. The Role of HDL, ABCA1 and ABCG1 Transporters in Cholesterol Efflux and Immune Responses. Arter. Thromb. Vasc. Biol. 2010, 30, 139–143. [Google Scholar] [CrossRef] [Green Version]
- Westerterp, M.; Gourion-Arsiquaud, S.; Murphy, A.J.; Shih, A.; Cremers, S.; Levine, R.L.; Tall, A.R.; Yvan-Charvet, L. Regulation of Hematopoietic Stem and Progenitor Cell Mobilization by Cholesterol Efflux Pathways. Cell Stem Cell 2012, 11, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Frodermann, V.; Rohde, D.; Courties, G.; Severe, N.; Schloss, M.J.; Amatullah, H.; McAlpine, C.S.; Cremer, S.; Hoyer, F.F.; Ji, F.; et al. Exercise Reduces Inflammatory Cell Production and Cardiovascular Inflammation via Instruction of Hematopoietic Progenitor Cells. Nat. Med. 2019, 25, 1761–1771. [Google Scholar] [CrossRef]
- Hoyer, F.F.; Zhang, X.; Coppin, E.; Vasamsetti, S.B.; Modugu, G.; Schloss, M.J.; Rohde, D.; McAlpine, C.S.; Iwamoto, Y.; Libby, P.; et al. Bone Marrow Endothelial Cells Regulate Myelopoiesis in Diabetes. Circulation 2020, 142, 244–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Nix, D.; Gregory, M.; Ciorba, M.; Ostrander, E.; Newberry, R.; Spencer, D.; Challen, A. Inflammatory Cytokines Promote Clonal Hematopoiesis with Specific Mutations in Ulcerative Colitis Patients. Exp. Hematol. 2019, 80, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Pietras, E.; Hernandez, G.; Kuldanek, S.; Jennifer, R.; Kirkpatrick, G.; Noetzli, L.; Acharya, S.; Adane, B.; Jordan, C.; DiPaola, J.; et al. Altered HSC Fate Underlies Aberrant Blood Phenotypes in Rheumatoid Arthritis. Exp. Hematol. 2017, 53, S71. [Google Scholar] [CrossRef]
- Hernandez, G.; Mills, T.S.; Rabe, J.L.; Chavez, J.S.; Kuldanek, S.; Kirkpatrick, G.; Noetzli, L.; Jubair, W.K.; Zanche, M.; Myers, J.R.; et al. Pro-Inflammatory Cytokine Blockade Attenuates Myeloid Expansion in a Murine Model of Rheumatoid Arthritis. Haematologica 2020, 105, 585–597. [Google Scholar] [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [Google Scholar] [CrossRef] [Green Version]
- Hemmati, S.; Haque, T.; Gritsman, K. Inflammatory Signaling Pathways in Preleukemic and Leukemic Stem Cells. Front. Oncol. 2017, 7, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abegunde, S.O.; Buckstein, R.; Wells, R.A.; Rauh, M.J. An Inflammatory Environment Containing TNFα Favors Tet2-Mutant Clonal Hematopoiesis. Exp. Hematol. 2018, 59, 60–65. [Google Scholar] [CrossRef]
- Zambetti, N.A.; Ping, Z.; Chen, S.; Kenswil, K.J.G.; Mylona, M.A.; Sanders, M.A.; Hoogenboezem, R.M.; Bindels, E.M.J.; Adisty, M.N.; Van Strien, P.M.H.; et al. Mesenchymal Inflammation Drives Genotoxic Stress in Hematopoietic Stem Cells and Predicts Disease Evolution in Human Pre-Leukemia. Cell Stem Cell 2016, 19, 613–627. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.J.; Scadden, D.T. The Bone Marrow Niche for Haematopoietic Stem Cells. Nature 2014, 505, 327–334. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, S.K.; Johnston, H.M.; Coverdale, J.A. Spatial Localization of Transplanted Hemopoietic Stem Cells: Inferences for the Localization of Stem Cell Niches. Blood 2001, 97, 2293–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lévesque, J.-P.; Helwani, F.M.; Winkler, I.G. The Endosteal ‘Osteoblastic’ Niche and Its Role in Hematopoietic Stem Cell Homing and Mobilization. Leukemia 2010, 24, 1979–1992. [Google Scholar] [CrossRef] [Green Version]
- Hooper, A.T.; Butler, J.M.; Nolan, D.J.; Kranz, A.; Iida, K.; Kobayashi, M.; Kopp, H.-G.; Shido, K.; Petit, I.; Yanger, K.; et al. Engraftment and Reconstitution of Hematopoiesis Is Dependent on VEGFR2-Mediated Regeneration of Sinusoidal Endothelial Cells. Cell Stem Cell 2009, 4, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Acar, M.; Kocherlakota, K.S.; Murphy, M.M.; Peyer, J.G.; Oguro, H.; Inra, C.N.; Jaiyeola, C.; Zhao, Z.; Luby-Phelps, K.; Morrison, S.J. Deep Imaging of Bone Marrow Shows Non-Dividing Stem Cells Are Mainly Perisinusoidal. Nature 2015, 526, 126–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, P.; Poulos, M.G.; Lazzari, E.; Gutkin, M.C.; Lopez, D.; Kloss, C.C.; Crowley, M.J.; Katsnelson, L.; Freire, A.G.; Greenblatt, M.B.; et al. Chronic Activation of Endothelial MAPK Disrupts Hematopoiesis via NFKB Dependent Inflammatory Stress Reversible by SCGF. Nat. Commun. 2020, 11, 666. [Google Scholar] [CrossRef]
- Prendergast, Á.M.; Kuck, A.; van Essen, M.; Haas, S.; Blaszkiewicz, S.; Essers, M.A.G. IFNα-Mediated Remodeling of Endothelial Cells in the Bone Marrow Niche. Haematologica 2017, 102, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Wu, Q.; Johnson, C.B.; Pham, G.; Kinder, J.M.; Olsson, A.; Slaughter, A.; May, M.; Weinhaus, B.; D’Alessandro, A.; et al. In Situ Mapping Identifies Distinct Vascular Niches for Myelopoiesis. Nature 2021, 590, 457. [Google Scholar] [CrossRef] [PubMed]
- Winkler, I.G.; Sims, N.A.; Pettit, A.R.; Barbier, V.; Nowlan, B.; Helwani, F.; Poulton, I.J.; van Rooijen, N.; Alexander, K.A.; Raggatt, L.J.; et al. Bone Marrow Macrophages Maintain Hematopoietic Stem Cell (HSC) Niches and Their Depletion Mobilizes HSCs. Blood 2010, 116, 4815–4828. [Google Scholar] [CrossRef] [Green Version]
- Chow, A.; Huggins, M.; Ahmed, J.; Hashimoto, D.; Lucas, D.; Kunisaki, Y.; Pinho, S.; Leboeuf, M.; Noizat, C.; van Rooijen, N.; et al. CD169+ Macrophages Provide a Niche Promoting Erythropoiesis under Homeostasis and Stress. Nat. Med. 2013, 19, 429–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, A.; Lucas, D.; Hidalgo, A.; Méndez-Ferrer, S.; Hashimoto, D.; Scheiermann, C.; Battista, M.; Leboeuf, M.; Prophete, C.; van Rooijen, N.; et al. Bone Marrow CD169+ Macrophages Promote the Retention of Hematopoietic Stem and Progenitor Cells in the Mesenchymal Stem Cell Niche. J. Exp. Med. 2011, 208, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Perry, J.M.; Marshall, H.; Venkatraman, A.; Qian, P.; He, X.C.; Ahamed, J.; Li, L. Megakaryocytes Maintain Homeostatic Quiescence and Promote Post-Injury Regeneration of Hematopoietic Stem Cells. Nat. Med. 2014, 20, 1321–1326. [Google Scholar] [CrossRef]
- Beaulieu, L.M.; Lin, E.; Mick, E.; Koupenova, M.; Weinberg, E.O.; Kramer, C.D.; Genco, C.A.; Tanriverdi, K.; Larson, M.G.; Benjamin, E.J.; et al. Interleukin 1 Receptor 1 and Interleukin 1β Regulate Megakaryocyte Maturation, Platelet Activation, and Transcript Profile During Inflammation in Mice and Humans. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 552. [Google Scholar] [CrossRef] [Green Version]
- Ghannam, S.; Bouffi, C.; Djouad, F.; Jorgensen, C.; Noël, D. Immunosuppression by Mesenchymal Stem Cells: Mechanisms and Clinical Applications. Stem Cell Res. Ther. 2010, 1, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isringhausen, S.; Mun, Y.; Kovtonyuk, L.; Kräutler, N.J.; Suessbier, U.; Gomariz, A.; Spaltro, G.; Helbling, P.M.; Wong, H.C.; Nagasawa, T.; et al. Chronic Viral Infections Persistently Alter Marrow Stroma and Impair Hematopoietic Stem Cell Fitness. J. Exp. Med. 2021, 218, e20192070. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Kondo, M.; Kelsoe, G. Inflammation and the Reciprocal Production of Granulocytes and Lymphocytes in Bone Marrow. J. Exp. Med. 2005, 201, 1771–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Qin, H.; Jiang, N.; Liu, G.; Wu, H.; Bai, L.; Yu, B.; Zhang, X. G-CSF Partially Mediates Bone Loss Induced by Staphylococcus Aureus Infection in Mice. Clin. Sci. 2019, 133, 1297–1308. [Google Scholar] [CrossRef] [PubMed]
- Asada, N.; Katayama, Y.; Sato, M.; Minagawa, K.; Wakahashi, K.; Kawano, H.; Kawano, Y.; Sada, A.; Ikeda, K.; Matsui, T.; et al. Matrix-Embedded Osteocytes Regulate Mobilization of Hematopoietic Stem/Progenitor Cells. Cell Stem Cell 2013, 12, 737–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamori, Y.; Katayama, Y.; Asada, N.; Minagawa, K.; Sato, M.; Okamura, A.; Shimoyama, M.; Nakagawa, K.; Okano, T.; Tanimoto, M.; et al. Role for Vitamin D Receptor in the Neuronal Control of the Hematopoietic Stem Cell Niche. Blood 2010, 116, 5528–5535. [Google Scholar] [CrossRef] [Green Version]
- Spiegel, A.; Kalinkovich, A.; Shivtiel, S.; Kollet, O.; Lapidot, T. Stem Cell Regulation via Dynamic Interactions of the Nervous and Immune Systems with the Microenvironment. Cell Stem Cell 2008, 3, 484–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiegel, A.; Shivtiel, S.; Kalinkovich, A.; Ludin, A.; Netzer, N.; Goichberg, P.; Azaria, Y.; Resnick, I.; Hardan, I.; Ben-Hur, H.; et al. Catecholaminergic Neurotransmitters Regulate Migration and Repopulation of Immature Human CD34+ Cells through Wnt Signaling. Nat. Immunol. 2007, 8, 1123–1131. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, D.; Xu, C.; Li, H.; Caron, K.M.; Frenette, P.S. Nociceptive Nerves Regulate Haematopoietic Stem Cell Mobilization. Nature 2021, 589, 591–596. [Google Scholar] [CrossRef]
- Katayama, Y.; Battista, M.; Kao, W.-M.; Hidalgo, A.; Peired, A.J.; Thomas, S.A.; Frenette, P.S. Signals from the Sympathetic Nervous System Regulate Hematopoietic Stem Cell Egress from Bone Marrow. Cell 2006, 124, 407–421. [Google Scholar] [CrossRef] [Green Version]
- Méndez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic Stem Cell Release Is Regulated by Circadian Oscillations. Nature 2008, 452, 442–447. [Google Scholar] [CrossRef]
- Maestroni, G.J.M. Adrenergic Modulation of Hematopoiesis. J. Neuroimmune Pharm. 2020, 15, 82–92. [Google Scholar] [CrossRef]
- Pongratz, G.; Straub, R.H. The Sympathetic Nervous Response in Inflammation. Arthritis Res. Ther. 2014, 16, 504. [Google Scholar] [CrossRef] [Green Version]
- Comazzetto, S.; Murphy, M.M.; Berto, S.; Jeffery, E.; Zhao, Z.; Morrison, S.J. Restricted Hematopoietic Progenitors and Erythropoiesis Require SCF from Leptin Receptor+ Niche Cells in the Bone Marrow. Cell Stem Cell 2019, 24, 477–486.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Zeng, X.; Hu, J.; Chen, Q. Characterization of Nestin, a Selective Marker for Bone Marrow Derived Mesenchymal Stem Cells. Stem Cells Int. 2015, 2015, e762098. [Google Scholar] [CrossRef] [Green Version]
- Fernández-García, M.; Yañez, R.M.; Sánchez-Domínguez, R.; Hernando-Rodriguez, M.; Peces-Barba, M.; Herrera, G.; O’Connor, J.E.; Segovia, J.C.; Bueren, J.A.; Lamana, M.L. Mesenchymal Stromal Cells Enhance the Engraftment of Hematopoietic Stem Cells in an Autologous Mouse Transplantation Model. Stem Cell Res. Ther. 2015, 6, 165. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Hu, L.; Zhang, Y.; Zhu, C.; Cheng, H.; Xie, X.; Shi, M.; Zhu, P.; Zhao, X.; Chen, W.; et al. PDGFB-Expressing Mesenchymal Stem Cells Improve Human Hematopoietic Stem Cell Engraftment in Immunodeficient Mice. Bone Marrow Transplant. 2020, 55, 1029–1040. [Google Scholar] [CrossRef] [Green Version]
- Court, A.C.; Le-Gatt, A.; Luz-Crawford, P.; Parra, E.; Aliaga-Tobar, V.; Bátiz, L.F.; Contreras, R.A.; Ortúzar, M.I.; Kurte, M.; Elizondo-Vega, R.; et al. Mitochondrial Transfer from MSCs to T Cells Induces Treg Differentiation and Restricts Inflammatory Response. EMBO Rep. 2020, 21, e48052. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Quintin, J.; van der Meer, J.W.M. Trained Immunity: A Memory for Innate Host Defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef] [Green Version]
- de Laval, B.; Maurizio, J.; Kandalla, P.K.; Brisou, G.; Simonnet, L.; Huber, C.; Gimenez, G.; Matcovitch-Natan, O.; Reinhardt, S.; David, E.; et al. C/EBPβ-Dependent Epigenetic Memory Induces Trained Immunity in Hematopoietic Stem Cells. Cell Stem Cell 2020, 26, 657–674.e8. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, E.; Sanz, J.; Dunn, J.L.; Khan, N.; Mendonça, L.E.; Pacis, A.; Tzelepis, F.; Pernet, E.; Dumaine, A.; Grenier, J.-C.; et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell 2018, 172, 176–190.e19. [Google Scholar] [CrossRef] [Green Version]
- Cirovic, B.; de Bree, L.C.J.; Groh, L.; Blok, B.A.; Chan, J.; van der Velden, W.J.F.M.; Bremmers, M.E.J.; van Crevel, R.; Händler, K.; Picelli, S.; et al. BCG Vaccination in Humans Elicits Trained Immunity via the Hematopoietic Progenitor Compartment. Cell Host Microbe 2020, 28, 322–334.e5. [Google Scholar] [CrossRef]
- Fidanza, M.; Seif, A.E.; Jo, S.; Kariminia, A.; Rolf, N.; Sly, L.M.; Grupp, S.A.; Reid, G.S.D. IFN-γ Directly Inhibits Murine B-Cell Precursor Leukemia-Initiating Cell Proliferation Early in Life. Eur J. Immunol. 2017, 47, 892–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buteyn, N.J.; Santhanam, R.; Merchand-Reyes, G.; Murugesan, R.A.; Dettorre, G.M.; Byrd, J.C.; Sarkar, A.; Vasu, S.; Mundy-Bosse, B.L.; Butchar, J.P.; et al. Activation of the Intracellular Pattern Recognition Receptor NOD2 Promotes Acute Myeloid Leukemia (AML) Cell Apoptosis and Provides a Survival Advantage in an Animal Model of AML. J. Immunol. 2020, 204, 1988–1997. [Google Scholar] [CrossRef]
- Testa, U.; Labbaye, C.; Castelli, G.; Pelosi, E. Oxidative Stress and Hypoxia in Normal and Leukemic Stem Cells. Exp. Hematol. 2016, 44, 540–560. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Hao, X.; Lai, X.; Liu, L.; Zhu, J.; Shao, H.; Huang, D.; Gu, H.; Zhang, T.; Yu, Z.; et al. Oxidative Phosphorylation Enhances the Leukemogenic Capacity and Resistance to Chemotherapy of B Cell Acute Lymphoblastic Leukemia. Sci. Adv. 2021, 7, eabd6280. [Google Scholar] [CrossRef] [PubMed]
- de Beauchamp, L.; Himonas, E.; Helgason, G.V. Mitochondrial Metabolism as a Potential Therapeutic Target in Myeloid Leukaemia. Leukemia 2021, 36, 1–12. [Google Scholar] [CrossRef]
- Chen, Y.; Liang, Y.; Luo, X.; Hu, Q. Oxidative Resistance of Leukemic Stem Cells and Oxidative Damage to Hematopoietic Stem Cells under Pro-Oxidative Therapy. Cell Death Dis. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Burt, R.; Dey, A.; Aref, S.; Aguiar, M.; Akarca, A.; Bailey, K.; Day, W.; Hooper, S.; Kirkwood, A.; Kirschner, K.; et al. Activated Stromal Cells Transfer Mitochondria to Rescue Acute Lymphoblastic Leukemia Cells from Oxidative Stress. Blood 2019, 134, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- DiNardo, C.D.; Ravandi, F.; Agresta, S.; Konopleva, M.; Takahashi, K.; Kadia, T.; Routbort, M.; Patel, K.P.; Brandt, M.; Pierce, S.; et al. Characteristics, Clinical Outcome, and Prognostic Significance of IDH Mutations in AML. Am. J. Hematol. 2015, 90, 732–736. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Inflammation/Stress | Cell Type | Regulators | Inflammatory Responses | Reference | |
|---|---|---|---|---|---|
| Chronic | Obesity | Adipocyte | TNF-α IL-1β IL-6 | Adipocytes secreted diverse inflammatory cytokines and accumulated adipocytes impaired reconstitution potentials, increased myelopoiesis, and suppressed lymphopoiesis of HSCs | [77,79,80,81] |
| Chronic | MAPK-induced inflammation | Endothelial cell | NF-κB | Impaired HSC survival and functionality | [101] |
| Acute | pI:C/IFN-α administration | Endothelial cell | VEGF | Vasculature expansion by haematopoietic and non-haematopoietic pathways | [102] |
| Acute | Listeria monocytogenes infection | Endothelial cell | M-CSF | Loss of endothelial-derived CSF1 disrupted localisation of myeloid progenitors in perisinusoidal niche and, in turn, promoted dendritic cell generation | [103] |
| Acute | G-CSF administration | Macrophage (Osteomac) | n.d. | G-CSF administration depleted BM macrophages, and in turn, suppressed HSC-supportive osteoblasts | [104] |
| Acute | Haemolytic anaemia | Macrophage (Osteomac) | n.d. | The presence of macrophages is critical to erythroid recovery | [105] |
| Acute | Macrophage depletion | Macrophage (Osteomac) | n.d. | Macrophage depletion suppresses MSCs’ expression of HSC retention genes | [106] |
| Acute | 5-FU | Megakaryocyte (MK) | FGF1 | MKs supported HSC regeneration by increasing FG1 secretion | [107] |
| Chronic | Obesity | Megakaryocyte (MK) | IL-1β | Obesity augmented MK and platelet function and upregulated their inflammatory gene expressions | [108] |
| Chronic | Porphyromonas gingivalis infection | Megakaryocyte (MK) | IL-1β | Increased platelet production | [108] |
| Acute | TNF-α, IFN-γ, IL-1α/β signals | Mesenchymal stem cell (MSC) | IDO NO PGE2 | Activated MSCs secreted immunosuppressive molecules inhibited T cell proliferation and activities | [28,109] |
| Acute | LCMV infection | Mesenchymal stem cell (MSC) | IFN-γ | LCMV infection disrupted structural morphology, network, and capability of HSC-supportive cytokine secretion of CAR cells | [110] |
| Acute | NP-CGG immunisation | Neutrophil | n.d. | Neutrophil emigration from BM to create a vacancy in BM to promote myeloid cell generation | [111] |
| Acute | Staphylococcus aureus infection | Osteoblast | G-CSF | Osteoblastic suppression by G-CSF impaired osteoblasts’ support to HSCs and promoted HSC mobilisation | [112,113,114] |
| Acute | Lithium treatment | Sympathetic nerve | β-catenin | Increased HSPC proliferation, mobilisation, and granulocyte colony formation | [115] |
| Acute | Adrenergic neurotransmitter treatment | Sympathetic nerve | β-catenin | Increased hCD34+ HSPC proliferation, mobilisation, and repopulating potential in vivo via canonical Wnt signalling pathway | [116] |
| Chronic | Neurotransmission ablation | Sympathetic nerve | G-CSF | Neurotransmission ablation suppressed HSC mobilisation and osteoblast function | [117,118] |
| Chronic | Altered circadian rhythms | Sympathetic nerve | CXCL12 | Altered adrenergic signals disrupted rhythmic CXCL12 oscillations in BM and in turn dysregulation circadian HSC mobilisation | [119] |
| Acute | Allograft transplant | Treg cells | IL-10 | The presence of Treg cells was critical to support the survival of allo-HSCs | [2] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ho, N.P.-Y.; Takizawa, H. Inflammation Regulates Haematopoietic Stem Cells and Their Niche. Int. J. Mol. Sci. 2022, 23, 1125. https://doi.org/10.3390/ijms23031125
Ho NP-Y, Takizawa H. Inflammation Regulates Haematopoietic Stem Cells and Their Niche. International Journal of Molecular Sciences. 2022; 23(3):1125. https://doi.org/10.3390/ijms23031125
Chicago/Turabian StyleHo, Nicole Pui-Yu, and Hitoshi Takizawa. 2022. "Inflammation Regulates Haematopoietic Stem Cells and Their Niche" International Journal of Molecular Sciences 23, no. 3: 1125. https://doi.org/10.3390/ijms23031125