
Proinflammatory Cytokines (IL-1, -6, -8, -15, -17, -18, -23, TNF-α) Single Nucleotide Polymorphisms in Rheumatoid Arthritis—A Literature Review

 ,
, 
Abstract
:1. Introduction
Search Strategy
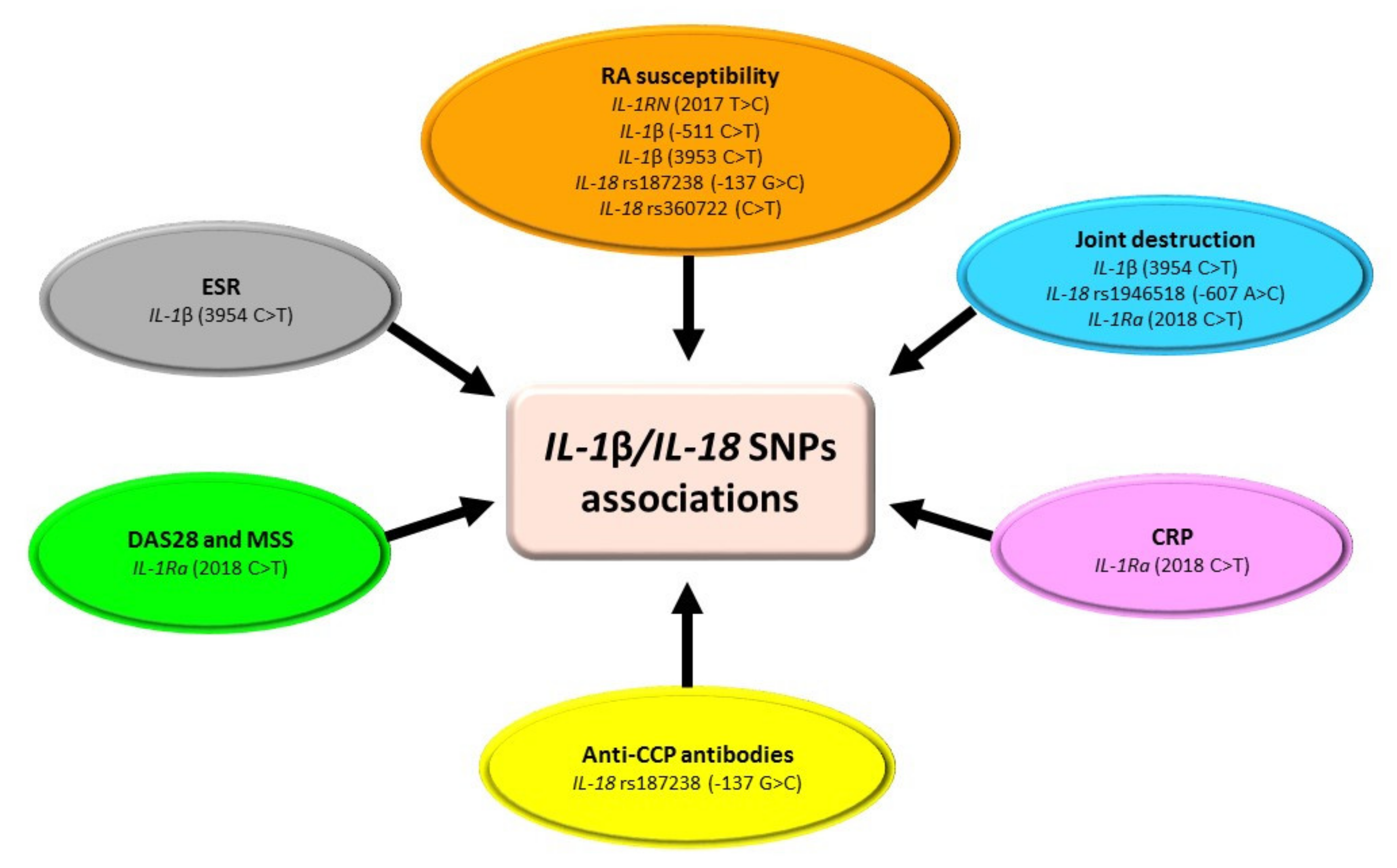
2. SNPs within IL-1 Family in RA
3. SNPs of IL-6 in RA
4. SNPs of IL-8 in RA
5. SNPs of IL-15 in RA
6. SNPs within IL-17 Family in RA
7. SNPs of IL-23R in RA
8. SNPs of TNF-α in RA
9. Conclusions
Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Márquez, A.; Martín, J.; Carmona, F.D. Emerging aspects of molecular biomarkers for diagnosis, prognosis and treatment response in rheumatoid arthritis. Expert Rev. Mol. Diagn. 2016, 16, 663–675. [Google Scholar] [CrossRef]
- Pavkova Goldbergova, M.; Nemec, P.; Lipkova, J.; Jarkovsky, J.; Gatterova, J.; Ambrozkova, D.; Vasku, A.; Soucek, M.; Pavek, N. Relation of IL-6, IL-13 and IL-15 gene polymorphisms to the rheumatoid factors, anti-CCP and other measures of rheumatoid arthritis activity. Int. J. Immunogenet. 2014, 41, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Saad, M.N.; Mabrouk, M.S.; Eldeib, A.M.; Shaker, O.G. Identification of rheumatoid arthritis biomarkers based on single nucleotide polymorphisms and haplotype blocks: A systematic review and meta-analysis. J. Adv. Res. 2016, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhaylenko, D.S.; Nemtsova, M.V.; Bure, I.V.; Kuznetsova, E.B.; Alekseeva, E.A.; Tarasov, V.V.; Lukashev, A.N.; Beloukhova, M.I.; Deviatkin, A.A.; Zamyatnin, A.A. Genetic polymorphisms associated with rheumatoid arthritis development and antirheumatic therapy response. Int. J. Mol. Sci. 2020, 21, 4911. [Google Scholar] [CrossRef] [PubMed]
- Brandt, B.; Rashidiani, S.; Bán, Á.; Rauch, T.A. DNA Methylation-Governed Gene Expression in Autoimmune Arthritis. Int. J. Mol. Sci. 2019, 20, 5646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moran-Moguel, M.C.; Petarra-del Rio, S.; Mayorquin-Galvan, E.E.; Zavala-Cerna, M.G. Rheumatoid Arthritis and miRNAs: A Critical Review through a Functional View. J. Immunol. Res. 2018, 2018, 2474529. [Google Scholar] [CrossRef]
- Chatzikyriakidou, A.; Voulgari, P.V.; Lambropoulos, A.; Drosos, A.A. Genetics in rheumatoid arthritis beyond HLA genes: What meta-analyses have shown? Semin. Arthritis Rheum. 2013, 43, 29–38. [Google Scholar] [CrossRef]
- Perricone, C.; Ceccarelli, F.; Valesini, G. An overview on the genetic of rheumatoid arthritis: A never-ending story. Autoimmun. Rev. 2011, 10, 599–608. [Google Scholar] [CrossRef]
- Amr, K.; El-Awady, R.; Raslan, H. Assessment of the −174G/C (Rs1800795) and −572G/C (rs1800796) Interleukin 6 gene polymorphisms in Egyptian patients with rheumatoid arthritis. Maced. J. Med. Sci. 2016, 4, 574–577. [Google Scholar] [CrossRef] [Green Version]
- Wielińska, J.; Dratwa, M.; Świerkot, J.; Korman, L.; Iwaszko, M.; Wysoczańska, B.; Bogunia-Kubik, K. Interleukin 6 gene polymorphism is associated with protein serum level and disease activity in Polish patients with rheumatoid arthritis. HLA 2018, 92, 38–41. [Google Scholar] [CrossRef]
- Pawlik, A.; Kurzawski, M.; Czerny, B.; Gawronska-Szklarz, B.; Drozdzik, M.; Herczynska, M. Interleukin-18 promoter polymorphism in patients with rheumatoid arthritis. Tissue Antigens 2006, 67, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Puren, A.J.; Fantuzzi, G.; Dinarello, C.A. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1β are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc. Natl. Acad. Sci. USA 1999, 96, 2256–2261. [Google Scholar] [CrossRef] [Green Version]
- Liang, D.; Ma, W.; Yao, C.; Liu, H.; Chen, X. Imbalance of interleukin 18 and interleukin 18 binding protein in patients with lupus nephritis. Cell. Mol. Immunol. 2006, 3, 303–306. [Google Scholar]
- Eisenberg, S.P.; Brewer, M.T.; Verderber, E.; Heimdal, P.; Brandhuber, B.J.; Thompson, R.C. Interleukin 1 receptor antagonist is a member of the interleukin 1 gene family: Evolution of a cytokine control mechanism. Proc. Natl. Acad. Sci. USA 1991, 88, 5232–5236. [Google Scholar] [CrossRef] [Green Version]
- Kay, J. The role of interleukin-1 in the pathogenesis of rheumatoid arthritis. Rheumatology 2004, 43, iii2–iii9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, P.; Pointon, J.J.; Chapman, K.; Roddam, A.; Wordsworth, B.P. Interleukin-1 promoter region polymorphism role in rheumatoid arthritis: A meta-analysis of IL-1B-511A/G variant reveals association with rheumatoid arthritis. Rheumatology 2008, 47, 1768–1770. [Google Scholar] [CrossRef] [Green Version]
- Arman, A.; Yilmaz, B.; Coker, A.; Inanc, N.; Direskeneli, H. Interleukin-1 receptor antagonist (IL-1RN) and interleukin-1B gene polymorphisms in Turkish patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2006, 24, 643–648. [Google Scholar]
- Kaijzel, E.L.; Van Dongen, H.; Bakker, A.M.; Breedveld, F.C.; Huizinga, T.W.J.; Verweij, C.L. Relationship of polymorphisms of the Interleukin-1 gene cluster to occurrence and severity of rheumatoid arthritis. Tissue Antigens 2002, 59, 122–126. [Google Scholar] [CrossRef]
- Buchs, N.; Di Giovine, F.S.; Silvestri, T.; Vannier, E.; Duff, G.W.; Miossec, P. IL-1B and IL-1Ra gene polymorphisms and disease severity in rheumatoid arthritis: Interaction with their plasma levels. Genes Immun. 2001, 2, 222–228. [Google Scholar] [CrossRef] [Green Version]
- Camargo, J.F.; Correa, P.A.; Castiblanco, J.; Anaya, J.M. Interleukin-1β polymorphisms in Colombian patients with autoimmune rheumatic diseases. Genes Immun. 2004, 5, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Tolusso, B.; Pietrapertosa, D.; Morelli, A.; De Santis, M.; Gremese, E.; Farina, G.; Carniello, S.G.; Del Frate, M.; Ferraccioli, G. IL-1B and IL-1RN gene polymorphisms in rheumatoid arthritis: Relationship with protein plasma levels and response to therapy. Pharmacogenomics 2006, 7, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Ismail, E.; Nofal, O.K.J.; Sakthiswary, R.; Shaharir, S.S.; Sridharan, R. The clinical significance of interleukin-1 receptor antagonist +2018 polymorphism in rheumatoid arthritis. PLoS ONE 2016, 11, e0153752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueda, B.; González-Gay, M.Á.; Mataran, L.; López-Nevot, M.Á.; Martín, J. Interleukin-18-promoter polymorphisms are not relevant in rheumatoid arthritis. Tissue Antigens 2005, 65, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, A.; Kurzawski, M.; Drozdzik, M.; Dziedziejko, V.; Safranow, K.; Herczynska, M. Interleukin18 gene IL18 promoter polymorphisms in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2009, 38, 159–165. [Google Scholar] [CrossRef]
- Ataie-Kachoie, P.; Pourgholami, M.H.; Richardson, D.R.; Morris, D.L. Gene of the month: Interleukin 6 (IL-6). J. Clin. Pathol. 2014, 67, 932–937. [Google Scholar] [CrossRef]
- Nishimoto, N.; Kishimoto, T. Interleukin 6: From bench to bedside. Nat. Clin. Pract. Rheumatol. 2006, 2, 619–626. [Google Scholar] [CrossRef]
- Schaper, F.; Rose-John, S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 2015, 26, 475–487. [Google Scholar] [CrossRef]
- Dittrich, A.; Hessenkemper, W.; Schaper, F. Systems biology of IL-6, IL-12 family cytokines. Cytokine Growth Factor Rev. 2015, 26, 595–602. [Google Scholar] [CrossRef]
- Malysheva, K.; de Rooij, K.; Löwik, C.W.G.M.; Baeten, D.L.; Rose-John, S.; Stoika, R.; Korchynskyi, O. Interleukin 6/Wnt interactions in rheumatoid arthritis: Interleukin 6 inhibits Wnt signaling in synovial fibroblasts and osteoblasts. Croat. Med. J. 2016, 57, 89–98. [Google Scholar] [CrossRef]
- Rose-john, S.; Elson, G.; Jones, S.A. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: Role in inflammation and cancer. J. Leukoc. Biol. 2006, 80, 227–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlik, A.; Wrzesniewska, J.; Florczak, M.; Gawronska-Szklarz, B.; Herczynska, M. IL-6 promoter polymorphism in patients with rheumatoid arthritis. Scand. J. Rheumatol. 2005, 34, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Arman, A.; Coker, A.; Sarioz, O.; Inanc, N.; Direskeneli, H. Lack of association between IL-6 gene polymorphisms and rheumatoid arthritis in Turkish population. Rheumatol. Int. 2012, 32, 2199–2201. [Google Scholar] [CrossRef] [PubMed]
- Zavaleta-Muñiz, S.A.; Martín-Márquez, B.T.; Gonzalez-Lopez, L.; Gonzalez-Montoya, N.G.; Díaz-Toscano, M.L.; Ponce-Guarneros, J.M.; Ruiz-Padilla, A.J.; Mercado, M.V.-D.; Maldonado-González, M.; Fafutis-Morris, M.; et al. The −174G/C and −572G/C Interleukin 6 Promoter Gene Polymorphisms in Mexican Patients with Rheumatoid Arthritis: A Case-Control Study. Clin. Dev. Immunol. 2013, 2013, 959084. [Google Scholar] [CrossRef]
- Shafia, S.; Dilafroze; Sofi, F.A.; Rasool, R.; Javeed, S.; Shah, Z.A. Rheumatoid arthritis and genetic variations in cytokine genes: A population-based study in Kashmir Valley. Immunol. Investig. 2014, 43, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Chai, W.; Ni, M.; Xu, M.; Lian, Z.; Shi, L.; Bai, Y.; Wang, Y. The Effects of Gene Polymorphisms in Interleukin-4 and Interleukin-6 on the Susceptibility of Rheumatoid Arthritis in a Chinese Population. Biomed Res. Int. 2014, 2014, 265435. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, A.; Yang, Y.; Si, Y.; Hao, D. Assessment of interleukin 6 gene polymorphisms with rheumatoid arthritis. Gene 2021, 765, 145070. [Google Scholar] [CrossRef]
- Koper, O.M.; Kamińska, J.; Sawicki, K.; Reszeć, J.; Rutkowski, R.; Jadeszko, M.; Mariak, Z.; Dymicka-Piekarska, V.; Kemona, H. Cerebrospinal fluid and serum IL-8, CCL2, and ICAM-1 concentrations in astrocytic brain tumor patients. Irish J. Med. Sci. 2018, 187, 767–775. [Google Scholar] [CrossRef] [Green Version]
- Kamińska, J.; Lyson, T.; Chrzanowski, R.; Sawicki, K.; Milewska, A.J.; Tylicka, M.; Zińczuk, J.; Matowicka-Karna, J.; Dymicka-Piekarska, V.; Mariak, Z.; et al. Ratio of IL-8 in CSF Versus Serum Is Elevated in Patients with Unruptured Brain Aneurysm. J. Clin. Med. 2020, 9, 1761. [Google Scholar] [CrossRef]
- Nashkevich, N.N.; Akalovich, S.; Louneva, N.; Heavner, G.A.; Voitenok, N.N. A monoclonal antibody and an enzyme immunoassay for human Ala-IL-877. J. Immunol. Methods 2002, 270, 37–51. [Google Scholar] [CrossRef]
- Fibbe, W.E.; Pruijt, J.F.M.; Velders, G.A.; Opdenakker, G.; Van Kooyk, Y.; Figdor, C.G.; Willemze, R. Biology of IL-8-induced stem cell mobilization. Ann. N. Y. Acad. Sci. 1999, 872, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Xie, K. Interleukin-8 and human cancer biology. Cytokine Growth Factor Rev. 2001, 12, 375–391. [Google Scholar] [CrossRef]
- Morita, T.; Shima, Y.; Fujimoto, K.; Tsuboi, H.; Saeki, Y.; Narazaki, M.; Ogata, A.; Kumanogoh, A. Anti-receptor activator of nuclear factor κB ligand antibody treatment increases osteoclastogenesis-promoting IL-8 in patients with rheumatoid arthritis. Int. Immunol. 2019, 31, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, A.; Joshua, V.; Hensvold, A.H.; Jin, T.; Sun, M.; Vivar, N.; Ytterberg, A.J.; Engström, M.; Fernandes-Cerqueira, C.; Amara, K.; et al. Identification of a novel chemokine-dependent molecular mechanism underlying Rheumatoid arthritisassociated autoantibody-mediated bone loss. Ann. Rheum. Dis. 2016, 75, 721–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, S.-F.; Huang, C.-M.; Lin, H.-C.; Chen, W.-C.; Tsai, C.-H.; Tsai, F.-J. Cytokine (IL-6) and chemokine (IL-8) gene polymorphisms among rheumatoid arthritis patients in Taiwan. Clin. Exp. Rheumatol. 2008, 26, 632–637. [Google Scholar]
- Emonts, M.; Hazes, M.J.M.W.; Houwing-Duistermaat, J.J.; van der Gaast-de Jongh, C.E.; de Vogel, L.; Han, H.K.H.; Wouters, J.M.G.W.; Laman, J.D.; Dolhain, R.J.E.M. Polymorphisms in genes controlling inflammation and tissue repair in rheumatoid arthritis: A case control study. BMC Med. Genet. 2011, 12, 36. [Google Scholar] [CrossRef] [Green Version]
- Chłopek, M.; Kowalik, A.; Góźdź, S.; Koziak, K. Rola interleukiny 15 w nowotworzeniu. Postepy Hig. Med. Dosw. 2017, 71, 5–19. [Google Scholar] [CrossRef]
- Zhu, X.; Marcus, W.D.; Xu, W.; Lee, H.; Han, K.; Egan, J.O.; Yovandich, J.L.; Rhode, P.R.; Wong, H.C. Novel Human Interleukin-15 Agonists. J. Immunol. 2009, 183, 3598–3607. [Google Scholar] [CrossRef] [Green Version]
- Perera, L.P.; Goldman, C.K.; Waldmann, T.A. IL-15 induces the expression of chemokines and their receptors in T lymphocytes. J. Immunol. 1999, 162, 2606–2612. [Google Scholar]
- Fehniger, T.A.; Caligiuri, M.A. Interleukin 15: Biology and relevance to human disease. Blood 2001, 97, 14–32. [Google Scholar] [CrossRef]
- Kurowska, W.; Przygodzka, M.; Jakubaszek, M.; Kwiatkowska, B.; Maslinski, W. Interleukin-15 as a Biomarker Candidate of Rheumatoid Arthritis Development. J. Clin. Med. 2020, 9, 1555. [Google Scholar] [CrossRef] [PubMed]
- Rueda, B.; López-Nevot, M.A.; González-Gay, M.A.; Balsa, A.; Pascual-Salcedo, D.; Garcia, A.; Gonzalez, A.; Martin, J. Molecular screening and association study of IL15 gene polymorphisms in rheumatoid arthritis. Cytokine 2007, 38, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Knevel, R.; Krabben, A.; Brouwer, E.; Posthumus, M.D.; Wilson, A.G.; Lindqvist, E.; Saxne, T.; De Rooy, D.; Daha, N.; Van Der Linden, M.P.M.; et al. Genetic variants in IL15 associate with progression of joint destruction in rheumatoid arthritis: A multicohort study. Ann. Rheum. Dis. 2012, 71, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, S.; Ikari, K.; Yano, K.; Toyama, Y.; Taniguchi, A.; Yamanaka, H.; Momohara, S. Lack of association between IL-15 genetic variants and progression of joint destruction in Japanese patients with rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 784–785. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M. Interleukin 17 is a chief orchestrator of immunity. Nat. Immunol. 2017, 18, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Moseley, T.A.; Haudenschild, D.R.; Rose, L.; Reddi, A.H. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev. 2003, 14, 155–174. [Google Scholar] [CrossRef]
- Iwakura, Y.; Ishigame, H.; Saijo, S.; Nakae, S. Functional Specialization of Interleukin-17 Family Members. Immunity 2011, 34, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Amatya, N.; Garg, A.V.; Gaffen, S.L. IL-17 Signaling: The Yin and the Yang. Trends Immunol. 2017, 38, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Chabaud, M.; Durand, J.M.; Buchs, N.; Page, G.; Frappart, L.; Miossec, P. Human Interleukin-17: A T cell–derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheumatol. 1999, 42, 963–970. [Google Scholar] [CrossRef]
- Van den Berg, W.B.; Miossec, P. IL-17 as a future therapeutic target for rheumatoid arthritis. Nat. Rev. Rheumatol. 2009, 5, 549–553. [Google Scholar] [CrossRef]
- Nordang, G.B.N.; Viken, M.K.; Hollis-moffatt, J.E.; Merriman, T.R.; Førre, Ø.T.; Helgetveit, K.; Kvien, T.K.; Lie, B.A. Association analysis of the interleukin 17A gene in Caucasian rheumatoid arthritis patients from Norway and New Zealand. Rheumatology 2009, 48, 367–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Zhang, H.; Yan, T.; Zhou, G.; Liu, R. Association between interleukin 17A polymorphisms and susceptibility to rheumatoid arthritis in a Chinese population. Gene 2015, 566, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Gomes da Silva, I.I.F.; Angelo, H.D.; Rushansky, E.; Mariano, M.H.; de Mascena Diniz Maia, M.; de Souza, P.R. Interleukin (IL)-23 Receptor, IL-17A and IL-17F Gene Polymorphisms in Brazilian Patients with Rheumatoid Arthritis. Arch. Immunol. Ther. Exp. 2017, 65, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.; Sheikh, N.; Mukhtar, M.; Saleem, T.; Akhtar, T.; Fatima, N.; Mehmood, R. Association of interleukin-17 gene polymorphisms with the onset of Rheumatoid Arthritis. Immunobiology 2021, 226, 152045. [Google Scholar] [CrossRef]
- Bogunia-Kubik, K.; Świerkot, J.; Malak, A.; Wysoczańska, B.; Nowak, B.; Białowąs, K.; Gębura, K.; Korman, L.; Wiland, P. IL-17A, IL-17F and IL-23R Gene Polymorphisms in Polish Patients with Rheumatoid Arthritis. Arch. Immunol. Ther. Exp. 2015, 63, 215–221. [Google Scholar] [CrossRef] [Green Version]
- Marwa, O.S.; Kalthoum, T.; Wajih, K.; Kamel, H. Association of IL17A and IL17F genes with rheumatoid arthritis disease and the impact of genetic polymorphisms on response to treatment. Immunol. Lett. 2017, 183, 24–36. [Google Scholar] [CrossRef]
- Paradowska-Gorycka, A.; Wojtecka-Lukasik, E.; Trefler, J.; Wojciechowska, B.; Lacki, J.K.; Maslinski, S. Association between IL-17F gene polymorphisms and susceptibility to and severity of rheumatoid arthritis (RA). Scand. J. Immunol. 2010, 72, 134–141. [Google Scholar] [CrossRef]
- Erkol İnal, E.; Görükmez, O.; Dündar, Ü.; Görükmez, Ö.; Yener, M.; Sağ, Ş.Ö.; Yakut, T. The Influence of Polymorphisms of Interleukin-17A and -17F Genes on Susceptibility and Activity of Rheumatoid Arthritis. Genet Test Mol Biomarkers 2015, 19, 461–464. [Google Scholar] [CrossRef]
- Dhaouadi, T.; Chahbi, M.; Haouami, Y.; Sfar, I.; Abdelmoula, L.; Ben Abdallah, T.; Gorgi, Y. IL-17A, IL-17RC polymorphisms and IL17 plasma levels in Tunisian patients with rheumatoid arthritis. PLoS One 2018, 13, e0194883. [Google Scholar] [CrossRef]
- Sun, L.; He, C.; Nair, L.; Yeung, J.; Egwuagu, C.E. Interleukin 12 (IL-12) family cytokines: Role in immune pathogenesis and treatment of CNS autoimmune disease. Cytokine 2015, 75, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Li, J.; Hu, Y.; Dai, K.; Gan, Y.; Zhao, J.; Huang, M.; Zhang, X. IL-23, but not IL-12, plays a critical role in inflammation-mediated bone disorders. Theranostics 2020, 10, 3925–3938. [Google Scholar] [CrossRef] [PubMed]
- Duvallet, E.; Semerano, L.; Assier, E.; Falgarone, G.; Boissier, M.C. Interleukin-23: A key cytokine in inflammatory diseases. Ann. Med. 2011, 43, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A Receptor for the Heterodimeric Cytokine IL-23 Is Composed of IL-12Rβ1 and a Novel Cytokine Receptor Subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Li, S.; Ying, S.; Tang, S.; Ding, Y.; Li, Y.; Qiao, J.; Fang, H. The IL-23/IL-17 Pathway in Inflammatory Skin Diseases: From Bench to Bedside. Front. Immunol. 2020, 11, 594735. [Google Scholar] [CrossRef] [PubMed]
- Shuttleworth, S.; Townsend, P.; Silva, F.; Cecil, A.; Hill, T.; Tomassi, C.; Rogers, H.; Harrison, R. Progress in the Development of Small Molecule Therapeutics Targeting Th17 Cell Function for the Treatment of Immune-Inflammatory Diseases. Prog. Med. Hist. 2011, 50, 109–133. [Google Scholar]
- Hillyer, P.; Larche, M.J.; Bowman, E.P.; McClanahan, T.K.; de Waal Malefyt, R.; Schewitz, L.P.; Giddins, G.; Feldmann, M.; Kastelein, R.A.; Brennan, F.M. Investigating the role of the interleukin-23/-17A axis in rheumatoid arthritis. Rheumatology 2009, 48, 1581–1589. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Kim, Y.J.; Park, B.L.; Bae, J.S.; Shin, H.D.; Bae, S.C. Lack of association between interleukin 23 receptor gene polymorphisms and rheumatoid arthritis susceptibility. Rheumatol. Int. 2009, 29, 781–786. [Google Scholar] [CrossRef]
- Orozco, G.; Rueda, B.; Robledo, G.; García, A.; Martín, J. Investigation of the IL23R gene in a Spanish rheumatoid arthritis cohort. Hum. Immunol. 2007, 68, 681–684. [Google Scholar] [CrossRef]
- Bridgewood, C.; Sharif, K.; Sherlock, J.; Watad, A.; McGonagle, D. Interleukin-23 pathway at the enthesis: The emerging story of enthesitis in spondyloarthropathy. Immunol. Rev. 2020, 294, 27–47. [Google Scholar] [CrossRef]
- Faragó, B.; Magyari, L.; Sáfrány, E.; Csöngei, V.; Járomi, L.; Horvatovich, K.; Sipeky, C.; Maász, A.; Radics, J.; Gyetvai, Á.; et al. Functional variants of interleukin-23 receptor gene confer risk for rheumatoid arthritis but not for systemic sclerosis. Ann. Rheum. Dis. 2008, 67, 248–250. [Google Scholar] [CrossRef]
- Hamdy, G.; Darweesh, H.; Khattab, E.A.; Fawzy, S.; Fawzy, E.; Sheta, M. Evidence of association of interleukin-23 receptor gene polymorphisms with Egyptian rheumatoid arthritis patients. Hum. Immunol. 2015, 76, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Safrany, E.; Melegh, B. Functional Variants of the Interleukin-23 Receptor Gene in Non-Gastrointestinal Autoimmune Diseases. Curr. Med. Chem. 2009, 16, 3766–3774. [Google Scholar] [CrossRef] [PubMed]
- Schottelius, A.J.G.; Moldawer, L.L.; Dinarello, C.A.; Asadullah, K.; Sterry, W.; Edwards, C.K. Biology of tumor necrosis factor-α–Implications for psoriaris. Exp. Dermatol. 2004, 13, 193–222. [Google Scholar] [CrossRef] [PubMed]
- Hajeer, A.H.; Hutchinson, I.V. TNF-α gene polymorphism: Clinical and biological implications. Microsc. Res. Tech. 2000, 50, 216–228. [Google Scholar] [CrossRef]
- Song, Y.; Jo, S.; Chung, J.Y.; Oh, Y.; Yoon, S.; Lee, Y.L.; Kim, S.S.; Yang, J.-H.; Jang, K.; Yang, C.-S.; et al. RNA interference-mediated suppression of TNF-α converting enzyme as an alternative anti-TNF-α therapy for rheumatoid arthritis. J. Control. Release 2021, 330, 1300–1312. [Google Scholar] [CrossRef]
- Bradley, J. TNF-mediated inflammatory disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Horiuchi, T.; Mitoma, H.; Harashima, S.-i.; Tsukamoto, H.; Shimoda, T. Transmembrane TNF-: Structure, function and interaction with anti-TNF agents. Rheumatology 2010, 49, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Möller, B.; Villiger, P.M. Inhibition of IL-1, IL-6, and TNF-α in immune-mediated inflammatory diseases. Springer Semin. Immunopathol. 2006, 27, 391–408. [Google Scholar] [CrossRef]
- Kaijzel, E.L.; van Krugten, M.V.; Brinkman, B.M.N.; Huizinga, T.W.J.; van der Straaten, T.; Hazes, J.M.W.; Ziegler-Heitbrock, H.W.L.; Nedospasov, S.A.; Breedveld, F.C.; Verweijl, C.L. Functional Analysis of a Human Tumor Necrosis Factor a (TNF-a) Promoter Polymorphism Related to Joint Damage in Rheumatoid Arthritis. Mol. Med. 1998, 4, 724–733. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Carreón, A.A.; Zúñiga, J.; Hernández-Pacheco, G.; Rodríguez-Pérez, J.M.; Pérez-Hernández, N.; Montes de Oca, J.V.; Cardiel, M.H.; Granados, J.; Vargas-Alarcón, G. Tumor necrosis factor-alpha −308 promoter polymorphism contributes independently to HLA alleles in the severity of rheumatoid arthritis in Mexicans. J. Autoimmun. 2005, 24, 63–68. [Google Scholar] [CrossRef]
- Ates, O.; Hatemi, G.; Hamuryudan, V.; Topal-Sarikaya, A. Tumor necrosis factor-alpha and Interleukin-10 gene promoter polymorphisms in Turkish rheumatoid arthritis patients. Clin. Rheumatol. 2008, 27, 1243–1248. [Google Scholar] [CrossRef] [PubMed]
- Gambhir, D.; Lawrence, A.; Aggarwal, A.; Misra, R.; Mandal, S.K.; Naik, S. Association of tumor necrosis factor alpha and IL-10 promoter polymorphisms with rheumatoid arthritis in North Indian population. Rheumatol. Int. 2010, 30, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Gheita, T.A.; Azkalany, G.S.; Gaber, W.; Mohey, A. Clinical significance of serum TNFα and -308 G/A promoter polymorphism in rheumatoid arthritis. Egypt. Rheumatol. 2015, 37, 49–54. [Google Scholar] [CrossRef] [Green Version]
- Lagha, A.; Zidi, S.; Stayoussef, M.; Gazouani, E.; Kochkar, R.; Kochbati, S.; Almawi, W.Y.; Yacoubi-Loueslati, B. Interleukin-1β, Interleukin1-Ra, Interleukin-10, and tumor necrosis factor-α polymorphisms in Tunisian patients with rheumatoid arthritis. Pathol. Biol. 2015, 63, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H. Meta-Analysis of Genetic Association Studies. Ann. Lab. Med. 2015, 35, 283–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Author | SNPs Analyzed | Nationality/ No. of Cases | Study Results |
|---|---|---|---|
| [20] | IL-1β (−511 C > T) IL-1β (3954 C > T) IL-1Ra (2018 C > T) | French/297 RA patients vs. 112 healthy controls |
|
| [19] | IL-1α (4845 G > T) IL-1β (3953 C > T) IL-1RN (5111 T > C) IL-1RN (2017 T > C) | Dutch/312 RA patients vs. 94 incident female RA patients vs. 245 healthy controls |
|
| [21] | IL-1β (−511 C > T) IL-1β (3953 C > T) | Colombian/ 172 RA patients vs. 392 healthy controls |
|
| [24] | IL-18 rs1946518 (−607 A > C) IL-18 rs187238 (−137 G > C) | Spanish/362 RA patients vs. 339 healthy controls |
|
| [18] | IL-1β (−511 C > T) IL-1β (3953 C > T) | Turkish/96 RA patients vs. 104 healthy controls |
|
| [22] | IL-1β (−511 C > T) IL-1β (3953 C > T) | Italian/126 RA patients vs. 178 healthy controls |
|
| [11] | IL-18 rs1946518 (−607 A > C) IL-18 rs187238 (−137 G > C) | Polish/309 RA patients vs. 305 healthy controls |
|
| [25] | IL-18 rs1946518 (−607 A > C) IL-18 rs187238 (−137 G > C) IL-18 rs360718 (T > G) IL-18 rs360721 (C > G) IL-18 rs360722 (C > T) IL-18 rs549908 (A > C) IL-18 rs5744292 (A > G) | Polish/404 RA patients vs. 148 healthy controls |
|
| [23] | IL-1Ra (2018 C > T) | Malaysians/77 RA patients vs. 18 healthy controls |
|
| Author | SNPs Analyzed | Ethnicity/ No. of Cases | Results |
|---|---|---|---|
| [32] | IL-6 rs1800795 (−174 G > C) | Polish/98 RA patients vs. 105 healthy controls |
|
| [33] | IL-6 rs1800795 (−174 G > C) IL-6 rs1800796 (−572 G > C) IL-6 rs1800797 (−597 G > A) | Turkish/178 RA patients vs. 247 healthy controls |
|
| [34] | IL-6 rs1800795 (−174 G > C) IL-6 rs1800796 (−572 G > C) | Mexican/ 137 RA patients vs. 102 healthy controls |
|
| [36] | IL-6 rs1800795 (−174 G > C) | Chines Han/752 RA patients vs. 798 healthy controls |
|
| [35] | IL-6 rs1800795 (−174 G > C) | Indian/150 RA patients vs. 200 healthy controls |
|
| [9] | IL-6 rs1800795 (−174 G > C) IL-6 rs1800796 (−572 G > C) | Egyptian/99 RA patients vs. 99 healthy controls |
|
| [10] | IL-6 rs1800795 (−174 G > C) | Polish/130 RA patients vs. 112 healthy controls |
|
| [37] | IL-6 rs1800796 (−572 G > C) IL-6 rs2069837 (A > G) IL-6 rs1524107 (C > T) IL-6 rs2069840 (G > C) | Chinese Han/508 RA patients vs. 494 healthy controls |
|
| Author | SNPs Analyzed | Nationality/ No. of Cases | Study Results |
|---|---|---|---|
| [52] | IL-15 rs4956403 (C > T) IL-15 rs3806798 (T > A) IL-15 rs7440292 (T > C) IL-15 rs1493012 (T > C) IL-15 rs1493013 (T > C) IL-15 rs2254514 (−267 C > T) IL-15 rs2857261 (367 A > G) IL-15 rs9282741 (C > T) IL-15 rs9282742 (T > C) IL-15 rs1057972 (14035 T > A) IL-15 rs9282743 (G > T) IL-15 rs10833 (C > T) IL-15 rs2291596 (C > T) | Spanish/645 RA patients vs. 656 healthy controls |
|
| [53] | IL-15 rs7667746 (G/A) IL-15 rs7665842 (G > A) IL-15 rs2322182 (A > G) IL-15 rs6821171 (C > A) IL-15 rs4371699 (A > C) | Four European cohorts (Dutch, British, Swedish)/1418 RA patients |
|
| [2] | IL-15 rs2857261 (367 A > G) IL-15 rs2254514 (−267 C > T) IL-15 rs1057972 (14035 T > A) | Czechs/156 RA patients vs. 200 healthy controls |
|
| [54] | IL-15 rs6821171 (C > A) IL-15 rs1521761 (T > A) | Japanese/865 RA patients |
|
| Author | SNPs Analyzed | Nationality/ No. of Cases | Study Results |
|---|---|---|---|
| [61] | IL-17A rs4711998 (A > G) IL-17A rs3819024 (A > G) IL-17A rs2275913 (−197 G > A) IL-17A rs7747909 (G > A) IL-17A rs8193036 (C > T) | Norwegian/ 950 RA patients vs. 933 healthy controls New Zealanders/580 RA patients vs. 504 healthy controls |
|
| [67] | IL-17F rs763780 (7488 A > G) IL-17F rs2397084 (7383 A > G) | Polish/220 RA patients vs. 106 healthy controls |
|
| [65] | IL-17A rs2275913 (−197 G > A) IL-17F rs763780 (7488 A > G) | Polish/89 RA patients vs. 125 healthy controls |
|
| [68] | IL-17A rs2275913 (−197 G > A) IL-17F rs763780 (7488 A > G) IL-17F rs2397084 (7383 A > G) | Turkish/161 RA patients vs. 88 healthy controls |
|
| [62] | IL-17A rs2275913 (−197 G > A) IL-17A rs3819024 (A > G) IL-17A rs3819025 (G > A) IL-17A rs4711998 (A > G) IL-17A rs8193036 (C > T) IL-17A rs8193037 (G > A) | Chinese/615 RA patients vs. 839 healthy controls |
|
| [63] | IL-17A rs2275913 (−197 G > A) IL-17F rs763780 (7488 A > G) | Brazilian/127 RA patients vs. 134 healthy controls |
|
| [66] | IL-17A rs2275913 (−197 G > A) IL-17F rs763780 (7488 A > G) IL-17F rs2397084 (7383 A > G) | Tunisians/108 RA patients vs. 202 healthy controls |
|
| [69] | IL-17A rs2275913 (−197 G > A) IL-17RC rs708567 (6313 G > A) | Tunisians/115 RA patients vs. 91 healthy controls |
|
| [64] | IL-17A rs2275913 (−197 G > A) IL-17F rs763780 (7488 A > G) IL-17F rs2397084 (7383 A > G) | Pakistans/50 RA patients vs. 50 healthy controls |
|
| Author | SNPs Analyzed | Nationality/ No. of Cases | Study Results |
|---|---|---|---|
| [78] | IL-23R rs1004819 (G > A) IL-23R rs7517847 (A > C) IL-23R rs10489629 (A > G) IL-23R rs11209026 (G > A) IL-23R rs1343151 (G > A) IL-23R rs10889677 (C > A) IL-23R rs11209032 (G > A) IL-23R rs1495965 (A > G) | Spanish/322 RA patients vs. 342 healthy controls |
|
| [80] | IL-23R rs10889677 (C > A) IL-23R rs2201841 (T > C) IL-23R rs1884444 (G > T) | Hungarian/ 412 RA patients vs. 220 healthy controls |
|
| [77] | IL-23R rs1004819 (A > G) IL-23R rs7517847 (T > G) IL-23R rs10489629 (T > C) IL-23R rs2201841 (G > A) IL-23R rs1343151(G > A) IL-23R rs11209032 (G > A) IL-23R rs1495965 (T > C) | Korean/1204 RA patients vs. 979 healthy controls |
|
| [65] | IL-23R rs11209026 (G > A) | Polish/89 RA patients vs. 125 healthy controls |
|
| [81] | IL-23R rs11209026 (G > A) IL-23R rs2201841(A > G) IL-23R rs10889677 (2199 C > A) | Egyptian/120 RA patients vs. 120 healthy controls |
|
| [63] | IL-23R rs10889677 (2199 C > A) | Brazilian/127 RA patient vs. 134 healthy controls |
|
| Author | SNPs Analyzed | Nationality/ No. of Cases | Study Results |
|---|---|---|---|
| [89] | TNF-α rs361525 (−238 G > A) TNF-α rs1800750 (−376 G > A) | Dutch Caucasians/ 101 RA patients vs. 403 healthy controls |
|
| [90] | TNF-α rs361525 (−238 G > A) TNF-α rs1800629 (−308 G > A) | Mexicans/137 RA patients vs. 169 healthy controls |
|
| [91] | TNF-α rs361525 (−238 G > A) TNF-α rs1800629 (−308 G > A) TNF-α rs1800750 (−376 G > A) | Turkish/98 RA patients vs. 122 healthy controls |
|
| [92] | TNF-α rs1800629 (−308 G > A) TNF-α rs1800630 (−863 C > A) | North Indians/222 RA patients vs. 208 controls |
|
| [93] | TNF-α rs1800629 (−308 G > A) | Egyptians/43 RA patients vs. 30 controls |
|
| [94] | TNF-α rs1800629 (−308 G > A) | Tunisians/104 RA patients vs. 150 healthy controls |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koper-Lenkiewicz, O.M.; Sutkowska, K.; Wawrusiewicz-Kurylonek, N.; Kowalewska, E.; Matowicka-Karna, J. Proinflammatory Cytokines (IL-1, -6, -8, -15, -17, -18, -23, TNF-α) Single Nucleotide Polymorphisms in Rheumatoid Arthritis—A Literature Review. Int. J. Mol. Sci. 2022, 23, 2106. https://doi.org/10.3390/ijms23042106
Koper-Lenkiewicz OM, Sutkowska K, Wawrusiewicz-Kurylonek N, Kowalewska E, Matowicka-Karna J. Proinflammatory Cytokines (IL-1, -6, -8, -15, -17, -18, -23, TNF-α) Single Nucleotide Polymorphisms in Rheumatoid Arthritis—A Literature Review. International Journal of Molecular Sciences. 2022; 23(4):2106. https://doi.org/10.3390/ijms23042106
Chicago/Turabian StyleKoper-Lenkiewicz, Olga M., Kinga Sutkowska, Natalia Wawrusiewicz-Kurylonek, Ewa Kowalewska, and Joanna Matowicka-Karna. 2022. "Proinflammatory Cytokines (IL-1, -6, -8, -15, -17, -18, -23, TNF-α) Single Nucleotide Polymorphisms in Rheumatoid Arthritis—A Literature Review" International Journal of Molecular Sciences 23, no. 4: 2106. https://doi.org/10.3390/ijms23042106