
The “Elastic Perspective” of SARS-CoV-2 Infection and the Role of Intrinsic and Extrinsic Factors


Abstract
1. Introduction
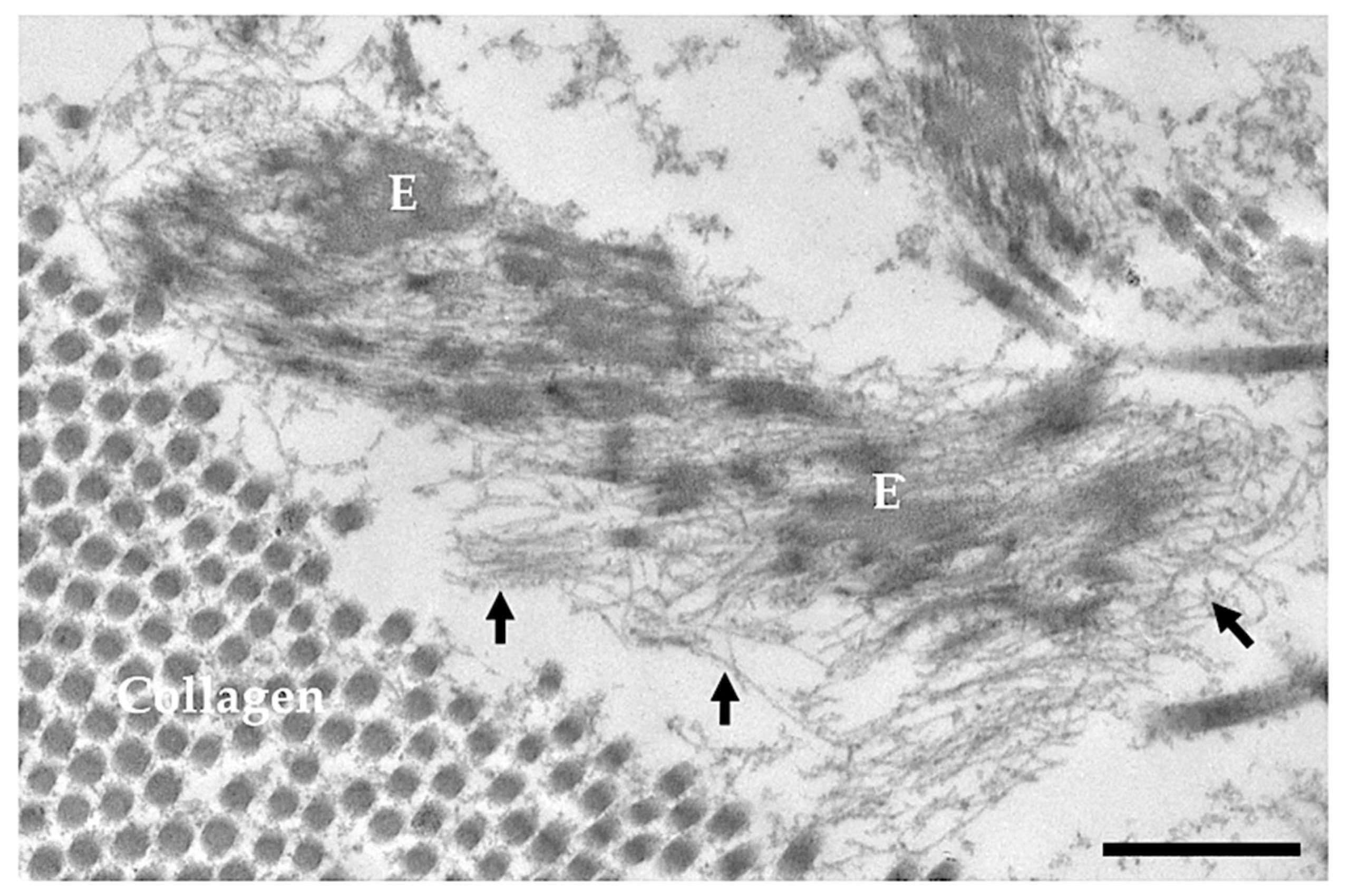
2. Elastic Fibres
2.1. Tropoelastin: Synthesis, Secretion, Coacervation and Cross-Linking
2.2. Microfibril Scaffold and Proteins Associated with Elastic Fibres
2.3. Elastic Fibre Degradation

{kind=link}
{kind=link}
| Gene Name | Protein Name | Uniprot Accession | Major Cellular Sources | TE/ ELN | FBN | Endogenous Inhibitors |
|---|---|---|---|---|---|---|
| Cysteine-endopeptidases | ||||||
| CTSB | Cathepsin B (EC 3.4.22.1) | P07858 | Ubiquitous | [67] | Cystatin A, B, C, S [61] | |
| CTSF | Cathepsin F (EC 3.4.22.41) | Q9UBX1 | Ubiquitous | [67] | Cystatin F [61] | |
| CTSK | Cathepsin K (EC 3.4.22.38) | P43235 | Fibroblast, macrophage, osteoclast | [67] | [68] | Cystatin F [61] |
| CTSL | Procathepsin L (EC 3.4.22.15) | P07711 | Ubiquitous | [67] | Cystatin A, B, C, D, E, M, F [61] | |
| CTSS | Cathepsin S (EC 3.4.22.27) | P25774 | Macrophage, SMC | [67] | Cystatin B, C, D, F [61] | |
| CTSV | Cathepsin L2 (EC 3.4.22.43) | O60911 | Tissue specific EC, macrophage | [67] | [68] | Cystatin E, M, F [61] |
| Metallo-endopeptidases | ||||||
| MMEL-1 | Membrane metallo- endopeptidase-like 1 (EC 3.4.24.11) | Q495T6 | Fibroblast | [69] | Peptides of the opiorphin family [70] | |
| MMP-2 | 72 kDa type IV collagenase (EC 3.4.24.24) | P08253 | Fibroblast, macrophage, neutrophil, T-cell, VEC | [71] | [72] | TIMP-1, -2, -3, -4 [62] |
| MMP-3 | Stromelysin-1 (EC 3.4.24.17) | P08254 | EC, lymphocytes, macrophage, SMC | [71] | [72] | TIMP-1, -2, -3, -4 [62] |
| MMP-7 | Matrilysin (EC 3.4.24.23) | P09237 | EC, macrophage | [71,73] | TIMP-1, -2, -3, -4 [62] | |
| MMP-9 | Matrix metalloproteinase-9 (EC 3.4.24.35) | P14780 | EC, fibroblast, macrophage, neutrophil | [71,73,74] | [72,75] | TIMP-1, -2, -3, -4 [62] |
| MMP-10 | Stromelysin-2 (EC 3.4.24.22) | P09238 | EC, macrophage, SMC | [71] | TIMP-1, -2, -3, -4 [62] | |
| MMP-12 | Macrophage metalloelastase (EC 3.4.24.65) | P39900 | Lung epithelial cells, macrophage, | [76,77] | [72,75] | TIMP-1, -2, -3, -4 [62] |
| MMP-13 | Collagenase 3 (EC 3.4.24.-) | P45452 | Fibroblast, macrophage, SMC, VEC | [72,75] | TIMP-1, -2, -3, -4 [62] | |
| MMP-14 | Matrix metalloproteinase-14 (EC 3.4.24.80) | P50281 | Fibroblast, macrophage, SMC, VEC | [78,79] | [72] | TIMP-1, -2, -3, -4 [62] |
| Serine-endopeptidases | ||||||
| CELA1 | Chymotrypsin-like elastase family member 1 (EC 3.4.21.36) | Q9UNI1 | Lung epithelial, intestinal, and immune cells | [80] | α1-anti-trypsin [80] | |
| CTSG | Cathepsin G (EC 3.4.21.20) | P08311 | Polymorphonuclear leucocytes | [81] | α1-anti-chymotrypsin, SLPI [63] | |
| ELANE | Neutrophil elastase (EC 3.4.21.37) | P08246 | Polymorphonuclear leucocytes | [82] | [83] | α1-anti-trypsin, α2-macroglobulin, elafin [63,64] |
| PRTN3 | Myeloblastin (EC 3.4.21.76) | P24158 | Polymorphonuclear leucocytes | [84] | Elafin [64] | |
3. SARS-CoV-2 Infection
4. Elastic Fibres and Elastases in SARS-CoV-2 Infection
5. Intrinsic and Extrinsic Factors Impairing Elastic Fibre Homeostasis and Their Impact on SARS-CoV-2 Infection
5.1. Aging
5.2. Oxidative Stress
5.3. Smoke
5.4. Vitamin K
5.5. Pollution
5.6. Mechanical Stress
6. Elastase Inhibitors: Any Valuable Perspective in SARS-CoV-2 Infection?
| Compound | Activity | Applications |
|---|---|---|
| Sivelestat (ONO-5046) | Selective, reversible and competitive neutrophil elastase inhibitor without effects on other proteases. | Approved for acute respiratory syndromes and proposed for COVID-19 [124]. |
| Roseltide | A plant derived peptide acting as a neutrophil elastase inhibitor. | Proposed for airway inflammatory diseases [219]. |
| Lonodelestat (POL6014) | A macrocycle based on the protein epitope mimetic technology acting as a potent and selective neutrophil elastase inhibitor. | Proposed for chronic inflammatory conditions and in phase 2 trial for patients with cystic fibrosis [220]. |
| Alvelestat (MPH966) | Neutrophil elastase inhibitor. | Proposed for bronchiolitis obliterans syndrome, emphysema, COPD and in phase 2 trial for COVID-19 patients [182,221]. |
| Brensocatib (INS1007) | Selective, competitive and reversible cathepsin C inhibitor that reduces neutrophil elastase activity. | In phase 3 trial for COVID-19 patients [222] |
| Prolastin | α1-antitrypsin. | Approved for self-administration AAT therapy to preserve functional lung tissue in AAT deficiency, COPD and proposed for COVID-19 [223]. |
| Elafin | Endogenously synthesized protein containing domains with antiproteolytic properties (i.e., vascular elastase). | Proposed for the treatment of inflammatory vascular, systemic and pulmonary diseases as COPD [224]. |
| Secretory leucocyte protease inhibitor (SLPI) | Unglycosylated natural protease inhibitor with a additional role as NET modulator. | Proposed for COPD, chronic lung diseases [224,225]. |
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Weibel, E.R. Functional Morphology of Lung Parenchyma. In Comprehensive Physiology; John Wiley & Sons, Ltd: Hoboken, NJ, USA, 2011; pp. 89–111. ISBN 978-0-470-65071-4. [Google Scholar]
- Birzle, A.M.; Hobrack, S.M.K.; Martin, C.; Uhlig, S.; Wall, W.A. Constituent-specific material behavior of soft biological tissue: Experimental quantification and numerical identification for lung parenchyma. Biomech. Model. Mechanobiol. 2019, 18, 1383–1400. [Google Scholar] [CrossRef]
- Mecham, R.P. Elastin in lung development and disease pathogenesis. Matrix Biol. 2018, 73, 6–20. [Google Scholar] [CrossRef]
- Vindin, H.J.; Oliver, B.G.; Weiss, A.S. Elastin in healthy and diseased lung. Curr. Opin. Biotechnol. 2021, 74, 15–20. [Google Scholar] [CrossRef]
- Rosenbloom, J.; Bashir, M.; Yeh, H.; Rosenbloom, J.; Ornstein-Goldstein, N.; Fazio, M.; Kahari, V.M.; Uitto, J. Regulation of elastin gene expression. Ann. N. Y. Acad. Sci. 1991, 624, 116–136. [Google Scholar] [CrossRef]
- Keeley, F.W. The synthesis of soluble and insoluble elastin in chicken aorta as a function of development and age. Effect of a high cholesterol diet. Can. J. Biochem. 1979, 57, 1273–1280. [Google Scholar] [CrossRef]
- Holzenberger, M.; Lièvre, C.A.; Robert, L. Tropoelastin gene expression in the developing vascular system of the chicken: An in situ hybridization study. Anat. Embryol. 1993, 188, 481–492. [Google Scholar] [CrossRef]
- Shapiro, S.D.; Endicott, S.K.; Province, M.A.; Pierce, J.A.; Campbell, E.J. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J. Clin. Investig. 1991, 87, 1828–1834. [Google Scholar] [CrossRef]
- Uitto, J. Biochemistry of the elastic fibers in normal connective tissues and its alterations in diseases. J. Investig. Dermatol. 1979, 72, 1–10. [Google Scholar] [CrossRef]
- Mikawa, Y.; Hamagami, H.; Shikata, J.; Yamamuro, T. Elastin in the human intervertebral disk. A histological and biochemical study comparing it with elastin in the human yellow ligament. Arch. Orthop. Trauma. Surg. 1986, 105, 343–349. [Google Scholar] [CrossRef]
- Rosenbloom, J. Elastin: Relation of protein and gene structure to disease. Lab. Investig. 1984, 51, 605–623. [Google Scholar]
- Kielty, C.M.; Sherratt, M.J.; Shuttleworth, C.A. Elastic fibres. J. Cell Sci. 2002, 115, 2817–2828. [Google Scholar] [CrossRef]
- Pasquali-Ronchetti, I.; Baccarani-Contri, M. Elastic fiber during development and aging. Microsc. Res. Tech. 1997, 38, 428–435. [Google Scholar] [CrossRef]
- Pasquali-Ronchetti, I.; Fornieri, C.; Baccarani-Contri, M.; Quaglino, D. Ultrastructure of elastin. Ciba Found. Symp. 1995, 192, 31–42; discussion 42–50. [Google Scholar]
- Reichheld, S.E.; Muiznieks, L.D.; Lu, R.; Sharpe, S.; Keeley, F.W. Sequence variants of human tropoelastin affecting assembly, structural characteristics and functional properties of polymeric elastin in health and disease. Matrix Biol. 2019, 84, 68–80. [Google Scholar] [CrossRef]
- Blanchevoye, C.; Floquet, N.; Scandolera, A.; Baud, S.; Maurice, P.; Bocquet, O.; Blaise, S.; Ghoneim, C.; Cantarelli, B.; Delacoux, F.; et al. Interaction between the elastin peptide VGVAPG and human elastin binding protein. J. Biol. Chem. 2013, 288, 1317–1328. [Google Scholar] [CrossRef]
- Hinek, A.; Keeley, F.W.; Callahan, J. Recycling of the 67-kDa elastin binding protein in arterial myocytes is imperative for secretion of tropoelastin. Exp. Cell Res. 1995, 220, 312–324. [Google Scholar] [CrossRef]
- Cox, B.A.; Starcher, B.C.; Urry, D.W. Communication: Coacervation of tropoelastin results in fiber formation. J. Biol. Chem. 1974, 249, 997–998. [Google Scholar] [CrossRef]
- Volpin, D.; Pasquali-Ronchetti, I. The ultrastruct of high-temperature coacervates from elastin. J. Ultrastruct. Res. 1977, 61, 295–302. [Google Scholar] [CrossRef]
- Kozel, B.A.; Rongish, B.J.; Czirok, A.; Zach, J.; Little, C.D.; Davis, E.C.; Knutsen, R.H.; Wagenseil, J.E.; Levy, M.A.; Mecham, R.P. Elastic fiber formation: A dynamic view of extracellular matrix assembly using timer reporters. J. Cell. Physiol. 2006, 207, 87–96. [Google Scholar] [CrossRef]
- Papke, C.L.; Yanagisawa, H. Fibulin-4 and fibulin-5 in elastogenesis and beyond: Insights from mouse and human studies. Matrix Biol. 2014, 37, 142–149. [Google Scholar] [CrossRef]
- Yeo, G.C.; Keeley, F.W.; Weiss, A.S. Coacervation of tropoelastin. Adv. Colloid Interface Sci. 2011, 167, 94–103. [Google Scholar] [CrossRef]
- Cirulis, J.T.; Bellingham, C.M.; Davis, E.C.; Hubmacher, D.; Reinhardt, D.P.; Mecham, R.P.; Keeley, F.W. Fibrillins, fibulins, and matrix-associated glycoprotein modulate the kinetics and morphology of in vitro self-assembly of a recombinant elastin-like polypeptide. Biochemistry 2008, 47, 12601–12613. [Google Scholar] [CrossRef]
- Vrhovski, B.; Jensen, S.; Weiss, A.S. Coacervation characteristics of recombinant human tropoelastin. Eur. J. Biochem. 1997, 250, 92–98. [Google Scholar] [CrossRef]
- Toonkool, P.; Jensen, S.A.; Maxwell, A.L.; Weiss, A.S. Hydrophobic domains of human tropoelastin interact in a context-dependent manner. J. Biol. Chem. 2001, 276, 44575–44580. [Google Scholar] [CrossRef]
- Muiznieks, L.D.; Cirulis, J.T.; van der Horst, A.; Reinhardt, D.P.; Wuite, G.J.L.; Pomès, R.; Keeley, F.W. Modulated growth, stability and interactions of liquid-like coacervate assemblies of elastin. Matrix Biol. 2014, 36, 39–50. [Google Scholar] [CrossRef]
- Boraldi, F.; Annovi, G.; Tiozzo, R.; Sommer, P.; Quaglino, D. Comparison of ex vivo and in vitro human fibroblast ageing models. Mech. Ageing Dev. 2010, 131, 625–635. [Google Scholar] [CrossRef]
- Reiser, K.; McCormick, R.J.; Rucker, R.B. Enzymatic and nonenzymatic cross-linking of collagen and elastin. FASEB J. 1992, 6, 2439–2449. [Google Scholar] [CrossRef]
- Narayanan, A.S.; Page, R.C.; Kuzan, F.; Cooper, C.G. Elastin cross-linking in vitro. Studies on factors influencing the formation of desmosines by lysyl oxidase action on tropoelastin. Biochem. J. 1978, 173, 857–862. [Google Scholar] [CrossRef]
- Schmelzer, C.E.H.; Heinz, A.; Troilo, H.; Lockhart-Cairns, M.P.; Jowitt, T.A.; Marchand, M.F.; Bidault, L.; Bignon, M.; Hedtke, T.; Barret, A.; et al. Lysyl oxidase-like 2 (LOXL2)-mediated cross-linking of tropoelastin. FASEB J. 2019, 33, 5468–5481. [Google Scholar] [CrossRef]
- Umeda, H.; Aikawa, M.; Libby, P. Liberation of desmosine and isodesmosine as amino acids from insoluble elastin by elastolytic proteases. Biochem. Biophys. Res. Commun. 2011, 411, 281–286. [Google Scholar] [CrossRef]
- Stoilov, I.; Starcher, B.C.; Mecham, R.P.; Broekelmann, T.J. Measurement of elastin, collagen, and total protein levels in tissues. Methods Cell Biol. 2018, 143, 133–146. [Google Scholar] [CrossRef]
- Turino, G.M.; Ma, S.; Lin, Y.Y.; Cantor, J.O.; Luisetti, M. Matrix elastin: A promising biomarker for chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 184, 637–641. [Google Scholar] [CrossRef]
- Zhang, H.; Hu, W.; Ramirez, F. Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils. J. Cell Biol. 1995, 129, 1165–1176. [Google Scholar] [CrossRef]
- Kinsey, R.; Williamson, M.R.; Chaudhry, S.; Mellody, K.T.; McGovern, A.; Takahashi, S.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin-1 microfibril deposition is dependent on fibronectin assembly. J. Cell Sci. 2008, 121, 2696–2704. [Google Scholar] [CrossRef]
- Sabatier, L.; Chen, D.; Fagotto-Kaufmann, C.; Hubmacher, D.; McKee, M.D.; Annis, D.S.; Mosher, D.F.; Reinhardt, D.P. Fibrillin assembly requires fibronectin. Mol. Biol. Cell 2009, 20, 846–858. [Google Scholar] [CrossRef]
- Reinboth, B.; Hanssen, E.; Cleary, E.G.; Gibson, M.A. Molecular Interactions of Biglycan and Decorin with Elastic Fiber Components: Biglycan Forms a Ternary Complex with Tropoelastin and Microfibril-Associated Glycoprotein. J. Biol. Chem. 2002, 277, 3950–3957. [Google Scholar] [CrossRef]
- Sakai, L.Y.; Keene, D.R.; Glanville, R.W.; Bächinger, H.P. Purification and partial characterization of fibrillin, a cysteine-rich structural component of connective tissue microfibrils. J. Biol. Chem. 1991, 266, 14763–14770. [Google Scholar] [CrossRef]
- Reinhardt, D.P.; Keene, D.R.; Corson, G.M.; Pöschl, E.; Bächinger, H.P.; Gambee, J.E.; Sakai, L.Y. Fibrillin-1: Organization in microfibrils and structural properties. J. Mol. Biol. 1996, 258, 104–116. [Google Scholar] [CrossRef]
- Rifkin, D.B.; Rifkin, W.J.; Zilberberg, L. LTBPs in biology and medicine: LTBP diseases. Matrix Biol. 2018, 71–72, 90–99. [Google Scholar] [CrossRef]
- Pilecki, B.; Holm, A.T.; Schlosser, A.; Moeller, J.B.; Wohl, A.P.; Zuk, A.V.; Heumüller, S.E.; Wallis, R.; Moestrup, S.K.; Sengle, G.; et al. Characterization of Microfibrillar-associated Protein 4 (MFAP4) as a Tropoelastin- and Fibrillin-binding Protein Involved in Elastic Fiber Formation. J. Biol. Chem. 2016, 291, 1103–1114. [Google Scholar] [CrossRef]
- Lemaire, R.; Bayle, J.; Mecham, R.P.; Lafyatis, R. Microfibril-associated MAGP-2 stimulates elastic fiber assembly. J. Biol. Chem. 2007, 282, 800–808. [Google Scholar] [CrossRef]
- Kasamatsu, S.; Hachiya, A.; Fujimura, T.; Sriwiriyanont, P.; Haketa, K.; Visscher, M.O.; Kitzmiller, W.J.; Bello, A.; Kitahara, T.; Kobinger, G.P.; et al. Essential role of microfibrillar-associated protein 4 in human cutaneous homeostasis and in its photoprotection. Sci. Rep. 2011, 1, 164. [Google Scholar] [CrossRef]
- Choudhury, R.; McGovern, A.; Ridley, C.; Cain, S.A.; Baldwin, A.; Wang, M.-C.; Guo, C.; Mironov, A.; Drymoussi, Z.; Trump, D.; et al. Differential regulation of elastic fiber formation by fibulin-4 and -5. J. Biol. Chem. 2009, 284, 24553–24567. [Google Scholar] [CrossRef]
- Hubmacher, D.; Apte, S.S. ADAMTS proteins as modulators of microfibril formation and function. Matrix Biol. J. Int. Soc. Matrix Biol. 2015, 47, 34–43. [Google Scholar] [CrossRef]
- Bressan, G.M.; Daga-Gordini, D.; Colombatti, A.; Castellani, I.; Marigo, V.; Volpin, D. Emilin, a component of elastic fibers preferentially located at the elastin-microfibrils interface. J. Cell Biol. 1993, 121, 201–212. [Google Scholar] [CrossRef]
- Colombatti, A.; Doliana, R.; Bot, S.; Canton, A.; Mongiat, M.; Mungiguerra, G.; Paron-Cilli, S.; Spessotto, P. The EMILIN protein family. Matrix Biol. 2000, 19, 289–301. [Google Scholar] [CrossRef]
- Thomson, J.; Singh, M.; Eckersley, A.; Cain, S.A.; Sherratt, M.J.; Baldock, C. Fibrillin microfibrils and elastic fibre proteins: Functional interactions and extracellular regulation of growth factors. Semin. Cell Dev. Biol. 2019, 89, 109–117. [Google Scholar] [CrossRef]
- Godwin, A.R.F.; Singh, M.; Lockhart-Cairns, M.P.; Alanazi, Y.F.; Cain, S.A.; Baldock, C. The role of fibrillin and microfibril binding proteins in elastin and elastic fibre assembly. Matrix Biol. 2019, 84, 17–30. [Google Scholar] [CrossRef]
- Schmelzer, C.E.H.; Duca, L. Elastic fibers: Formation, function, and fate during aging and disease. FEBS J. 2021. [Google Scholar] [CrossRef]
- Heinz, A. Elastic fibers during aging and disease. Ageing Res. Rev. 2021, 66, 101255. [Google Scholar] [CrossRef]
- Jobin, P.G.; Butler, G.S.; Overall, C.M. New intracellular activities of matrix metalloproteinases shine in the moonlight. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 2043–2055. [Google Scholar] [CrossRef]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and Outs of Cathepsins: Physiological Function and Role in Disease Management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta BBA-Proteins Proteom. 2012, 1824, 68–88. [Google Scholar] [CrossRef]
- Schmelzer, C.E.H.; Getie, M.; Neubert, R.H.H. Mass spectrometric characterization of human skin elastin peptides produced by proteolytic digestion with pepsin and thermitase. J. Chromatogr. A 2005, 1083, 120–126. [Google Scholar] [CrossRef]
- Collins, J.F.; Fine, R. The enzymatic digestion of elastin at acidic pH. Biochim. Biophys. Acta 1981, 657, 295–303. [Google Scholar] [CrossRef]
- Loeven, W.A. Susceptibility of various kinds of elastin to elastolytic enzymes, trypsin and chymotrypsin. Clin. Chim. Acta 1970, 27, 521–533. [Google Scholar] [CrossRef]
- Getie, M.; Schmelzer, C.E.H.; Neubert, R.H.H. Characterization of peptides resulting from digestion of human skin elastin with elastase. Proteins 2005, 61, 649–657. [Google Scholar] [CrossRef]
- Kozel, B.A.; Wachi, H.; Davis, E.C.; Mecham, R.P. Domains in tropoelastin that mediate elastin deposition in vitro and in vivo. J. Biol. Chem. 2003, 278, 18491–18498. [Google Scholar] [CrossRef]
- Heinz, A.; Schräder, C.U.; Baud, S.; Keeley, F.W.; Mithieux, S.M.; Weiss, A.S.; Neubert, R.H.H.; Schmelzer, C.E.H. Molecular-level characterization of elastin-like constructs and human aortic elastin. Matrix Biol. 2014, 38, 12–21. [Google Scholar] [CrossRef]
- Soond, S.M.; Kozhevnikova, M.V.; Townsend, P.A.; Zamyatnin, A.A. Cysteine Cathepsin Protease Inhibition: An update on its Diagnostic, Prognostic and Therapeutic Potential in Cancer. Pharmaceuticals 2019, 12, 87. [Google Scholar] [CrossRef]
- Troeberg, L.; Nagase, H. Analysis of TIMP expression and activity. Methods Mol. Med. 2007, 135, 251–267. [Google Scholar] [CrossRef]
- Zani, M.-L.; Baranger, K.; Guyot, N.; Dallet-Choisy, S.; Moreau, T. Protease inhibitors derived from elafin and SLPI and engineered to have enhanced specificity towards neutrophil serine proteases. Protein Sci. 2009, 18, 579–594. [Google Scholar] [CrossRef]
- Guyot, N.; Butler, M.W.; McNally, P.; Weldon, S.; Greene, C.M.; Levine, R.L.; O’Neill, S.J.; Taggart, C.C.; McElvaney, N.G. Elafin, an elastase-specific inhibitor, is cleaved by its cognate enzyme neutrophil elastase in sputum from individuals with cystic fibrosis. J. Biol. Chem. 2008, 283, 32377–32385. [Google Scholar] [CrossRef]
- Tetley, T.D. New perspectives on basic mechanisms in lung disease. Proteinase imbalance: Its role in lung disease. Thorax 1993, 48, 560–565. [Google Scholar] [CrossRef]
- Shi, G.P.; Sukhova, G.K.; Grubb, A.; Ducharme, A.; Rhode, L.H.; Lee, R.T.; Ridker, P.M.; Libby, P.; Chapman, H.A. Cystatin C deficiency in human atherosclerosis and aortic aneurysms. J. Clin. Investig. 1999, 104, 1191–1197. [Google Scholar] [CrossRef]
- Yasuda, Y.; Li, Z.; Greenbaum, D.; Bogyo, M.; Weber, E.; Brömme, D. Cathepsin V, a novel and potent elastolytic activity expressed in activated macrophages. J. Biol. Chem. 2004, 279, 36761–36770. [Google Scholar] [CrossRef]
- Kirschner, R.; Hubmacher, D.; Iyengar, G.; Kaur, J.; Fagotto-Kaufmann, C.; Brömme, D.; Bartels, R.; Reinhardt, D.P. Classical and neonatal Marfan syndrome mutations in fibrillin-1 cause differential protease susceptibilities and protein function. J. Biol. Chem. 2011, 286, 32810–32823. [Google Scholar] [CrossRef]
- Mora Huertas, A.C.; Schmelzer, C.E.H.; Luise, C.; Sippl, W.; Pietzsch, M.; Hoehenwarter, W.; Heinz, A. Degradation of tropoelastin and skin elastin by neprilysin. Biochimie 2018, 146, 73–78. [Google Scholar] [CrossRef]
- Feygina, E.E.; Katrukha, A.G.; Semenov, A.G. Neutral Endopeptidase (Neprilysin) in Therapy and Diagnostics: Yin and Yang. Biochemistry 2019, 84, 1346–1358. [Google Scholar] [CrossRef]
- Murphy, G.; Cockett, M.I.; Ward, R.V.; Docherty, A.J. Matrix metalloproteinase degradation of elastin, type IV collagen and proteoglycan. A quantitative comparison of the activities of 95 kDa and 72 kDa gelatinases, stromelysins-1 and -2 and punctuated metalloproteinase (PUMP). Biochem. J. 1991, 277, 277–279. [Google Scholar] [CrossRef]
- Ashworth, J.L.; Murphy, G.; Rock, M.J.; Sherratt, M.J.; Shapiro, S.D.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin degradation by matrix metalloproteinases: Implications for connective tissue remodelling. Biochem. J. 1999, 340, 171–181. [Google Scholar] [CrossRef]
- Heinz, A.; Jung, M.C.; Duca, L.; Sippl, W.; Taddese, S.; Ihling, C.; Rusciani, A.; Jahreis, G.; Weiss, A.S.; Neubert, R.H.H.; et al. Degradation of tropoelastin by matrix metalloproteinases--cleavage site specificities and release of matrikines. FEBS J. 2010, 277, 1939–1956. [Google Scholar] [CrossRef]
- Katsuda, S.; Okada, Y.; Okada, Y.; Imai, K.; Nakanishi, I. Matrix metalloproteinase-9 (92-kd gelatinase/type IV collagenase equals gelatinase B) can degrade arterial elastin. Am. J. Pathol. 1994, 145, 1208–1218. [Google Scholar]
- Hindson, V.J.; Ashworth, J.L.; Rock, M.J.; Cunliffe, S.; Shuttleworth, C.A.; Kielty, C.M. Fibrillin degradation by matrix metalloproteinases: Identification of amino- and carboxy-terminal cleavage sites. FEBS Lett. 1999, 452, 195–198. [Google Scholar] [CrossRef]
- Shapiro, S.D.; Kobayashi, D.K.; Ley, T.J. Cloning and characterization of a unique elastolytic metalloproteinase produced by human alveolar macrophages. J. Biol. Chem. 1993, 268, 23824–23829. [Google Scholar] [CrossRef]
- Taddese, S.; Weiss, A.S.; Neubert, R.H.H.; Schmelzer, C.E.H. Mapping of macrophage elastase cleavage sites in insoluble human skin elastin. Matrix Biol. 2008, 27, 420–428. [Google Scholar] [CrossRef]
- Xiong, W.; Knispel, R.; MacTaggart, J.; Greiner, T.C.; Weiss, S.J.; Baxter, B.T. Membrane-type 1 matrix metalloproteinase regulates macrophage-dependent elastolytic activity and aneurysm formation in vivo. J. Biol. Chem. 2009, 284, 1765–1771. [Google Scholar] [CrossRef]
- Miekus, N.; Luise, C.; Sippl, W.; Baczek, T.; Schmelzer, C.E.H.; Heinz, A. MMP-14 degrades tropoelastin and elastin. Biochimie 2019, 165, 32–39. [Google Scholar] [CrossRef]
- Joshi, R.; Heinz, A.; Fan, Q.; Guo, S.; Monia, B.; Schmelzer, C.E.H.; Weiss, A.S.; Batie, M.; Parameshwaran, H.; Varisco, B.M. Role for Cela1 in Postnatal Lung Remodeling and Alpha-1 Antitrypsin–Deficient Emphysema. Am. J. Respir. Cell Mol. Biol. 2018, 59, 167–178. [Google Scholar] [CrossRef]
- Boudier, C.; Godeau, G.; Hornebeck, W.; Robert, L.; Bieth, J.G. The elastolytic activity of cathepsin G: An ex vivo study with dermal elastin. Am. J. Respir. Cell Mol. Biol. 1991, 4, 497–503. [Google Scholar] [CrossRef]
- Schmelzer, C.E.H.; Jung, M.C.; Wohlrab, J.; Neubert, R.H.H.; Heinz, A. Does human leukocyte elastase degrade intact skin elastin? FEBS J. 2012, 279, 4191–4200. [Google Scholar] [CrossRef]
- Kielty, C.M.; Woolley, D.E.; Whittaker, S.P.; Shuttleworth, C.A. Catabolism of intact fibrillin microfibrils by neutrophil elastase, chymotrypsin and trypsin. FEBS Lett. 1994, 351, 85–89. [Google Scholar] [CrossRef]
- Kao, R.C.; Wehner, N.G.; Skubitz, K.M.; Gray, B.H.; Hoidal, J.R. Proteinase A distinct human polymorphonuclear leukocyte proteinase that produces emphysema in hamsters. J. Clin. Investig. 1988, 82, 1963–1973. [Google Scholar] [CrossRef]
- Hauck, M.; Seres, I.; Kiss, I.; Saulnier, J.; Mohacsi, A.; Wallach, J.; Fulop, T. Effects of synthesized elastin peptides on human leukocytes. Biochem. Mol. Biol. Int. 1995, 37, 45–55. [Google Scholar]
- Péterszegi, G.; Mandet, C.; Texier, S.; Robert, L.; Bruneval, P. Lymphocytes in human atherosclerotic plaque exhibit the elastin-laminin receptor: Potential role in atherogenesis. Atherosclerosis 1997, 135, 103–107. [Google Scholar] [CrossRef]
- Nowak, D.; Główczyńska, I.; Piasecka, G. Chemotactic activity of elastin-derived peptides for human polymorphonuclear leukocytes and their effect on hydrogen peroxide and myeloperoxidase release. Arch. Immunol. Ther. Exp. 1989, 37, 741–748. [Google Scholar]
- Hance, K.A.; Tataria, M.; Ziporin, S.J.; Lee, J.K.; Thompson, R.W. Monocyte chemotactic activity in human abdominal aortic aneurysms: Role of elastin degradation peptides and the 67-kD cell surface elastin receptor. J. Vasc. Surg. 2002, 35, 254–261. [Google Scholar] [CrossRef]
- Antonicelli, F.; Bellon, G.; Debelle, L.; Hornebeck, W. Elastin-elastases and inflamm-aging. Curr. Top. Dev. Biol. 2007, 79, 99–155. [Google Scholar] [CrossRef]
- Houghton, A.M.; Quintero, P.A.; Perkins, D.L.; Kobayashi, D.K.; Kelley, D.G.; Marconcini, L.A.; Mecham, R.P.; Senior, R.M.; Shapiro, S.D. Elastin fragments drive disease progression in a murine model of emphysema. J. Clin. Investig. 2006, 116, 753–759. [Google Scholar] [CrossRef]
- Guo, G.; Gehle, P.; Doelken, S.; Martin-Ventura, J.L.; von Kodolitsch, Y.; Hetzer, R.; Robinson, P.N. Induction of macrophage chemotaxis by aortic extracts from patients with Marfan syndrome is related to elastin binding protein. PLoS ONE 2011, 6, e20138. [Google Scholar] [CrossRef]
- Simionescu, A.; Philips, K.; Vyavahare, N. Elastin-derived peptides and TGF-beta1 induce osteogenic responses in smooth muscle cells. Biochem. Biophys. Res. Commun. 2005, 334, 524–532. [Google Scholar] [CrossRef]
- Tamburro, A.M.; Pepe, A.; Bochicchio, B.; Quaglino, D.; Ronchetti, I.P. Supramolecular amyloid-like assembly of the polypeptide sequence coded by exon 30 of human tropoelastin. J. Biol. Chem. 2005, 280, 2682–2690. [Google Scholar] [CrossRef] [PubMed]
- Bochicchio, B.; Pepe, A.; Flamia, R.; Lorusso, M.; Tamburro, A.M. Investigating the amyloidogenic nanostructured sequences of elastin: Sequence encoded by exon 28 of human tropoelastin gene. Biomacromolecules 2007, 8, 3478–3486. [Google Scholar] [CrossRef] [PubMed]
- Boraldi, F.; Moscarelli, P.; Bochicchio, B.; Pepe, A.; Salvi, A.M.; Quaglino, D. Heparan sulfates facilitate harmless amyloidogenic fibril formation interacting with elastin-like peptides. Sci. Rep. 2018, 8, 3115. [Google Scholar] [CrossRef]
- Moscarelli, P.; Boraldi, F.; Bochicchio, B.; Pepe, A.; Salvi, A.M.; Quaglino, D. Structural characterization and biological properties of the amyloidogenic elastin-like peptide (VGGVG). Matrix Biol. 2014, 36, 15–27. [Google Scholar] [CrossRef]
- Le Page, A.; Khalil, A.; Vermette, P.; Frost, E.H.; Larbi, A.; Witkowski, J.M.; Fulop, T. The role of elastin-derived peptides in human physiology and diseases. Matrix Biol. 2019, 84, 81–96. [Google Scholar] [CrossRef]
- Pierre, A.; Lemaire, F.; Meghraoui-Kheddar, A.; Audonnet, S.; Héry-Huynh, S.; Le Naour, R. Impact of aging on inflammatory and immune responses during elastin peptide-induced murine emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol. 2019, 316, L608–L620. [Google Scholar] [CrossRef]
- Janssen, R.; Vermeer, C. Vitamin K deficit and elastolysis theory in pulmonary elasto-degenerative diseases. Med. Hypotheses 2017, 108, 38–41. [Google Scholar] [CrossRef]
- Starcher, B.C.; Urry, D.W. Elastin coacervate as a matrix for calcification. Biochem. Biophys. Res. Commun. 1973, 53, 210–216. [Google Scholar] [CrossRef]
- Boraldi, F.; Moscarelli, P.; Lofaro, F.D.; Sabia, C.; Quaglino, D. The mineralization process of insoluble elastin fibrillar structures: Ionic environment vs degradation. Int. J. Biol. Macromol. 2020, 149, 693–706. [Google Scholar] [CrossRef]
- Boraldi, F.; Lofaro, F.D.; Losi, L.; Quaglino, D. Dermal Alterations in Clinically Unaffected Skin of Pseudoxanthoma elasticum Patients. J. Clin. Med. 2021, 10, 500. [Google Scholar] [CrossRef] [PubMed]
- Daamen, W.F.; Quaglino, D. Signaling pathways in elastic tissues. Cell. Signal. 2019, 63, 109364. [Google Scholar] [CrossRef] [PubMed]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Prado, E.; Simbaña-Rivera, K.; Gómez-Barreno, L.; Rubio-Neira, M.; Guaman, L.P.; Kyriakidis, N.C.; Muslin, C.; Jaramillo, A.M.G.; Barba-Ostria, C.; Cevallos-Robalino, D.; et al. Clinical, molecular, and epidemiological characterization of the SARS-CoV-2 virus and the Coronavirus Disease 2019 (COVID-19), a comprehensive literature review. Diagn. Microbiol. Infect. Dis. 2020, 98, 115094. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, T.; Pigazzini, S.; Degenhardt, F.; Cordioli, M.; Butler-Laporte, G.; Maya-Miles, D.; Bujanda, L.; Bouysran, Y.; Niemi, M.E.; Palom, A.; et al. Age-dependent impact of the major common genetic risk factor for COVID-19 on severity and mortality. J. Clin. Investig. 2021, 131, e152386. [Google Scholar] [CrossRef] [PubMed]
- Yuki, K.; Fujiogi, M.; Koutsogiannaki, S. COVID-19 pathophysiology: A review. Clin. Immunol. 2020, 215, 108427. [Google Scholar] [CrossRef]
- Larici, A.R.; Cicchetti, G.; Marano, R.; Merlino, B.; Elia, L.; Calandriello, L.; Del Ciello, A.; Farchione, A.; Savino, G.; Infante, A.; et al. Multimodality imaging of COVID-19 pneumonia: From diagnosis to follow-up. A comprehensive review. Eur. J. Radiol. 2020, 131, 109217. [Google Scholar] [CrossRef]
- Osuchowski, M.F.; Winkler, M.S.; Skirecki, T.; Cajander, S.; Shankar-Hari, M.; Lachmann, G.; Monneret, G.; Venet, F.; Bauer, M.; Brunkhorst, F.M.; et al. The COVID-19 puzzle: Deciphering pathophysiology and phenotypes of a new disease entity. Lancet Respir. Med. 2021, 9, 622–642. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.W.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020, 27, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.G.; Javed, A.; Akter, S.; Saha, S. SARS-CoV-2 host diversity: An update of natural infections and experimental evidence. J. Microbiol. Immunol. Infect. 2021, 54, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Takeda, M. Proteolytic activation of SARS-CoV-2 spike protein. Microbiol. Immunol. 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- De Biasi, S.; Meschiari, M.; Gibellini, L.; Bellinazzi, C.; Borella, R.; Fidanza, L.; Gozzi, L.; Iannone, A.; Lo Tartaro, D.; Mattioli, M.; et al. Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat. Commun. 2020, 11, 3434. [Google Scholar] [CrossRef] [PubMed]
- De Biasi, S.; Lo Tartaro, D.; Meschiari, M.; Gibellini, L.; Bellinazzi, C.; Borella, R.; Fidanza, L.; Mattioli, M.; Paolini, A.; Gozzi, L.; et al. Expansion of plasmablasts and loss of memory B cells in peripheral blood from COVID-19 patients with pneumonia. Eur. J. Immunol. 2020, 50, 1283–1294. [Google Scholar] [CrossRef] [PubMed]
- Gangaev, A.; Ketelaars, S.L.C.; Isaeva, O.I.; Patiwael, S.; Dopler, A.; Hoefakker, K.; De Biasi, S.; Gibellini, L.; Mussini, C.; Guaraldi, G.; et al. Identification and characterization of a SARS-CoV-2 specific CD8+ T cell response with immunodominant features. Nat. Commun. 2021, 12, 2593. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; De Biasi, S.; Paolini, A.; Borella, R.; Boraldi, F.; Mattioli, M.; Lo Tartaro, D.; Fidanza, L.; Caro-Maldonado, A.; Meschiari, M.; et al. Altered bioenergetics and mitochondrial dysfunction of monocytes in patients with COVID-19 pneumonia. EMBO Mol. Med. 2020, 12, e13001. [Google Scholar] [CrossRef]
- Effah, C.Y.; Drokow, E.K.; Agboyibor, C.; Ding, L.; He, S.; Liu, S.; Akorli, S.Y.; Nuamah, E.; Sun, T.; Zhou, X.; et al. Neutrophil-Dependent Immunity During Pulmonary Infections and Inflammations. Front. Immunol. 2021, 12, 689866. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Hogg, J.C.; Walker, B.A. Polymorphonuclear leucocyte traffic in lung inflammation. Thorax 1995, 50, 819–820. [Google Scholar] [CrossRef]
- Lien, D.C.; Wagner, W.W.; Capen, R.L.; Haslett, C.; Hanson, W.L.; Hofmeister, S.E.; Henson, P.M.; Worthen, G.S. Physiological neutrophil sequestration in the lung: Visual evidence for localization in capillaries. J. Appl. Physiol. 1987, 62, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Tam, F.W.; Clague, J.; Dixon, C.M.; Stuttle, A.W.; Henderson, B.L.; Peters, A.M.; Lavender, J.P.; Ind, P.W. Inhaled platelet-activating factor causes pulmonary neutrophil sequestration in normal humans. Am. Rev. Respir. Dis. 1992, 146, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Thierry, A.R.; Roch, B. Neutrophil Extracellular Traps and By-Products Play a Key Role in COVID-19: Pathogenesis, Risk Factors, and Therapy. J. Clin. Med. 2020, 9, 2942. [Google Scholar] [CrossRef] [PubMed]
- Borella, R.; De Biasi, S.; Paolini, A.; Boraldi, F.; Tartaro, D.L.; Mattioli, M.; Fidanza, L.; Neroni, A.; Caro-Maldonado, A.; Meschiari, M.; et al. Metabolic reprograming shapes neutrophil functions in severe COVID-19. Eur. J. Immunol. 2021. [Google Scholar] [CrossRef]
- Barbosa da Cruz, D.; Helms, J.; Aquino, L.R.; Stiel, L.; Cougourdan, L.; Broussard, C.; Chafey, P.; Riès-Kautt, M.; Meziani, F.; Toti, F.; et al. DNA-bound elastase of neutrophil extracellular traps degrades plasminogen, reduces plasmin formation, and decreases fibrinolysis: Proof of concept in septic shock plasma. FASEB J. 2019, 33, 14270–14280. [Google Scholar] [CrossRef]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Vasarmidi, E.; Tsitoura, E.; Spandidos, D.A.; Tzanakis, N.; Antoniou, K.M. Pulmonary fibrosis in the aftermath of the COVID-19 era (Review). Exp. Ther. Med. 2020, 20, 2557–2560. [Google Scholar] [CrossRef]
- Yang, Y.; Ding, L.; Bao, T.; Li, Y.; Ma, J.; Li, Q.; Gao, Z.; Song, S.; Wang, J.; Zhao, J.; et al. Network Pharmacology and Experimental Assessment to Explore the Pharmacological Mechanism of Qimai Feiluoping Decoction Against Pulmonary Fibrosis. Front. Pharmacol. 2021, 12, 770197. [Google Scholar] [CrossRef]
- Vaz de Paula, C.B.; Nagashima, S.; Liberalesso, V.; Collete, M.; da Silva, F.P.G.; Oricil, A.G.G.; Barbosa, G.S.; da Silva, G.V.C.; Wiedmer, D.B.; da Silva Dezidério, F.; et al. COVID-19: Immunohistochemical Analysis of TGF-β Signaling Pathways in Pulmonary Fibrosis. Int. J. Mol. Sci. 2021, 23, 168. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, Z.; Li, J.-W.; Tan, K.; Yang, W.; Zhao, H.; Wang, G.-Q. Discharge may not be the end of treatment: Pay attention to pulmonary fibrosis caused by severe COVID-19. J. Med Virol. 2021, 93, 1378–1386. [Google Scholar] [CrossRef]
- Cha, C.; Baek, G. Symptoms and management of long COVID: A scoping review. J. Clin. Nurs. 2021. [Google Scholar] [CrossRef] [PubMed]
- King, C.S.; Mannem, H.; Kukreja, J.; Aryal, S.; Tang, D.; Singer, J.P.; Bharat, A.; Behr, J.; Nathan, S.D. Lung Transplantation for Patients With COVID-19. Chest 2022, 161, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Writing Committee for the COMEBAC Study Group; Morin, L.; Savale, L.; Pham, T.; Colle, R.; Figueiredo, S.; Harrois, A.; Gasnier, M.; Lecoq, A.-L.; Meyrignac, O.; et al. Four-Month Clinical Status of a Cohort of Patients After Hospitalization for COVID-19. JAMA 2021, 325, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Caruso, D.; Guido, G.; Zerunian, M.; Polidori, T.; Lucertini, E.; Pucciarelli, F.; Polici, M.; Rucci, C.; Bracci, B.; Nicolai, M.; et al. Post-Acute Sequelae of COVID-19 Pneumonia: Six-month Chest CT Follow-up. Radiology 2021, 301, E396–E405. [Google Scholar] [CrossRef]
- Leng, L.; Cao, R.; Ma, J.; Mou, D.; Zhu, Y.; Li, W.; Lv, L.; Gao, D.; Zhang, S.; Gong, F.; et al. Pathological features of COVID-19-associated lung injury: A preliminary proteomics report based on clinical samples. Signal Transduct. Target. Ther. 2020, 5, 240. [Google Scholar] [CrossRef]
- Cîrstea, A.E.; Buzulică, R.L.; Pirici, D.; Ceauşu, M.C.; Iman, R.V.; Gheorghe, O.M.; Neamţu, S.D.; Stanca, L.; Ene, R.; Kumar-Singh, S.; et al. Histopathological findings in the advanced natural evolution of the SARS-CoV-2 infection. Rom. J. Morphol. Embryol. 2020, 61, 209–218. [Google Scholar] [CrossRef]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807–815. [Google Scholar] [CrossRef]
- Tanni, S.E.; Fabro, A.T.; de Albuquerque, A.; Ferreira, E.V.M.; Verrastro, C.G.Y.; Sawamura, M.V.Y.; Ribeiro, S.M.; Baldi, B.G. Pulmonary fibrosis secondary to COVID-19: A narrative review. Expert Rev. Respir. Med. 2021, 15, 791–803. [Google Scholar] [CrossRef]
- Falleni, M.; Tosi, D.; Savi, F.; Chiumello, D.; Bulfamante, G. Endothelial-Mesenchymal Transition in COVID-19 lung lesions. Pathol. Res. Pract. 2021, 221, 153419. [Google Scholar] [CrossRef]
- Pandolfi, L.; Bozzini, S.; Frangipane, V.; Percivalle, E.; De Luigi, A.; Violatto, M.B.; Lopez, G.; Gabanti, E.; Carsana, L.; D’Amato, M.; et al. Neutrophil Extracellular Traps Induce the Epithelial-Mesenchymal Transition: Implications in Post-COVID-19 Fibrosis. Front. Immunol. 2021, 12, 663303. [Google Scholar] [CrossRef]
- D’Urso, M.; Kurniawan, N.A. Mechanical and Physical Regulation of Fibroblast-Myofibroblast Transition: From Cellular Mechanoresponse to Tissue Pathology. Front. Bioeng. Biotechnol. 2020, 8, 609653. [Google Scholar] [CrossRef] [PubMed]
- Delpino, M.V.; Quarleri, J. SARS-CoV-2 Pathogenesis: Imbalance in the Renin-Angiotensin System Favors Lung Fibrosis. Front. Cell. Infect. Microbiol. 2020, 10, 340. [Google Scholar] [CrossRef] [PubMed]
- Ojo, A.S.; Balogun, S.A.; Williams, O.T.; Ojo, O.S. Pulmonary Fibrosis in COVID-19 Survivors: Predictive Factors and Risk Reduction Strategies. Pulm. Med. 2020, 2020, 6175964. [Google Scholar] [CrossRef] [PubMed]
- Compagnone, N.; Palumbo, D.; Cremona, G.; Vitali, G.; De Lorenzo, R.; Calvi, M.R.; Del Prete, A.; Baiardo Redaelli, M.; Calamarà, S.; Belletti, A.; et al. Residual lung damage following ARDS in COVID-19 ICU survivors. Acta Anaesthesiol. Scand. 2021. [Google Scholar] [CrossRef]
- Toshima, M.; Ohtani, Y.; Ohtani, O. Three-dimensional architecture of elastin and collagen fiber networks in the human and rat lung. Arch. Histol. Cytol. 2004, 67, 31–40. [Google Scholar] [CrossRef]
- Naeije, R.; Caravita, S. Phenotyping long COVID. Eur. Respir. J. 2021, 58, 2101763. [Google Scholar] [CrossRef]
- Akgun, E.; Tuzuner, M.B.; Sahin, B.; Kilercik, M.; Kulah, C.; Cakiroglu, H.N.; Serteser, M.; Unsal, I.; Baykal, A.T. Proteins associated with neutrophil degranulation are upregulated in nasopharyngeal swabs from SARS-CoV-2 patients. PLoS ONE 2020, 15, e0240012. [Google Scholar] [CrossRef]
- Vargas-Alarcón, G.; Posadas-Sánchez, R.; Ramírez-Bello, J. Variability in genes related to SARS-CoV-2 entry into host cells (ACE2, TMPRSS2, TMPRSS11A, ELANE, and CTSL) and its potential use in association studies. Life Sci. 2020, 260, 118313. [Google Scholar] [CrossRef]
- Zhou, B.; Thao, T.T.N.; Hoffmann, D.; Taddeo, A.; Ebert, N.; Labroussaa, F.; Pohlmann, A.; King, J.; Steiner, S.; Kelly, J.N.; et al. SARS-CoV-2 spike D614G change enhances replication and transmission. Nature 2021, 592, 122–127. [Google Scholar] [CrossRef]
- Pokhrel, S.; Kraemer, B.R.; Lee, L.; Samardzic, K.; Mochly-Rosen, D. Increased elastase sensitivity and decreased intramolecular interactions in the more transmissible 501Y.V1 and 501Y.V2 SARS-CoV-2 variants’ spike protein-an in silico analysis. PLoS ONE 2021, 16, e0251426. [Google Scholar] [CrossRef]
- Bhattacharyya, C.; Das, C.; Ghosh, A.; Singh, A.K.; Mukherjee, S.; Majumder, P.P.; Basu, A.; Biswas, N.K. SARS-CoV-2 mutation 614G creates an elastase cleavage site enhancing its spread in high AAT-deficient regions. Infect. Genet. Evol. 2021, 90, 104760. [Google Scholar] [CrossRef] [PubMed]
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185. [Google Scholar] [CrossRef]
- Burgener, S.S.; Schroder, K. Neutrophil Extracellular Traps in Host Defense. Cold Spring Harb. Perspect. Biol. 2020, 12, a037028. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Aziz, M.; Wang, P. The vitals of NETs. J. Leukoc. Biol. 2021, 110, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Abboud, R.T.; Wallace, A.M.; English, J.C.; Coxson, H.O.; Finley, R.J.; Shumansky, K.; Paré, P.D.; Sandford, A.J. Alveolar macrophage proteinase/antiproteinase expression in lung function and emphysema. Eur. Respir. J. 2014, 43, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Sagel, S.D.; Wagner, B.D.; Anthony, M.M.; Emmett, P.; Zemanick, E.T. Sputum biomarkers of inflammation and lung function decline in children with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2012, 186, 857–865. [Google Scholar] [CrossRef] [PubMed]
- Heinz, A. Elastases and elastokines: Elastin degradation and its significance in health and disease. Crit. Rev. Biochem. Mol. Biol. 2020, 55, 252–273. [Google Scholar] [CrossRef]
- Visser, M.P.J.; Dofferhoff, A.S.M.; van den Ouweland, J.M.W.; van Daal, H.; Kramers, C.; Schurgers, L.J.; Janssen, R.; Walk, J. Effects of Vitamin D and K on Interleukin-6 in COVID-19. Front. Nutr. 2022, 8, 1282. [Google Scholar] [CrossRef]
- Turino, G.M. The lung parenchyma--a dynamic matrix. J. Burns Amberson lecture. Am. Rev. Respir. Dis. 1985, 132, 1324–1334. [Google Scholar] [CrossRef]
- Gregory, A.D.; Kliment, C.R.; Metz, H.E.; Kim, K.-H.; Kargl, J.; Agostini, B.A.; Crum, L.T.; Oczypok, E.A.; Oury, T.A.; Houghton, A.M. Neutrophil elastase promotes myofibroblast differentiation in lung fibrosis. J. Leukoc. Biol. 2015, 98, 143–152. [Google Scholar] [CrossRef]
- Zhou, L.; Gao, R.; Hong, H.; Li, X.; Yang, J.; Shen, W.; Wang, Z.; Yang, J. Emodin inhibiting neutrophil elastase-induced epithelial-mesenchymal transition through Notch1 signalling in alveolar epithelial cells. J. Cell. Mol. Med. 2020, 24, 11998–12007. [Google Scholar] [CrossRef] [PubMed]
- Noseda, M.; Fu, Y.; Niessen, K.; Wong, F.; Chang, L.; McLean, G.; Karsan, A. Smooth Muscle alpha-actin is a direct target of Notch/CSL. Circ. Res. 2006, 98, 1468–1470. [Google Scholar] [CrossRef] [PubMed]
- De, D.; Pawar, N.; Gupta, A.N. Glucose-induced structural changes and anomalous diffusion of elastin. Colloids Surf. B Biointerfaces 2020, 188, 110776. [Google Scholar] [CrossRef] [PubMed]
- Silverstein, M.C.; Bilici, K.; Morgan, S.W.; Wang, Y.; Zhang, Y.; Boutis, G.S. 13C, 2h NMR studies of structural and dynamical modifications of glucose-exposed porcine aortic elastin. Biophys. J. 2015, 108, 1758–1772. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, Y.; Hahn, J.; Zhang, Y. Mechanical Properties of Arterial Elastin With Water Loss. J. Biomech. Eng. 2018, 140, 041012. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Litvinova, M.; Liang, Y.; Wang, Y.; Wang, W.; Zhao, S.; Wu, Q.; Merler, S.; Viboud, C.; Vespignani, A.; et al. Changes in contact patterns shape the dynamics of the COVID-19 outbreak in China. Science 2020, 368, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Parisi, A.; Brand, S.P.C.; Hilton, J.; Aziza, R.; Keeling, M.J.; Nokes, D.J. Spatially resolved simulations of the spread of COVID-19 in three European countries. PLoS Comput. Biol. 2021, 17, e1009090. [Google Scholar] [CrossRef]
- Hilton, J.; Keeling, M.J. Estimation of country-level basic reproductive ratios for novel Coronavirus (SARS-CoV-2/COVID-19) using synthetic contact matrices. PLoS Comput. Biol. 2020, 16, e1008031. [Google Scholar] [CrossRef]
- Rahman, A.; Kuddus, M.A. Modelling the Transmission Dynamics of COVID-19 in Six High-Burden Countries. BioMed Res. Int. 2021, 2021, 5089184. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Sellegounder, D.; Zafari, P.; Rajabinejad, M.; Taghadosi, M.; Kapahi, P. Advanced glycation end products (AGEs) and its receptor, RAGE, modulate age-dependent COVID-19 morbidity and mortality. A review and hypothesis. Int. Immunopharmacol. 2021, 98, 107806. [Google Scholar] [CrossRef] [PubMed]
- Kehribar, D.; Cihangiroglu, M.; Sehmen, E.; Avci, B.; Capraz, A.; Yildirim Bilgin, A.; Gunaydin, C.; Ozgen, M. The receptor for advanced glycation end product (RAGE) pathway in COVID-19. Biomarkers 2021, 26, 114–118. [Google Scholar] [CrossRef]
- Rumende, C.M.; Susanto, E.C.; Sitorus, T.P. The Management of Pulmonary Fibrosis in COVID-19. Acta Med. Indones. 2021, 53, 233–241. [Google Scholar] [PubMed]
- Chiappalupi, S.; Salvadori, L.; Vukasinovic, A.; Donato, R.; Sorci, G.; Riuzzi, F. Targeting RAGE to prevent SARS-CoV-2-mediated multiple organ failure: Hypotheses and perspectives. Life Sci. 2021, 272, 119251. [Google Scholar] [CrossRef] [PubMed]
- Deslee, G.; Woods, J.C.; Moore, C.M.; Liu, L.; Conradi, S.H.; Milne, M.; Gierada, D.S.; Pierce, J.; Patterson, A.; Lewit, R.A.; et al. Elastin expression in very severe human COPD. Eur. Respir. J. 2009, 34, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Krettek, A.; Sukhova, G.K.; Libby, P. Elastogenesis in human arterial disease: A role for macrophages in disordered elastin synthesis. Art. Thromb. Vasc. Biol. 2003, 23, 582–587. [Google Scholar] [CrossRef]
- Cantor, J.O.; Shteyngart, B.; Cerreta, J.M.; Ma, S.; Turino, G.M. Synergistic effect of hydrogen peroxide and elastase on elastic fiber injury in vitro. Exp. Biol. Med. 2006, 231, 107–111. [Google Scholar] [CrossRef]
- Akhtar, K.; Broekelmann, T.J.; Miao, M.; Keeley, F.W.; Starcher, B.C.; Pierce, R.A.; Mecham, R.P.; Adair-Kirk, T.L. Oxidative and nitrosative modifications of tropoelastin prevent elastic fiber assembly in vitro. J. Biol. Chem. 2010, 285, 37396–37404. [Google Scholar] [CrossRef]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of Oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A Review. Protein J. 2020, 39, 644–656. [Google Scholar] [CrossRef]
- van Eijk, L.E.; Tami, A.; Hillebrands, J.-L.; den Dunnen, W.F.A.; de Borst, M.H.; van der Voort, P.H.J.; Bulthuis, M.L.C.; Veloo, A.C.M.; Wold, K.I.; Vincenti González, M.F.; et al. Mild Coronavirus Disease 2019 (COVID-19) Is Marked by Systemic Oxidative Stress: A Pilot Study. Antioxidants 2021, 10, 2022. [Google Scholar] [CrossRef]
- Guéant, J.-L.; Guéant-Rodriguez, R.-M.; Fromonot, J.; Oussalah, A.; Louis, H.; Chery, C.; Gette, M.; Gleye, S.; Callet, J.; Raso, J.; et al. Elastase and exacerbation of neutrophil innate immunity are involved in multi-visceral manifestations of COVID-19. Allergy 2021, 76, 1846–1858. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Arunachalam, G.; Hwang, J.-W.; Chung, S.; Sundar, I.K.; Kinnula, V.L.; Crapo, J.D.; Rahman, I. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc. Natl. Acad. Sci. USA 2010, 107, 15571–15576. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, N.H.; Tran, G.-B.; Nguyen, C.T. Anti-oxidative effects of superoxide dismutase 3 on inflammatory diseases. J. Mol. Med. 2020, 98, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Rodgman, A.; Perfetti, T.A. The Chemical Components of Tobacco and Tobacco Smoke, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2013; ISBN 978-1-4665-1548-2. [Google Scholar]
- Dietrich, M.; Block, G.; Norkus, E.P.; Hudes, M.; Traber, M.G.; Cross, C.E.; Packer, L. Smoking and exposure to environmental tobacco smoke decrease some plasma antioxidants and increase gamma-tocopherol in vivo after adjustment for dietary antioxidant intakes. Am. J. Clin. Nutr. 2003, 77, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Just, M.; Ribera, M.; Monsó, E.; Lorenzo, J.C.; Ferrándiz, C. Effect of smoking on skin elastic fibres: Morphometric and immunohistochemical analysis. Br. J. Dermatol. 2007, 156, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Blue, M.L.; Janoff, A. Possible mechanisms of emphysema in cigarette smokers. Release of elastase from human polymorphonuclear leukocytes by cigarette smoke condensate in vitro. Am. Rev. Respir. Dis. 1978, 117, 317–325. [Google Scholar] [CrossRef]
- Beatty, K.; Matheson, N.; Travis, J. Kinetic and chemical evidence for the inability of oxidized alpha 1-proteinase inhibitor to protect lung elastin from elastolytic degradation. Hoppe. Seylers Z. Physiol. Chem. 1984, 365, 731–736. [Google Scholar] [CrossRef]
- Francès, C.; Boisnic, S.; Hartmann, D.J.; Dautzenberg, B.; Branchet, M.C.; Charpentier, Y.L.; Robert, L. Changes in the elastic tissue of the non-sun-exposed skin of cigarette smokers. Br. J. Dermatol. 1991, 125, 43–47. [Google Scholar] [CrossRef]
- Kuperman, A.S.; Riker, J.B. The variable effect of smoking on pulmonary function. Chest 1973, 63, 655–660. [Google Scholar] [CrossRef]
- Shifren, A.; Durmowicz, A.G.; Knutsen, R.H.; Hirano, E.; Mecham, R.P. Elastin protein levels are a vital modifier affecting normal lung development and susceptibility to emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L778–L787. [Google Scholar] [CrossRef]
- Nicholl, I.D.; Bucala, R. Advanced glycation endproducts and cigarette smoking. Cell. Mol. Biol. 1998, 44, 1025–1033. [Google Scholar] [PubMed]
- Iles, K.E.; Dickinson, D.A.; Wigley, A.F.; Welty, N.E.; Blank, V.; Forman, H.J. HNE increases HO-1 through activation of the ERK pathway in pulmonary epithelial cells. Free Radic. Biol. Med. 2005, 39, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Kroening, P.R.; Barnes, T.W.; Pease, L.; Limper, A.; Kita, H.; Vassallo, R. Cigarette smoke-induced oxidative stress suppresses generation of dendritic cell IL-12 and IL-23 through ERK-dependent pathways. J. Immunol. 2008, 181, 1536–1547. [Google Scholar] [CrossRef] [PubMed]
- Byun, K.; Yoo, Y.; Son, M.; Lee, J.; Jeong, G.-B.; Park, Y.M.; Salekdeh, G.H.; Lee, B. Advanced glycation end-products produced systemically and by macrophages: A common contributor to inflammation and degenerative diseases. Pharmacol. Ther. 2017, 177, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Hippisley-Cox, J.; Young, D.; Coupland, C.; Channon, K.M.; Tan, P.S.; Harrison, D.A.; Rowan, K.; Aveyard, P.; Pavord, I.D.; Watkinson, P.J. Risk of severe COVID-19 disease with ACE inhibitors and angiotensin receptor blockers: Cohort study including 8.3 million people. Heart 2020, 106, 1503–1511. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.S.; Siddiqi, T.J.; Khan, M.S.; Patel, U.K.; Shahid, I.; Ahmed, J.; Kalra, A.; Michos, E.D. Is there a smoker’s paradox in COVID-19? BMJ Evid. Based Med. 2021, 26, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Besaratinia, A. COVID-19: A pandemic converged with global tobacco epidemic and widespread vaping-state of the evidence. Carcinogenesis 2021, 42, 1009–1022. [Google Scholar] [CrossRef]
- Smith, J.C.; Sausville, E.L.; Girish, V.; Yuan, M.L.; Vasudevan, A.; John, K.M.; Sheltzer, J.M. Cigarette Smoke Exposure and Inflammatory Signaling Increase the Expression of the SARS-CoV-2 Receptor ACE2 in the Respiratory Tract. Dev. Cell 2020, 53, 514–529.e3. [Google Scholar] [CrossRef]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef]
- Clift, A.K.; von Ende, A.; Tan, P.S.; Sallis, H.M.; Lindson, N.; Coupland, C.A.C.; Munafò, M.R.; Aveyard, P.; Hippisley-Cox, J.; Hopewell, J.C. Smoking and COVID-19 outcomes: An observational and Mendelian randomisation study using the UK Biobank cohort. Thorax 2022, 77, 65–73. [Google Scholar] [CrossRef]
- Korzeniowska, A.; Ręka, G.; Bilska, M.; Piecewicz-Szczęsna, H. The smoker’s paradox during the COVID-19 pandemic? The influence of smoking and vaping on the incidence and course of SARS-CoV-2 virus infection as well as possibility of using nicotine in the treatment of COVID-19-Review of the literature. Przegl. Epidemiol. 2021, 75, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, E.C.; Winsett, D.W.; Diamond, L. Augmentation of elastase-induced emphysema by cigarette smoke. Description of a model and a review of possible mechanisms. Am. Rev. Respir. Dis. 1985, 132, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Janoff, A. Investigations into the biochemical mechanisms of pulmonary emphysema: Effects of cigarette smoke on enzymes and anti-enzymes in the lung. Respiration 1986, 50 (Suppl. S1), 13–25. [Google Scholar] [CrossRef] [PubMed]
- DiNicolantonio, J.J.; Bhutani, J.; O’Keefe, J.H. The health benefits of vitamin K. Open Heart 2015, 2, e000300. [Google Scholar] [CrossRef]
- Nigwekar, S.U.; Thadhani, R. Long-Term Anticoagulation for Patients Receiving Dialysis. Circulation 2018, 138, 1530–1533. [Google Scholar] [CrossRef] [PubMed]
- Dofferhoff, A.S.M.; Piscaer, I.; Schurgers, L.J.; Visser, M.P.J.; van den Ouweland, J.M.W.; de Jong, P.A.; Gosens, R.; Hackeng, T.M.; van Daal, H.; Lux, P.; et al. Reduced Vitamin K Status as a Potentially Modifiable Risk Factor of Severe Coronavirus Disease. Clin. Infect. Dis. 2021, 73, e4039–e4046. [Google Scholar] [CrossRef]
- Quaglino, D.; Boraldi, F.; Lofaro, F.D. The biology of vascular calcification. Int. Rev. Cell Mol. Biol. 2020, 354, 261–353. [Google Scholar] [CrossRef]
- Singh, N.; Singh, S. Interstitial Lung Diseases and Air Pollution: Narrative Review of Literature. Pulm. Ther. 2021, 7, 89–100. [Google Scholar] [CrossRef]
- Winterbottom, C.J.; Shah, R.J.; Patterson, K.C.; Kreider, M.E.; Panettieri, R.A.; Rivera-Lebron, B.; Miller, W.T.; Litzky, L.A.; Penning, T.M.; Heinlen, K.; et al. Exposure to Ambient Particulate Matter Is Associated With Accelerated Functional Decline in Idiopathic Pulmonary Fibrosis. Chest 2018, 153, 1221–1228. [Google Scholar] [CrossRef]
- Ali, N.; Islam, F. The Effects of Air Pollution on COVID-19 Infection and Mortality-A Review on Recent Evidence. Front. Public Health 2020, 8, 580057. [Google Scholar] [CrossRef]
- Mehmood, K.; Bao, Y.; Abrar, M.M.; Petropoulos, G.P.; Saifullah; Soban, A.; Saud, S.; Khan, Z.A.; Khan, S.M.; Fahad, S. Spatiotemporal variability of COVID-19 pandemic in relation to air pollution, climate and socioeconomic factors in Pakistan. Chemosphere 2021, 271, 129584. [Google Scholar] [CrossRef] [PubMed]
- Setti, L.; Passarini, F.; De Gennaro, G.; Barbieri, P.; Licen, S.; Perrone, M.G.; Piazzalunga, A.; Borelli, M.; Palmisani, J.; Di Gilio, A.; et al. Potential role of particulate matter in the spreading of COVID-19 in Northern Italy: First observational study based on initial epidemic diffusion. BMJ Open 2020, 10, e039338. [Google Scholar] [CrossRef] [PubMed]
- Wirtz, H.R.; Dobbs, L.G. The effects of mechanical forces on lung functions. Respir. Physiol. 2000, 119, 1–17. [Google Scholar] [CrossRef]
- Jesudason, R.; Sato, S.; Parameswaran, H.; Araujo, A.D.; Majumdar, A.; Allen, P.G.; Bartolák-Suki, E.; Suki, B. Mechanical forces regulate elastase activity and binding site availability in lung elastin. Biophys. J. 2010, 99, 3076–3083. [Google Scholar] [CrossRef]
- Cantor, J.O.; Keller, S.; Cerreta, J.M.; Turino, G.M. Synergistic effect of elastase and 60 percent oxygen on pulmonary airspace enlargement. Ann. N. Y. Acad. Sci. 1991, 624, 318–320. [Google Scholar] [CrossRef]
- Tsai, Y.-F.; Hwang, T.-L. Neutrophil elastase inhibitors: A patent review and potential applications for inflammatory lung diseases (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 1145–1158. [Google Scholar] [CrossRef] [PubMed]
- Loo, S.; Kam, A.; Xiao, T.; Nguyen, G.K.T.; Liu, C.F.; Tam, J.P. Identification and Characterization of Roseltide, a Knottin-type Neutrophil Elastase Inhibitor Derived from Hibiscus sabdariffa. Sci. Rep. 2016, 6, 39401. [Google Scholar] [CrossRef]
- Barth, P.; Bruijnzeel, P.; Wach, A.; Sellier Kessler, O.; Hooftman, L.; Zimmermann, J.; Naue, N.; Huber, B.; Heimbeck, I.; Kappeler, D.; et al. Single dose escalation studies with inhaled POL6014, a potent novel selective reversible inhibitor of human neutrophil elastase, in healthy volunteers and subjects with cystic fibrosis. J. Cyst. Fibros. 2020, 19, 299–304. [Google Scholar] [CrossRef]
- Li, H.; Zhou, X.; Tan, H.; Hu, Y.; Zhang, L.; Liu, S.; Dai, M.; Li, Y.; Li, Q.; Mao, Z.; et al. Neutrophil extracellular traps contribute to the pathogenesis of acid-aspiration-induced ALI/ARDS. Oncotarget 2018, 9, 1772–1784. [Google Scholar] [CrossRef]
- Shen, X.B.; Chen, X.; Zhang, Z.Y.; Wu, F.F.; Liu, X.H. Cathepsin C inhibitors as anti-inflammatory drug discovery: Challenges and opportunities. Eur. J. Med. Chem. 2021, 225, 113818. [Google Scholar] [CrossRef]
- Herth, F.J.F.; Sandhaus, R.A.; Turner, A.M.; Sucena, M.; Welte, T.; Greulich, T. Alpha 1 Antitrypsin Therapy in Patients with Alpha 1 Antitrypsin Deficiency: Perspectives from a Registry Study and Practical Considerations for Self-Administration During the COVID-19 Pandemic. Int. J. Chron. Obstr. Pulm. Dis. 2021, 16, 2983–2996. [Google Scholar] [CrossRef] [PubMed]
- Zani, M.-L.; Tanga, A.; Saidi, A.; Serrano, H.; Dallet-Choisy, S.; Baranger, K.; Moreau, T. SLPI and trappin-2 as therapeutic agents to target airway serine proteases in inflammatory lung diseases: Current and future directions. Biochem. Soc. Trans. 2011, 39, 1441–1446. [Google Scholar] [CrossRef] [PubMed]
- Cambier, S.; Metzemaekers, M.; Carvalho, A.C.; Nooyens, A.; Jacobs, C.; Vanderbeke, L.; Malengier-Devlies, B.; Gouwy, M.; Heylen, E.; Meersseman, P.; et al. Atypical response to bacterial co-infection and persistent neutrophilic broncho-alveolar inflammation distinguish critical COVID-19 from influenza. JCI Insight 2021, 7, e155055. [Google Scholar] [CrossRef] [PubMed]
- Sahebnasagh, A.; Saghafi, F.; Safdari, M.; Khataminia, M.; Sadremomtaz, A.; Talaei, Z.; Rezai Ghaleno, H.; Bagheri, M.; Habtemariam, S.; Avan, R. Neutrophil elastase inhibitor (sivelestat) may be a promising therapeutic option for management of acute lung injury/acute respiratory distress syndrome or disseminated intravascular coagulation in COVID-19. J. Clin. Pharm. Ther. 2020, 45, 1515–1519. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, R.; Lei, Z.; Guo, X.; Yang, Y.; Tian, J.; Huang, L. Efficacy, safety, and pharmacoeconomics of sivelestat sodium in the treatment of septic acute respiratory distress syndrome: A retrospective cohort study. Ann. Palliat. Med. 2021, 10, 11910–11917. [Google Scholar] [CrossRef]
- Maki, C.; Inoue, Y.; Ishihara, T.; Hirano, Y.; Kondo, Y.; Sueyoshi, K.; Okamoto, K.; Tanaka, H. Evaluation of appropriate indications for the use of sivelestat sodium in acute respiratory distress syndrome: A retrospective cohort study. Acute Med. Surg. 2020, 7, e471. [Google Scholar] [CrossRef]
- Mohamed, M.M.A.; El-Shimy, I.A.; Hadi, M.A. Neutrophil Elastase Inhibitors: A potential prophylactic treatment option for SARS-CoV-2-induced respiratory complications? Crit. Care 2020, 24, 311. [Google Scholar] [CrossRef]
- Korkmaz, B.; Lesner, A.; Marchand-Adam, S.; Moss, C.; Jenne, D.E. Lung Protection by Cathepsin C Inhibition: A New Hope for COVID-19 and ARDS? J. Med. Chem. 2020, 63, 13258–13265. [Google Scholar] [CrossRef]
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha1-Antitrypsin Deficiency. N. Engl. J. Med. 2020, 382, 1443–1455. [Google Scholar] [CrossRef]
- de Loyola, M.B.; Dos Reis, T.T.A.; de Oliveira, G.X.L.M.; da Fonseca Palmeira, J.; Argañaraz, G.A.; Argañaraz, E.R. Alpha-1-antitrypsin: A possible host protective factor against COVID-19. Rev. Med. Virol. 2021, 31, e2157. [Google Scholar] [CrossRef]
- Wettstein, L.; Weil, T.; Conzelmann, C.; Müller, J.A.; Groß, R.; Hirschenberger, M.; Seidel, A.; Klute, S.; Zech, F.; Prelli Bozzo, C.; et al. Alpha-1 antitrypsin inhibits TMPRSS2 protease activity and SARS-CoV-2 infection. Nat. Commun. 2021, 12, 1726. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Hippensteel, J.; Leavitt, A.; Maloney, J.P.; Beckham, D.; Garcia, C.; Li, Q.; Freed, B.M.; Ordway, D.; Sandhaus, R.A.; et al. Hypothesis: Alpha-1-antitrypsin is a promising treatment option for COVID-19. Med. Hypotheses 2021, 146, 110394. [Google Scholar] [CrossRef] [PubMed]
- Guney, C.; Akar, F. Epithelial and Endothelial Expressions of ACE2: SARS-CoV-2 Entry Routes. J. Pharm. Pharm. Sci. 2021, 24, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Ferrarotti, I.; Ottaviani, S.; Balderacchi, A.M.; Barzon, V.; De Silvestri, A.; Piloni, D.; Mariani, F.; Corsico, A.G. COVID-19 infection in severe Alpha 1-antitrypsin deficiency: Looking for a rationale. Respir. Med. 2021, 183, 106440. [Google Scholar] [CrossRef] [PubMed]
- Shimi, G.; Sohrab, G.; Pourvali, K.; Ghorbani, A.; Balam, F.H.; Rostami, K.; Zand, H. Correlation of Low Levels of α-1 Antitrypsin and Elevation of Neutrophil to Lymphocyte Ratio with Higher Mortality in Severe COVID-19 Patients. Mediat. Inflamm. 2021, 2021, 5555619. [Google Scholar] [CrossRef]
- Vianello, A.; Braccioni, F. Geographical Overlap Between Alpha-1 Antitrypsin Deficiency and COVID-19 Infection in Italy: Casual or Causal? Arch. Bronconeumol. 2020, 56, 609–610. [Google Scholar] [CrossRef]
- Douglas, T.C.; Hannila, S.S. Working from within: How secretory leukocyte protease inhibitor regulates the expression of pro-inflammatory genes. Biochem. Cell Biol. 2021, 99, 1–8. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boraldi, F.; Lofaro, F.D.; Cossarizza, A.; Quaglino, D. The “Elastic Perspective” of SARS-CoV-2 Infection and the Role of Intrinsic and Extrinsic Factors. Int. J. Mol. Sci. 2022, 23, 1559. https://doi.org/10.3390/ijms23031559
Boraldi F, Lofaro FD, Cossarizza A, Quaglino D. The “Elastic Perspective” of SARS-CoV-2 Infection and the Role of Intrinsic and Extrinsic Factors. International Journal of Molecular Sciences. 2022; 23(3):1559. https://doi.org/10.3390/ijms23031559
Chicago/Turabian StyleBoraldi, Federica, Francesco Demetrio Lofaro, Andrea Cossarizza, and Daniela Quaglino. 2022. "The “Elastic Perspective” of SARS-CoV-2 Infection and the Role of Intrinsic and Extrinsic Factors" International Journal of Molecular Sciences 23, no. 3: 1559. https://doi.org/10.3390/ijms23031559
APA StyleBoraldi, F., Lofaro, F. D., Cossarizza, A., & Quaglino, D. (2022). The “Elastic Perspective” of SARS-CoV-2 Infection and the Role of Intrinsic and Extrinsic Factors. International Journal of Molecular Sciences, 23(3), 1559. https://doi.org/10.3390/ijms23031559