Abstract
Primary aldosteronism (PA) is the most common cause of secondary hypertension. A growing body of evidence has suggested that, beyond its well-known effects on blood pressure and electrolyte balance, aldosterone excess can exert pro-inflammatory, pro-oxidant and pro-fibrotic effects on the kidney, blood vessels and heart, leading to potentially harmful pathophysiological consequences. In clinical studies, PA has been associated with an increased risk of cardiovascular, cerebrovascular, renal and metabolic complication compared to essential hypertension, including atrial fibrillation (AF) and aortic ectasia. An increased prevalence of AF in patients with PA has been demonstrated in several clinical studies. Aldosterone excess seems to be involved in the pathogenesis of AF by inducing cardiac structural and electrical remodeling that in turn predisposes to arrhythmogenicity. The association between PA and aortic ectasia is less established, but several studies have demonstrated an effect of aldosterone on aortic stiffness, vascular smooth muscle cells and media composition that, in turn, might lead to an increased risk of aortic dilation and dissection. In this review, we focus on the current evidence regarding the potential role of aldosterone excess in the pathogenesis of AF and aortic ectasia.
1. Introduction
Primary aldosteronism (PA) is a clinical syndrome characterized by arterial hypertension (AH) and electrolyte imbalance due to an autonomous overproduction of aldosterone by one or both adrenal glands, independently of renin levels. Laparoscopic adrenalectomy is the treatment of choice in unilateral PA, whereas medical treatment with mineralocorticoid receptor (MR) antagonists (MRAs) represents the preferred option in bilateral forms [1,2].
PA is the most common cause of secondary hypertension. Its prevalence in hypertensive patients ranges between 5% and 15% in most studies [3,4] and increases with the severity of hypertension, reaching up to 29.1% in patients with resistant hypertension [5,6].
Beyond its well-known effect on sodium reabsorption in the distal nephron, a growing body of evidence has demonstrated that aldosterone levels that are inappropriate for salt intake can also exert pro-inflammatory, pro-oxidant and pro-fibrotic effects on the kidney, blood vessels and heart, leading to potentially harmful pathophysiological consequences [7].
In clinical studies, PA has been associated with an increased risk of cerebrovascular events, myocardial infarction, left ventricular (LV) hypertrophy (LVH), atrial fibrillation (AF), increased carotid intima-media thickness, aortic ectasia, metabolic alterations and renal impairment as compared with essential hypertension (EH) [6,8,9] (Figure 1). An appropriate surgical or medical treatment of PA not only improves blood pressure control and corrects hypokalemia in most patients, but also seems to reverse target organ damage and reduce the increased risk of cardiovascular, cerebrovascular and renal complications [10,11,12,13,14,15].
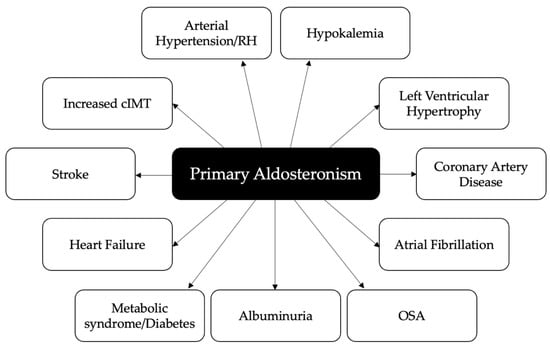
Figure 1.
Cardiovascular, cerebrovascular, metabolic and renal complications associated with primary aldosteronism. RH: resistant hypertension; cIMT: carotid intima-media thickness; OSA: obstructive sleep apnea.
In the present review, we focus on the potential role of aldosterone excess in the pathophysiology of AF and aortic ectasia.
2. Primary Aldosteronism and Atrial Fibrillation
AF is the most common sustained cardiac arrhythmia in adults and is associated with increased morbidity and mortality. Its prevalence increases with age and ranges between 2% and 4% in the adult general population [16].
AF is initiated by focal ectopic firing and is maintained by re-entry mechanisms in a vulnerable atrial substrate. The ectopic firing seems to arise from myocyte sleeves within the pulmonary veins and is triggered by a diastolic leak of Ca2+ from the sarcoplasmic reticulum that in turn determines myocyte depolarization due to an inward Na+ current via Na+-Ca2+ exchanger. The re-entry mechanism is promoted by slow conduction velocity of the depolarizing wavefront and a shortened refractory period of cardiac myocytes. The presence of structural and electrophysiological atrial abnormalities favors the self-perpetuation of AF by promoting re-entry [17].
In recent years, accumulating evidence has suggested a role for the renin-angiotensin-aldosterone system (RAAS) in the pathophysiology of AF and a potential association between AF and PA.
In a study by Goette et al., restoration of sinus rhythm by electrical cardioversion was associated with a fall in serum aldosterone in patients with persistent AF [18]. Similarly, in a prospective study conducted in 45 consecutive patients with non-valvular persistent AF and preserved LV systolic function, the authors found a positive correlation between the fall in aldosterone concentration 24 h after cardioversion and maintenance of sinus rhythm during a 30-day follow up [19].
In 2005, Milliez et al. reported a 12.1-fold greater risk of AF in 124 patients with PA as compared with 465 EH controls (7.3% vs. 0.6%); i multivariate analysis, PA remained an independent predictor of AF [20]. In a prospective study by Rossi et al. of 323 hypertensive patients who were systematically screened for PA, at baseline, patients with PA had a 7.2-fold greater prevalence of FA as compared to patients with EH (6.5% vs. 0.9%); at follow-up, the AF-free survival was significantly lower in the PA than in the EH group [11]. These data were confirmed in a meta-analysis by Monticone et al., who reported a 3.52-fold greater risk of AF in PA patients as compared to EH controls over a median follow up of 8.8 years [8].
On the other hand, in a population of 149 patients with a history of AF who were screened for PA using the aldosterone-to-renin ratio, a conclusive diagnosis of PA was made in 2.6% [21]. In a retrospective nationwide case-control study by the same group, the prevalence of PA in the AF group was relatively low (0.056%), but patients with AF had a significantly higher risk of being diagnosed with PA as compared with controls (OR 1.65). The low prevalence of PA reported in this study may be, at least in part, due to the retrospective design that might have led to an underestimation of PA cases in the absence of a systematic screening [22]. A much higher prevalence of PA was found in a prospective study recruiting 411 consecutive hypertensive patients with AF: PA was diagnosed in 42% of patients who showed no known cause of the arrhythmia, thus suggesting that PA is highly prevalent in hypertensive patients with unexplained AF [23].
Three different meta-analyses suggested a reduction in AF risk in MRA-treated patients as compared with controls, especially in the setting of heart failure (HF) [24,25,26]. As regards to patients with PA, in a large-scale prospective observational cohort study with an 11.8-year median follow-up, Rossi et al. reported a significantly worse AF-free survival in medically treated PA patients as compared with EH patients, whereas adrenalectomy was associated with AF-free survival similar to that of optimally treated EH patients, thus suggesting that removing the cause of aldosterone excess is better than controlling the MR-mediated effects of aldosterone [27].
Aldosterone has been shown to promote AF in animal models. Reil et al. demonstrated that the infusion of aldosterone in rats favors the development of AF after transesophageal atrial burst stimulation, independently of its effects on ventricular function or atrial pressure [28]. In another study by Lammers et al. in a rat model in which AF was induced by transesophageal burst pacing, aldosterone administration doubled the time until AF spontaneously converted into sinus rhythm [29].
On the other hand, Tsai et al. demonstrated that patients with AF had a significantly higher atrial MR expression as compared with those in sinus rhythm and that rapid depolarization increased MR expression through a Ca2+-dependent mechanism in HL-1 atrial myocytes, thus suggesting that AF itself can increase the sensitivity of cardiomyocytes to aldosterone [30]. Moreover, aldosterone itself has been shown to upregulate MR expression in cultured HL-1 cardiomyocytes, thus reinforcing its effects on the heart [31]. Since the MR not only binds aldosterone, but also cortisol, it can be argued that some effects mediated by MR activation may be attributable to cortisol. However, Lavall et al. reported that 11â-hydroxysteroid dehydrogenase type 2, an enzyme which converts cortisol to inactive metabolites allowing aldosterone binding to the MR, is upregulated in the left atrial myocardium of patients with AF, thereby suggesting that MR activation in this setting is mainly due to aldosterone [32].
As regards to the potential physiopathological mechanism linking aldosterone excess to FA, aldosterone is thought to be involved in the genesis and perpetuation of AF not only by causing AH and electrolyte imbalance, but also by inducing inflammation, oxidative stress, fibrosis and electrophysiological changes; all these mechanisms contribute to structural and electrical atrial remodeling that are known to predispose to AF [33] (Figure 2).
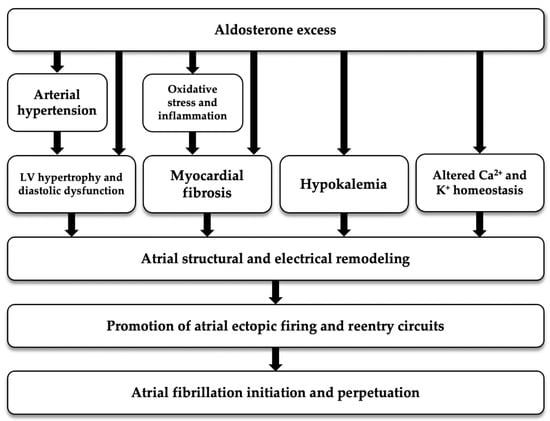
Figure 2.
Potential pathogenetic mechanism linking aldosterone excess with atrial fibrillation. LV: left ventricular.
2.1. Arterial Hypertension and Left Ventricular Hypertrophy
AH is a known risk factor for the development of AF [16]. AH causes LVH and diastolic dysfunction that in turn induce an increase in LV end-diastolic pressure and left atrial (LA) pressure, leading to atrial structural changes (dilation, hypertrophy, interstitial fibrosis, inflammatory infiltrates and apoptosis) with subsequent reduction and heterogeneity of conduction velocity [17]. Moreover, aldosterone induces LV remodeling and hypertrophy independently of its effects on blood pressure, since several studies have reported a greater increase in LV mass in patients with PA as compared with EH [8,11,34] probably because of direct pro-fibrotic, pro-oxidant and pro-inflammatory effects [35].
2.2. Hypokalemia
Hypokalemia is a common manifestation of PA, occurring in 9–37% of affected patients, and is a direct consequence of the activation of MR in the distal tubules and collecting duct of the nephron, where it mediates sodium reabsorption in exchange for potassium and hydrogen ions [2].
Hypokalemia itself can induce resting membrane hyperpolarization, Na+-K+ ATPase inhibition and suppression of K+ channel conductance, resulting in prolongation of the action potential duration, reduced repolarization reserve, early afterdepolarization, delayed afterdepolarization and automaticity, which may all participate in the genesis and perpetuation of AF [36].
2.3. Aldosterone and Atrial Fibrosis
As mentioned before, aldosterone is a well-known pro-fibrotic factor, and it exerts its effects both on the atria and on the ventricles [37].
Atrial fibrosis is a common finding in AF and is characterized by myocyte degeneration, fibroblast proliferation and interstitial expansion due to abnormal accumulation of fibrillar collagen, predisposing to arrhythmias by disrupting electrical conduction [38].
Sun et al. demonstrated that chronic administration of aldosterone to uninephrectomized rats in the presence of a high-salt diet leads to myocyte loss and interstitial collagen deposition in both right and left atria [39].
In a study by Lavall et al., aldosterone increased the expression of CTGF in vitro, probably by upregulating the RhoA/Rho kinase pathway via MR activation in cardiomyocytes. Overactivity of the RhoA/Rho kinase pathway has been observed in patients with aldosterone-producing adenomas and appears to be involved in the pathogenesis of arterial hypertension, by increasing peripheral vascular resistance via modulation of Ca2+ sensitivity in vascular smooth muscle cells (VSMCs) [40,41]. In the same study, aldosterone also increased the expression of miRNA-21, a microRNA that is associated with fibroblast survival and cardiac fibrosis, and lysyl oxidase, a key enzyme for collagen cross-linking, whereas the MR antagonists BR-4628 and spironolactone prevented these alterations [32].
In another study by Tsai et al. in HL-1 atrial cells, aldosterone mediated its fibrotic effects via MR and the MAPK intracellular signaling cascade, leading to an increase in the expression of collagen 1A and 3A, transforming growth factor (TGF)-â1 and á-smooth muscle actin (SMA) [31].
In a rat PA model, Reil et al. reported an increase in atrial fibroblasts and interstitial collagen, a reduction in active matrix metalloproteinase 13 and hypertrophy of atrial myocytes, as compared with controls [28].
MRAs have been shown to at least partially reverse aldosterone-induced atrial fibrosis in animal models. In a study by Milliez et al., the administration of spironolactone in the setting of HF induced a significant decrease in atrial fibrosis in a rat model [42]. These data were confirmed in a study by Yang et al. in congestive HF dogs, in which spironolactone reversed atrial fibrosis, decreased atrial refractory period and shortened conduction time [43]. Similarly, spironolactone treatment prevented arrhythmogenic atrial dilatation and reversed atrial fibrosis in a canine model of atrial fibrillation by normalizing the balance between matrix metalloproteinase (MMP)-9 and tissue inhibitors of metalloproteinase (TIMP)-1, whose alteration is associated with atrial fibrosis [44].
2.4. Aldosterone, Oxidative Stress and Inflammation
Increased oxidative stress and mitochondrial dysfunction have been reported in the atria of patients with AF. The main sources of oxidative stress in the atria are the mitochondrial electron transport chain, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase and uncoupled nitric oxide synthase (NOS) [45].
The pro-oxidant effect of aldosterone has been demonstrated in several studies, and oxidative stress seems to be an important cofactor in aldosterone-induced cardiac inflammation and fibrosis (Figure 3). In a study by Johar et al. in a rat model, the chronic infusion of aldosterone increased NADPH oxidase activity, as well as the expression of pro-fibrotic genes and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-êB), inducing interstitial cardiac fibrosis [46]. In another study on a rat model by Sun et al., chronic exposure to aldosterone and salt was accompanied by a time-dependent sustained activation of NADPH oxidase with 3-nitrotyrosine generation and NF-êB activation in endothelial and inflammatory cells in the right and left ventricle, creating a pro-inflammatory and pro-fibrogenic environment [47].
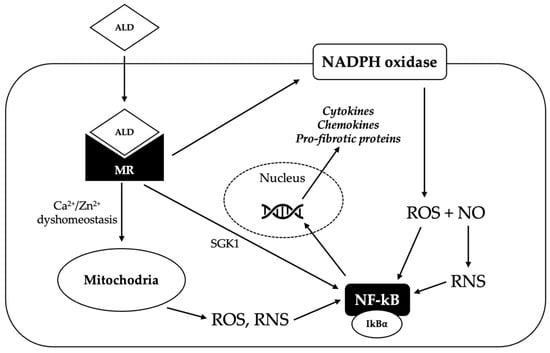
Figure 3.
Potential mechanisms underlying the pro-oxidant effect of aldosterone. Aldosterone (ALD) diffuses through the plasma membrane to the cytosol, where it binds the mineralocorticoid receptor (MR), inducing its dimerization and activation. Activated MR promotes the generation of reactive oxygen species (ROS) via nicotinamide adenine dinucleotide phosphate (NADPH) oxidase; ROS react with nitric oxide (NO) to produce reactive nitrogen species (RNS). MR activation can also trigger the production of ROS and RNS in the mitochondria, probably by inducing Ca2+/Zn2+ dyshomeostasis. Increased ROS and RNS levels cause the release of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-êB) from its repressor NF-êB inhibitor alpha (IkBá); NF-êB can therefore translocate in the nucleus, where it binds to a specific promoter and induces the transcription of pro-inflammatory and pro-fibrotic genes. Activated MR can also directly activate NF-êB, probably via the serum/glucocorticoid regulated kinase 1 (SGK1).
Similarly, the administration of eplerenone in mice with chronic pressure overload was associated with a reduction in myocardial oxidative stress, as assessed by a reduction in 3-nitrotyrosine, as well as a diminished expression of intercellular adhesion molecule-1 and a reduced infiltration by macrophages [48].
In another a study by Kagiyama et al. in angiotensin II receptor knock-out mice, the exposure to aldosterone and salt increased cardiac expression of CTGF and NADPH components via the Rho kinase pathway, as well as the levels of nitrotyrosine and 4-hydroxy-2-nonenal, which are markers of oxidative damage. The administration of eplerenone antagonized these effects [49].
Moreover, in a study by Kamalov et al., aldosterone induced oxidative stress, as demonstrated by increased levels of 3-nitrotyrosine, 4-hydroxy-2-nonenal, malondialdehyde, H2O2 and oxidized glutathione, by inducing a dyshomeostasis of intracellular Ca2+ and Zn2+ in cardiac myocytes and mitochondria of rats [50].
Finally, aldosterone has been shown to induce Ca2+/calmodulin-dependent protein kinase II (CaMKII) oxidation via NADPH oxidase activation [51]. Oxidized CaMKII was found to be increased in atria from AF patients compared with patients in sinus rhythm [52].
Chronic inflammation, which is strictly related to oxidative stress, is another well-known pro-fibrotic factor and can contribute to the development of an AF-prone atrial substrate. Several studies have suggested a MR-mediated effect of aldosterone on inflammatory cell adhesion and infiltration in different tissues, such as the kidney, the vasculature and the adipose tissue [45].
As we have already mentioned, MR activation promotes inflammation by stimulating the generation of ROS, which in turn activate pro-inflammatory transcription factors such as NF-êB. Aldosterone has also been shown to directly activate the NF-êB pathway signaling in the kidney, leading to increased expression of several chemokines and interleukins, probably via the serum and glucocorticoid-inducible kinase 1 [53]. In addition to stimulating the formation of ROS, aldosterone may promote vascular inflammation by stimulating endothelial expression of adhesion molecules that, in turn, promote the recruitment of leukocytes [51].
As regards to the pro-inflammatory effects of aldosterone in the heart, Rocha et al. reported that the myocardial and coronary injury observed in angiotensin II/salt-treated rats, which was mainly inflammatory in nature and associated with the expression of cyclooxygenase 2 and osteopontin, was at least in part mediated by aldosterone, since treatment with eplerenone attenuated the cardiac and vascular damage [54].
2.5. Aldosterone and Electrical Remodeling
Abnormal Ca2+ handling is thought to be a possible mechanism underlying AF-generating ectopic foci [17]. Aldosterone has been shown to directly influence Ca2+ and K+ currents in cardiomyocytes in animal models, possibly participating in the electrophysiological changes underlying the development and perpetuation of cardiac arrhythmias. How the effects of aldosterone on Ca2+ and K+ currents might predispose to AF is, however, yet to be established.
In a study by Perrier et al. in a mouse model, elevated plasma aldosterone concentrations were associated with an increase in L-type channel Ca2+ current (ICa) in ventricular cardiomyocytes, and this effect was attributable to changes in Ca2+ channel expression rather than modulation of channel activity [55]. These data were consistent with a previous study by Bénitah et al., in which aldosterone induced an increase in whole-cell ICa in isolated adult rat ventricular myocytes via the MR, probably by upregulating the expression of the á1c gene, which encodes for the á-subunit of the L-type voltage-dependent Ca2+ channel. The administration of spironolactone was able to blunt the aldosterone-induced increase in ICa density [56]. Similarly, in another study on cultured neonatal rat ventricular cardiomyocytes, aldosterone amplified L- and T-type ICa, resulting in a positive chronotropic effect. This was associated with an increase in the mRNA coding for á1c and â2 subunits of cardiac L-type channel and an increased expression of T-type channel á-1H isoform. The administration of spironolactone inhibited the aldosterone-induced increase in beating frequency [57]. A more recent study by Mesquita et al. showed that aldosterone increases the expression of L-type channel á1c subunit N-terminal transcripts at both mRNA and protein levels through P1-promoter activation [58]. In the aforementioned study by Tsai et al. in HL-1 atrial myocytes, aldosterone increased expression of the á-1G and á-1H subunits of the T-type Ca2+ channel and thus increased the T-type ICa and intracellular Ca2+ load through a genomic pathway. In the same study, aldosterone decreased the rapidly activating delayed rectifier K+ current (IKr), thereby altering repolarization [30]. In a study by Laszlo et al. in a rabbit model, the administration of spironolactone induced a decrease in L-type channel ICa, but did not influence transient outward K+ current Ito in atrial myocytes [59]. In another study, Ouvrard-Pascaud et al. demonstrated that aldosterone upregulates L-type ICa and downregulates Ito in ventricular cardiomyocytes in a transgenic mouse model with cardiac-specific overexpression of the human MR, thereby prolonging the action potential and increasing the risk of arrhythmias [60].
Conversely, in a study in a rat model by Lammers et al., aldosterone infusion induced a significant shortening of the action potential in atrial cardiomyocytes. This was associated with an increased expression of K+ channel subunits Kir2.1 and Kv1.5 that carry the inward rectifier K+ current Ik1 and the ultra-rapid activating delayed rectifier K+ current Ikur, respectively, both involved in the repolarization process. These effects were prevented by the administration of spironolactone [29]. The shortening of the action potential is associated with a reduction in the effective refractory period that, in turn, favors re-entry mechanisms underlying FA perpetuation [17].
Moreover, aldosterone has been shown to promote the occurrence of delayed afterdepolarization, a fundamental mechanism underlying several arrhythmias, by causing an abnormal diastolic opening of ryanodine receptors (RyR) that in turn determines an altered Ca2+ release from the sarcoplasmic reticulum in isolated adult rat ventricular cardiomyocytes. These changes were associated with downregulation of FKBP12 and FKBP12.6 that act as regulatory proteins of the RyR complex [61]. As we have already mentioned, the diastolic leak of Ca2+ from the sarcoplasmic reticulum activates an inward Na+ current via Na+-Ca2+ exchanger, resulting in spontaneous myocyte depolarization [17].
The slowing of conduction velocity, along with the shortening of the effective refractory period, contributes to the development of the re-entry mechanisms that underlie the perpetuation of AF [17]. In a study by Qu et al. in a rat model of cardiac hypertrophy, the administration of spironolactone reversed pathological gap junction remodeling and the associated slowing of impulse propagation, thereby improving conduction velocity, without influencing hypertrophy [62].
To summarize, aldosterone excess can contribute to the genesis and perpetuation of AF by inducing atrial structural and electrical remodeling. These consequences are not only dependent on the well-known influence of an aldosterone overproduction on blood pressure levels and K+ balance, but also on direct pro-oxidant, pro-inflammatory, pro-fibrotic and electrophysiological effects of aldosterone on cardiomyocytes.
3. Primary Aldosteronism and Aortic Ectasia
Aldosterone has been shown to have detrimental effects on blood vessels in the presence of a high salt intake, by inducing inflammation, oxidative stress, fibrosis and endothelial dysfunction [7].
The expression of MRs by aortic endothelial and VSMCs was reported almost 30 years ago [63]. However, there is little evidence on the association between PA and proximal aortic ectasia, a condition that is associated with an increased risk of aortic rupture and dissection [64].
In a rat model, the infusion of aldosterone and salt induced abdominal and thoracic aortic aneurysm formation and rupture in an age-dependent manner, whereas the administration of MRA significantly attenuated this effect [65].
In clinical studies, patients with PA have been shown to have larger ascending aortas and a higher prevalence of aortic ectasia as compared with patients with EH. In a prospective study on 45 patients with PA compared to 47 patients with EH, Zavatta et al. reported that patients with PA had larger ascending aortic diameters than those with EH before starting any specific treatment, even after considering possible confounding factors such as age, sex, body surface area, duration of hypertension, evidence of subclinical hypercortisolism and number of hypertensive drugs. After a mean follow-up of 3 years, patients with PA did not show significant changes in ascending aortic diameter, irrespective of the type of treatment (medical treatment with MRA vs. unilateral adrenalectomy), probably because a longer period of observation is necessary to detect significant differences [66]. In another study by Parasiliti-Caprino et al. in 110 patients with resistant hypertension, PA was associated with an increased prevalence of aortic ectasia as compared with patients with EH (22% vs. 6%), and this association remained statistically significant even in multivariate analysis, using age as a covariate [6]. In addition, proximal aortic aneurysm and aortic dissection have been described as potential complications in patients with PA in several case reports [67,68,69,70,71,72].
Measures of aortic stiffness, such as pulse wave velocity (PWV), have been shown to be independently associated with future thoracic aortic aneurysm expansion [73]. Several studies have reported that patients with PA have increased arterial stiffness, as indicated by higher values of PWV and augmentation index (AIx) as compared with patients with EH with comparable blood pressure levels [74,75]. Moreover, in patients with non-ischemic dilated cardiomyopathy, treatment with eplerenone induced a reduction of aortic stiffness, an increase in aortic distensibility and systolic aortic strain, independently of its effects on blood pressure [76]. Finally, aldosterone excess has also been associated with impaired endothelium-dependent vascular reactivity measured by flow-mediated dilation (FMD), which has been used as a surrogate marker of vascular health [40,77,78] and appears to be reduced in patients with ascending aorta dilation in the setting of Marfan syndrome and bicuspid aortic valve [79,80,81].
3.1. Effects of Aldosterone on Vascular Smooth Muscle Cells
In histological examination, ascending aortic aneurysm are characterized by focal loss of VSMCs, although areas of VSMCs hyperplasia that have been interpreted as an adaptive response to increased wall stress have also been reported [64].
Aldosterone has been shown to induce aortic VSMC apoptosis in rat models. A study by Yan et al. on a rat PA model showed that aldosterone can induce vascular cell apoptosis in vivo independently of blood pressure levels. This effect seems to be mediated by MR activation and an increased Bax/Bcl-2 ratio that induces apoptosis via the mitochondrial pathway [82]. Another study by Wei et al. showed that excess aldosterone promotes NADPH oxidase activation and reactive oxygen species production, which, in turn, impairs Akt serine phosphorylation and consequently causes activation of apoptotic signaling pathways, leading to vascular cell apoptosis in a rat model. MR antagonism with low-dose spironolactone prevented aldosterone-induced apoptosis by restoring Akt serine phosphorylation, independently of its effect on blood pressure [83].
Conversely, an in vitro study by Schwerdt et al. showed that increased cell loss in rat aorta VSMCs after exposure to aldosterone and salt reflected higher necrosis rates rather than higher apoptotic rates [84].
In contrast with the aforementioned evidence, other studies have shown that aldosterone promotes aortic VSMCs proliferation in rats [85].
3.2. Effects of Aldosterone on Extracellular Matrix
The histological examination of thoracic aortic aneurysms shows a maladaptive remodeling of the extracellular matrix, with disruption and loss of elastic fibers, accumulation of proteoglycans, altered collagen synthesis and fibrosis [64,86].
Several studies have investigated the effects of aldosterone on the composition of aortic extracellular matrix. In a study by Benetos et al. in spontaneously hypertensive rats, the administration of spironolactone prevented aortic collagen accumulation and reduced the collagen-to-elastin ratio, independent of blood pressure changes [87]. In another study by the same research group on old normotensive rats, the administration of spironolactone significantly decreased the collagen content in the carotid artery but not in the thoracic aorta [88]. Finally, the same authors reported that infusion of aldosterone in salt-drinking rats increased aortic stiffness, with elevated fibronectin but no change in collagen density [89].
In another study on a rat model, Iglarz et al. reported that a 6-week aldosterone infusion induced collagen deposition in the aortic media [90]. Similarly, Yan et al. showed that aldosterone can induce aortic fibrosis in a rat model by increasing the collagen production, probably via the upregulation of TGF-â1 expression. This profibrotic effect was prevented by MR antagonism with eplerenone, independently of blood pressure variations [82].
Moreover, Park et al. demonstrated that aortic collagen and the media cross-sectional area were significantly increased in aldosterone-infused rats and that the administration of an endothelin-1 (ET-1) type A receptor antagonist prevented these effects, thereby suggesting a role for ET-1 in fibrosis of large vessels in conditions associated with mineralocorticoid excess [91].
In a study by Harvey et al. on the abdominal aorta of stroke-prone spontaneously hypertensive rats, treatment with canrenoic acid blunted the MR-mediated activation of pro-inflammatory and pro-fibrotic signaling molecules (p66Shc and p38 MAPK) and prevented the expression of Nox1 and collagen I mRNA [92].
In another study on spontaneously hypertensive rats, the administration of eplerenone significantly reduced connective tissue growth factor (CTGF) mRNA expression in the aorta. In the same study, the authors also investigated the direct effect of aldosterone on VSMCs, showing that aldosterone increases CTGF production in a dose-related manner and that this effect is prevented by spironolactone [93].
Finally, an in vitro study by Gekle et al. in human aortic VSMCs reported that aldosterone, in combination with oxidative stress induced by H2O2, led to increased collagen synthesis that was reversed by MRA, leading to the idea that aldosterone sensitizes the cells to oxidative stress-induced collagen secretion [94].
In conclusion, aldosterone seems to modify the composition of the aortic extracellular matrix and to damage aortic VSMCs. These effects might alter the structural and functional properties of the aorta, thereby leading to increased arterial stiffness and weakening of the arterial wall, conditions that can in turn predispose to aortic enlargement and dissection.
4. Conclusions
In the past few decades, a growing body of evidence has suggested a detrimental effect of aldosterone excess on several target organs, such as the kidney, the vasculature and the heart. This is not only dependent on aldosterone influence on blood pressure levels, but also on its established pro-fibrotic, pro-inflammatory and pro-oxidant effects.
In clinical studies, PA has been associated with an increased risk of several cardiovascular, metabolic and renal complications as compared with EH, further validating this hypothesis. In this review, we summarized the current evidence on the potential role of aldosterone excess in AF and aortic ectasia.
An increased risk of AF has been demonstrated in patients with PA. As regards to the pathophysiological basis of this association, experimental studies have suggested a direct effect of aldosterone excess on cardiac structural and electrical remodeling, potentially leading to an AF-prone substrate. This association has some therapeutic implications, since some studies have demonstrated a beneficial role of MRAs on the risk of developing AF in the setting of HF. Concerning patients with PA, while adrenalectomy appears to be effective in reducing AF risk, further studies are needed to evaluate the role of MRAs.
Although evidence of the association of PA with aortic ectasia is limited, in clinical studies, patients with PA have been shown to have larger ascending aortas and a higher prevalence of proximal aortic ectasia as compared with patients with EH. Preclinical studies have demonstrated a direct effect of aldosterone on aortic VSMCs, media composition and wall stiffness, that in turn might lead to aortic wall weakening, dilation and increased risk of dissection. However, further research is needed to validate this hypothesis and to clarify if MR antagonism or surgical treatment may have a protective role in proximal aortic enlargement in patients with PA.
In conclusion, in patients with PA, aldosterone-mediated organ damage seems to be more extensive than previously thought, requiring a more holistic approach in the diagnosis and management of this disease and its potential complications.
Author Contributions
Conceptualization, M.M. and M.P.-C.; writing—original draft preparation, M.B. and C.L.; writing—review and editing, F.B. and F.P.; visualization, E.G.; supervision, E.G. and M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We wish to acknowledge the European Reference Network for rare endocrine conditions (Endo-ERN), of which several authors of this publication are members (project ID no. 739543).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Funder, J.W.; Carey, R.M.; Mantero, F.; Murad, M.H.; Reincke, M.; Shibata, H.; Stowasser, M.; Young, W.F.J. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J. Clin. Endocrinol. Metab. 2016, 101, 1889–1916. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.P.; Bisogni, V.; Bacca, A.V.; Belfiore, A.; Cesari, M.; Concistrè, A.; Del Pinto, R.; Fabris, B.; Fallo, F.; Fava, C.; et al. The 2020 Italian Society of Arterial Hypertension (SIIA) practical guidelines for the management of primary aldosteronism. Int. J. Cardiol. Hypertens. 2020, 5, 100029. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.P.; Bernini, G.; Caliumi, C.; Desideri, G.; Fabris, B.; Ferri, C.; Ganzaroli, C.; Giacchetti, G.; Letizia, C.; Maccario, M.; et al. A Prospective Study of the Prevalence of Primary Aldosteronism in 1125 Hypertensive Patients. J. Am. Coll. Cardiol. 2006, 48, 2293–2300. [Google Scholar] [CrossRef] [PubMed]
- Monticone, S.; Burrello, J.; Tizzani, D.; Bertello, C.; Viola, A.; Buffolo, F.; Gabetti, L.; Mengozzi, G.; Williams, T.A.; Rabbia, F.; et al. Prevalence and Clinical Manifestations of Primary Aldosteronism Encountered in Primary Care Practice. J. Am. Coll. Cardiol. 2017, 69, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Siddiqui, M.; Calhoun, D.A.; Carey, R.M.; Hopkins, P.N.; Williams, G.H.; Vaidya, A. The Unrecognized Prevalence of Primary Aldosteronism: A Cross-sectional Study. Ann. Intern. Med. 2020, 173, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Parasiliti-Caprino, M.; Lopez, C.; Prencipe, N.; Lucatello, B.; Settanni, F.; Giraudo, G.; Rossato, D.; Mengozzi, G.; Ghigo, E.; Benso, A.; et al. Prevalence of primary aldosteronism and association with cardiovascular complications in patients with resistant and refractory hypertension. J. Hypertens. 2020, 38, 1841–1848. [Google Scholar] [CrossRef] [PubMed]
- Briet, M.; Schiffrin, E.L. Vascular actions of aldosterone. J. Vasc. Res. 2013, 50, 89–99. [Google Scholar] [CrossRef]
- Monticone, S.; D’Ascenzo, F.; Moretti, C.; Williams, T.A.; Veglio, F.; Gaita, F.; Mulatero, P. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: A systematic review and meta-analysis. Lancet Diabetes Endocrinol. 2018, 6, 41–50. [Google Scholar] [CrossRef]
- Monticone, S.; Sconfienza, E.; D’Ascenzo, F.; Buffolo, F.; Satoh, F.; Sechi, L.A.; Veglio, F.; Mulatero, P. Renal damage in primary aldosteronism: A systematic review and meta-analysis. J. Hypertens. 2020, 38, 3–12. [Google Scholar] [CrossRef]
- Williams, T.A.; Lenders, J.W.M.; Mulatero, P.; Burrello, J.; Rottenkolber, M.; Adolf, C.; Satoh, F.; Amar, L.; Quinkler, M.; Deinum, J.; et al. Outcomes after adrenalectomy for unilateral primary aldosteronism: An international consensus on outcome measures and analysis of remission rates in an international cohort. Lancet Diabetes Endocrinol. 2017, 5, 689–699. [Google Scholar] [CrossRef]
- Rossi, G.P.; Cesari, M.; Cuspidi, C.; Maiolino, G.; Cicala, M.V.; Bisogni, V.; Mantero, F.; Pessina, A.C. Long-term control of arterial hypertension and regression of left ventricular hypertrophy with treatment of primary aldosteronism. Hypertension 2013, 62, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Catena, C.; Colussi, G.L.; Nadalini, E.; Chiuch, A.; Baroselli, S.; Lapenna, R.; Sechi, L.A. Cardiovascular outcomes in patients with primary aldosteronism after treatment. Arch. Intern. Med. 2008, 168, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Wu, V.-C.; Wang, S.-M.; Chang, C.-H.; Hu, Y.-H.; Lin, L.-Y.; Lin, Y.-H.; Chueh, S.-C.J.; Chen, L.; Wu, K.-D. Long term outcome of Aldosteronism after target treatments. Sci. Rep. 2016, 6, 32103. [Google Scholar] [CrossRef] [PubMed]
- Hundemer, G.L.; Curhan, G.C.; Yozamp, N.; Wang, M.; Vaidya, A. Cardiometabolic outcomes and mortality in medically treated primary aldosteronism: A retrospective cohort study. Lancet Diabetes Endocrinol. 2018, 6, 51–59. [Google Scholar] [CrossRef]
- Hundemer, G.L.; Curhan, G.C.; Yozamp, N.; Wang, M.; Vaidya, A. Renal Outcomes in Medically and Surgically Treated Primary Aldosteronism. Hypertension 2018, 72, 658–666. [Google Scholar] [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Bax, J.J.; Boriani, G.; Dan, G.A.; Fauchier, L.; Kalman, J.M.; Lane, D.A.; Lettino, M.; et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS). Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef]
- Staerk, L.; Sherer, J.A.; Ko, D.; Benjamin, E.J.; Helm, R.H. Atrial Fibrillation: Epidemiology, Pathophysiology, Clinical Outcomes. Circ. Res. 2017, 120, 1501–1517. [Google Scholar] [CrossRef]
- Goette, A.; Hoffmanns, P.; Enayati, W.; Meltendorf, U.; Geller, J.C.; Klein, H.U. Effect of successful electrical cardioversion on serum aldosterone in patients with persistent atrial fibrillation. Am. J. Cardiol. 2001, 88, 906–909. [Google Scholar] [CrossRef]
- Wozakowska-Kaplon, B.; Bartkowiak, R.; Janiszewska, G. A decrease in serum aldosterone level is associated with maintenance of sinus rhythm after successful cardioversion of atrial fibrillation. PACE Pacing Clin. Electrophysiol. 2010, 33, 561–565. [Google Scholar] [CrossRef]
- Milliez, P.; Girerd, X.; Plouin, P.F.; Blacher, J.; Safar, M.E.; Mourad, J.J. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J. Am. Coll. Cardiol. 2005, 45, 1243–1248. [Google Scholar] [CrossRef]
- Mourtzinis, G.; Ebrahimi, A.; Gustafsson, H.; Johannsson, G.; Manhem, K. Aldosterone to Renin Ratio as a Screening Instrument for Primary Aldosteronism in a Middle-Aged Population with Atrial Fibrillation. Horm. Metab. Res. 2017, 49, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Mourtzinis, G.; Adamsson Eryd, S.; Rosengren, A.; Björck, L.; Adiels, M.; Johannsson, G.; Manhem, K. Primary aldosteronism and thyroid disorders in atrial fibrillation: A Swedish nationwide case–control study. Eur. J. Prev. Cardiol. 2018, 25, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Seccia, T.M.; Letizia, C.; Muiesan, M.L.; Lerco, S.; Cesari, M.; Bisogni, V.; Petramala, L.; Maiolino, G.; Volpin, R.; Rossi, G.P. Atrial fibrillation as presenting sign of primary aldosteronism: Results of the Prospective Appraisal on the Prevalence of Primary Aldosteronism in Hypertensive (PAPPHY) Study. J. Hypertens. 2020, 38, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Korantzopoulos, P.; Shao, Q.; Zhang, Z.; Letsas, K.P.; Li, G. Mineralocorticoid receptor antagonists and atrial fibrillation: A meta-analysis. Europace 2016, 18, 672–678. [Google Scholar] [CrossRef]
- Neefs, J.; van den Berg, N.W.E.; Limpens, J.; Berger, W.R.; Boekholdt, S.M.; Sanders, P.; de Groot, J.R. Aldosterone Pathway Blockade to Prevent Atrial Fibrillation: A Systematic Review and Meta-Analysis. Int. J. Cardiol. 2017, 231, 155–161. [Google Scholar] [CrossRef]
- Alexandre, J.; Dolladille, C.; Douesnel, L.; Font, J.; Dabrowski, R.; Shavit, L.; Legallois, D.; Funck-Brentano, C.; Champ-Rigot, L.; Ollitrault, P.; et al. Effects of Mineralocorticoid Receptor Antagonists on Atrial Fibrillation Occurrence: A Systematic Review, Meta-Analysis, and Meta-Regression to Identify Modifying Factors. J. Am. Heart Assoc. 2019, 8, e013267. [Google Scholar] [CrossRef]
- Rossi, G.P.; Maiolino, G.; Flego, A.; Belfiore, A.; Bernini, G.; Fabris, B.; Ferri, C.; Giacchetti, G.; Letizia, C.; Maccario, M.; et al. Adrenalectomy Lowers Incident Atrial Fibrillation in Primary Aldosteronism Patients at Long Term. Hypertension 2018, 71, 585–591. [Google Scholar] [CrossRef]
- Reil, J.C.; Hohl, M.; Selejan, S.; Lipp, P.; Drautz, F.; Kazakow, A.; Münz, B.M.; Müller, P.; Steendijk, P.; Reil, G.H.; et al. Aldosterone promotes atrial fibrillation. Eur. Heart J. 2012, 33, 2098–2108. [Google Scholar] [CrossRef]
- Lammers, C.; Dartsch, T.; Brandt, M.C.; Rottländer, D.; Halbach, M.; Peinkofer, G.; Ockenpoehler, S.; Weiergraeber, M.; Schneider, T.; Reuter, H.; et al. Spironolactone prevents aldosterone induced increased duration of atrial fibrillation in rat. Cell. Physiol. Biochem. 2012, 29, 833–840. [Google Scholar] [CrossRef]
- Tsai, C.T.; Chiang, F.T.; Tseng, C.D.; Hwang, J.J.; Kuo, K.T.; Wu, C.K.; Yu, C.C.; Wang, Y.C.; Lai, L.P.; Lin, J.L. Increased Expression of Mineralocorticoid Receptor in Human Atrial Fibrillation and a Cellular Model of Atrial Fibrillation. J. Am. Coll. Cardiol. 2010, 55, 758–770. [Google Scholar] [CrossRef]
- Tsai, C.F.; Yang, S.F.; Chu, H.J.; Ueng, K.C. Cross-talk between mineralocorticoid receptor/angiotensin II type 1 receptor and mitogen-activated protein kinase pathways underlies aldosterone-induced atrial fibrotic responses in HL-1 cardiomyocytes. Int. J. Cardiol. 2013, 169, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Lavall, D.; Selzer, C.; Schuster, P.; Lenski, M.; Adam, O.; Schäfers, H.J.; Böhm, M.; Laufs, U. The mineralocorticoid receptor promotes fibrotic remodeling in atrial fibrillation. J. Biol. Chem. 2014, 289, 6656–6668. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.T.; Tsai, C.H.; Chen, Z.W.; Chang, Y.Y.; Wu, V.C.; Hung, C.S.; Lin, Y.H. Atrial Fibrillation in Primary Aldosteronism. Horm. Metab. Res. 2020, 52, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Catena, C.; Colussi, G.; Brosolo, G.; Novello, M.; Sechi, L.A. Aldosterone and Left Ventricular Remodeling. Horm. Metab. Res. 2015, 47, 981–986. [Google Scholar] [CrossRef]
- Tsai, C.H.; Pan, C.T.; Chang, Y.Y.; Chen, Z.W.; Wu, V.C.; Hung, C.S.; Lin, Y.H. Left ventricular remodeling and dysfunction in primary aldosteronism. J. Hum. Hypertens. 2021, 35, 131–147. [Google Scholar] [CrossRef]
- Weiss, J.N.; Qu, Z.; Shivkumar, K. Electrophysiology of hypokalemia and hyperkalemia. Circ. Arrhythm. Electrophysiol. 2017, 10, e004667. [Google Scholar] [CrossRef]
- Azibani, F.; Fazal, L.; Chatziantoniou, C.; Samuel, J.L.; Delcayre, C. Aldosterone mediates cardiac fibrosis in the setting of hypertension. Curr. Hypertens. Rep. 2013, 15, 395–400. [Google Scholar] [CrossRef]
- Burstein, B.; Nattel, S. Atrial Fibrosis: Mechanisms and Clinical Relevance in Atrial Fibrillation. J. Am. Coll. Cardiol. 2008, 51, 802–809. [Google Scholar] [CrossRef]
- Sun, Y.; Ramires, F.J.A.; Weber, K.T. Fibrosis of atria and great vessels in response to angiotensin II or aldosterone infusion. Cardiovasc. Res. 1997, 35, 138–147. [Google Scholar] [CrossRef][Green Version]
- Matsumoto, T.; Oki, K.; Kajikawa, M.; Nakashima, A.; Maruhashi, T.; Iwamoto, Y.; Iwamoto, A.; Oda, N.; Hidaka, T.; Kihara, Y.; et al. Effect of aldosterone-producing adenoma on endothelial function and Rho-associated kinase activity in patients with primary aldosteronism. Hypertension 2015, 65, 841–848. [Google Scholar] [CrossRef]
- Somlyo, A.P. Signal transduction. Rhomantic interludes raise blood pressure. Nature 1997, 389, 908–911. [Google Scholar] [CrossRef] [PubMed]
- Milliez, P.; DeAngelis, N.; Rucker-Martin, C.; Leenhardt, A.; Vicaut, E.; Robidel, E.; Beaufils, P.; Delcayre, C.; Hatem, S.N. Spironolactone reduces fibrosis of dilated atria during heart failure in rats with myocardial infarction. Eur. Heart J. 2005, 26, 2193–2199. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.S.; Han, W.; Zhou, H.Y.; Dong, G.; Wang, B.C.; Huo, H.; Wei, N.; Cao, Y.; Zhou, G.; Xiu, C.H.; et al. Effects of spironolactone on electrical and structural remodeling of atrium in congestive heart failure dogs. Chin. Med. J. 2008, 121, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Li, J.; Li, W.; Li, Y.; Shan, H.; Gong, Y.; Yang, B. Effects of spironolactone on atrial structural remodelling in a canine model of atrial fibrillation produced by prolonged atrial pacing. Br. J. Pharmacol. 2010, 159, 1584–1594. [Google Scholar] [CrossRef] [PubMed]
- Mayyas, F.; Alzoubi, K.H.; Van Wagoner, D.R. Impact of aldosterone antagonists on the substrate for atrial fibrillation: Aldosterone promotes oxidative stress and atrial structural/electrical remodeling. Int. J. Cardiol. 2013, 168, 5135–5142. [Google Scholar] [CrossRef] [PubMed]
- Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M.; Johar, S.; Cave, A.C.; Narayanapanicker, A.; Grieve, D.J.; Shah, A.M. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. 2006, 20, 1546–1548. [Google Scholar] [CrossRef]
- Sun, Y.; Zhang, J.; Lu, L.; Chen, S.S.; Quinn, M.T.; Weber, K.T. Aldosterone-induced inflammation in the rat heart: Role of oxidative stress. Am. J. Pathol. 2002, 161, 1773–1781. [Google Scholar] [CrossRef]
- Kuster, G.M.; Kotlyar, E.; Rude, M.K.; Siwik, D.A.; Liao, R.; Colucci, W.S.; Sam, F. Mineralocorticoid receptor inhibition ameliorates the transition to myocardial failure and decreases oxidative stress and inflammation in mice with chronic pressure overload. Circulation 2005, 111, 420–427. [Google Scholar] [CrossRef]
- Kagiyama, S.; Matsumura, K.; Goto, K.; Otsubo, T.; Iida, M. Role of Rho kinase and oxidative stress in cardiac fibrosis induced by aldosterone and salt in angiotensin type 1a receptor knockout mice. Regul. Pept. 2010, 160, 133–139. [Google Scholar] [CrossRef]
- Kamalov, G.; Deshmukh, P.A.; Baburyan, N.Y.; Gandhi, M.S.; Johnson, P.L.; Ahokas, R.A.; Bhattacharya, S.K.; Sun, Y.; Gerling, I.C.; Weber, K.T. Coupled calcium and zinc dyshomeostasis and oxidative stress in cardiac myocytes and mitochondria of rats with chronic aldosteronism. J. Cardiovasc. Pharmacol. 2009, 53, 414–423. [Google Scholar] [CrossRef]
- He, B.J.; Joiner, M.L.A.; Singh, M.V.; Luczak, E.D.; Swaminathan, P.D.; Koval, O.M.; Kutschke, W.; Allamargot, C.; Yang, J.; Guan, X.; et al. Oxidation of CaMKII determines the cardiotoxic effects of aldosterone. Nat. Med. 2011, 17, 1610–1618. [Google Scholar] [CrossRef] [PubMed]
- Purohit, A.; Rokita, A.G.; Guan, X.; Chen, B.; Koval, O.M.; Voigt, N.; Neef, S.; Sowa, T.; Gao, Z.; Luczak, E.D.; et al. Oxidized Ca2+/Calmodulin-Dependent Protein Kinase II Triggers Atrial Fibrillation. Circulation 2013, 128, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Brown, N.J. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat. Rev. Nephrol. 2013, 9, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Rocha, R.; Martin-Berger, C.L.; Yang, P.; Scherrer, R.; Delyani, J.; McMahon, E. Selective aldosterone blockade prevents angiotensin II/salt-induced vascular inflammation in the rat heart. Endocrinology 2002, 143, 4828–4836. [Google Scholar] [CrossRef]
- Perrier, R.; Richard, S.; Sainte-Marie, Y.; Rossier, B.C.; Jaisser, F.; Hummler, E.; Bénitah, J.-P. A direct relationship between plasma aldosterone and cardiac L-type Ca2+ current in mice. J. Physiol. 2005, 569, 153–162. [Google Scholar] [CrossRef]
- Bénitah, J.P.; Vassort, G. Aldosterone upregulates Ca2+ current in adult rat cardiomyocytes. Circ. Res. 1999, 85, 1139–1145. [Google Scholar] [CrossRef]
- Lalevée, N.; Rebsamen, M.C.; Barrère-Lemaire, S.; Perrier, E.; Nargeot, J.; Bénitah, J.-P.; Rossier, M.F. Aldosterone increases T-type calcium channel expression and in vitro beating frequency in neonatal rat cardiomyocytes. Cardiovasc. Res. 2005, 67, 216–224. [Google Scholar] [CrossRef]
- Mesquita, T.R.; Auguste, G.; Falcón, D.; Ruiz-Hurtado, G.; Salazar-Enciso, R.; Sabourin, J.; Lefebvre, F.; Viengchareun, S.; Kobeissy, H.; Lechène, P.; et al. Specific Activation of the Alternative Cardiac Promoter of Cacna1c by the Mineralocorticoid Receptor. Circ. Res. 2018, 122, e49–e61. [Google Scholar] [CrossRef]
- Laszlo, R.; Bentz, K.; Konior, A.; Eick, C.; Schreiner, B.; Kettering, K.; Schreieck, J. Effects of selective mineralocorticoid receptor antagonism on atrial ion currents and early ionic tachycardia-induced electrical remodelling in rabbits. Naunyn. Schmiedeberg’s Arch. Pharmacol. 2010, 382, 347–356. [Google Scholar] [CrossRef]
- Ouvrard-Pascaud, A.; Sainte-Marie, Y.; Bénitah, J.P.; Perrier, R.; Soukaseum, C.; Nguyen Dinh Cat, A.A.; Royer, A.; Le Quang, K.; Charpentier, F.; Demolombe, S.; et al. Conditional mineralocorticoid receptor expression in the heart leads to life-threatening arrhythmias. Circulation 2005, 111, 3025–3033. [Google Scholar] [CrossRef]
- Gómez, A.M.; Rueda, A.; Sainte-Marie, Y.; Pereira, L.; Zissimopoulos, S.; Zhu, X.; Schaub, R.; Perrier, E.; Perrier, R.; Latouche, C.; et al. Mineralocorticoid modulation of cardiac ryanodine receptor activity is associated with downregulation of FK506-binding proteins. Circulation 2009, 119, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Qu, J.; Volpicelli, F.M.; Garcia, L.I.; Sandeep, N.; Zhang, J.; Márquez-Rosado, L.; Lampe, P.D.; Fishman, G.I. Gap junction remodeling and spironolactone-dependent reverse remodeling in the hypertrophied heart. Circ. Res. 2009, 104, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Lombès, M.; Oblin, M.E.; Gasc, J.M.; Baulieu, E.E.; Farman, N.; Bonvalet, J.P. Immunohistochemical and biochemical evidence for a cardiovascular mineralocorticoid receptor. Circ. Res. 1992, 71, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Hiratzka, L.F.; Bakris, G.L.; Beckman, J.A.; Bersin, R.M.; Carr, V.F.; Casey, D.E.; Eagle, K.A.; Hermann, L.K.; Isselbacher, E.M.; Kazerooni, E.A.; et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: Executive summary: A report of the american college of cardiology foundation/american heart association task force on pra. Circulation 2010, 121, 266–369. [Google Scholar] [CrossRef]
- Liu, S.; Xie, Z.; Daugherty, A.; Cassis, L.A.; Pearson, K.J.; Gong, M.C.; Guo, Z. Mineralocorticoid receptor agonists induce mouse aortic aneurysm formation and rupture in the presence of high salt. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1568–1579. [Google Scholar] [CrossRef]
- Zavatta, G.; Di Dalmazi, G.; Pizzi, C.; Bracchetti, G.; Mosconi, C.; Balacchi, C.; Pagotto, U.; Vicennati, V. Larger ascending aorta in primary aldosteronism: A 3-year prospective evaluation of adrenalectomy vs. medical treatment. Endocrine 2019, 63, 470–475. [Google Scholar] [CrossRef]
- Tartière, J.-M.; Kesri, L.; Mourad, J.-J.; Safar, M.; Blacher, J. Primary aldosteronism. A risk factor for aortic dissection? J. Mal. Vasc. 2003, 28, 185–189. [Google Scholar]
- Shimizu, A.; Aoi, W.; Akahoshi, M.; Utsunomiya, T.; Doi, Y.; Suzuki, S.; Kuramochi, M.; Hashiba, K. Elevation of plasma renin activity during pregnancy and rupture of a dissecting aortic aneurysm in a patient with primary aldosteronism. Jpn. Heart J. 1983, 24, 995–1006. [Google Scholar] [CrossRef]
- Safi, A.M.; Kwan, T.; Afflu, E.; Alam, M.; Anderson, J.E.; Clark, L.T. Coronary artery aneurysms, aortic dissection, and hypertension secondary to primary aldosteronism: A rare triad—A case report. Angiology 1999, 50, 503–508. [Google Scholar] [CrossRef]
- Lam, K.Y.; Lo, C.Y. The clinicopathologic significance of unilateral adrenal cortical hyperplasia: Report of an unusual case and a review of the literature. Endocr. Pathol. 1999, 10, 243–249. [Google Scholar] [CrossRef]
- Ahmed, S.H.; Husain, N.M.; Khawaja, S.N.; Massey, C.V.; Pettyjohn, F.S. Is primary hyperaldosteronism a risk factor for aortic dissection? Cardiology 2007, 108, 48–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Luo, F.; Fan, P.; Meng, X.; Yang, K.; Zhou, X. Is primary aldosteronism a potential risk factor for aortic dissection? A case report and literature review. BMC Endocr. Disord. 2020, 20, 115. [Google Scholar] [CrossRef] [PubMed]
- Boczar, K.E.; Boodhwani, M.; Beauchesne, L.; Dennie, C.; Chan, K.L.; Wells, G.A.; Coutinho, T. Aortic Stiffness, Central Blood Pressure, and Pulsatile Arterial Load Predict Future Thoracic Aortic Aneurysm Expansion. Hypertension 2020, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Štrauch, B.; Petrák, O.; Wichterle, D.; Zelinka, T.; Holaj, R.; Widimský, J. Increased Arterial Wall Stiffness in Primary Aldosteronism in Comparison with Essential Hypertension. Am. J. Hypertens. 2006, 19, 909–914. [Google Scholar] [CrossRef]
- Bernini, G.; Galetta, F.; Franzoni, F.; Bardini, M.; Taurino, C.; Bernardini, M.; Ghiadoni, L.; Bernini, M.; Santoro, G.; Salvetti, A. Arterial stiffness, intima-media thickness and carotid artery fibrosis in patients with primary aldosteronism. J. Hypertens. 2008, 26, 2399–2405. [Google Scholar] [CrossRef]
- Vizzardi, E.; Pina, P.D.; Caretta, G.; Bonadei, I.; Sciatti, E.; Lombardi, C.; D’Aloia, A.; Curnis, A.; Metra, M. The effect of aldosterone-antagonist therapy on aortic elastic properties in patients with nonischemic dilated cardiomyopathy. J. Cardiovasc. Med. 2015, 16, 597–602. [Google Scholar] [CrossRef]
- Nishizaka, M.K.; Zaman, M.A.; Green, S.A.; Renfroe, K.Y.; Calhoun, D.A. Impaired endothelium-dependent flow-mediated vasodilation in hypertensive subjects with hyperaldosteronism. Circulation 2004, 109, 2857–2861. [Google Scholar] [CrossRef]
- Demirkiran, A.; Everaars, H.; Elitok, A.; van de Ven, P.M.; Smulders, Y.M.; Dreijerink, K.M.; Tanakol, R.; Ozcan, M. Hypertension with primary aldosteronism is associated with increased carotid intima-media thickness and endothelial dysfunction. J. Clin. Hypertens. 2019, 21, 932–941. [Google Scholar] [CrossRef]
- Takata, M.; Amiya, E.; Watanabe, M.; Omori, K.; Imai, Y.; Fujita, D.; Nishimura, H.; Kato, M.; Morota, T.; Nawata, K.; et al. Impairment of flow-mediated dilation correlates with aortic dilation in patients with Marfan syndrome. Heart Vessel. 2014, 29, 478–485. [Google Scholar] [CrossRef]
- Tzemos, N.; Lyseggen, E.; Silversides, C.; Jamorski, M.; Tong, J.H.; Harvey, P.; Floras, J.; Siu, S. Endothelial function, carotid-femoral stiffness, and plasma matrix metalloproteinase-2 in men with bicuspid aortic valve and dilated aorta. J. Am. Coll. Cardiol. 2010, 55, 660–668. [Google Scholar] [CrossRef]
- Wang, Y.B.; Li, Y.; Deng, Y.B.; Liu, Y.N.; Zhang, J.; Sun, J.; Zhu, Y.; Li, L.; Tang, Q.Y.; Zhou, W. Enlarged Size and Impaired Elastic Properties of the Ascending Aorta are Associated with Endothelial Dysfunction and Elevated Plasma Matrix Metalloproteinase-2 Level in Patients with Bicuspid Aortic Valve. Ultrasound Med. Biol. 2018, 44, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Ouyang, J.; Wang, C.; Wu, Z.; Ma, X.; Li, H.; Xu, H.; Hu, Z.; Li, J.; Wang, B.; et al. Aortic cell apoptosis in rat primary aldosteronism model. J. Huazhong Univ. Sci. Technol. Med. Sci. 2010, 30, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Whaley-Connell, A.T.; Habibi, J.; Rehmer, J.; Rehmer, N.; Patel, K.; Hayden, M.; DeMarco, V.; Ferrario, C.M.; Ibdah, J.A.; et al. Mineralocorticoid receptor antagonism attenuates vascular apoptosis and injury via rescuing protein kinase B activation. Hypertension 2009, 53, 158–165. [Google Scholar] [CrossRef] [PubMed]
- Schwerdt, G.; Frisch, A.; Mildenberger, S.; Hilgenfeld, T.; Grossmann, C.; Gekle, M. Influence of aldosterone and salt or ouabain in A10 rat aorta smooth muscle cells. J. Vasc. Res. 2012, 49, 231–241. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, C.; Lu, Y.; Gong, H.; Wu, Z.; Ma, X.; Li, H.; Wang, B.; Zhang, X. Mineralocorticoid receptor antagonism protects the aorta from vascular smooth muscle cell proliferation and collagen deposition in a rat model of adrenal aldosterone-producing adenoma. J. Physiol. Biochem. 2018, 74, 17–24. [Google Scholar] [CrossRef]
- Surman, T.L.; Abrahams, J.M.; Manavis, J.; Finnie, J.; O’Rourke, D.; Reynolds, K.J.; Edwards, J.; Worthington, M.G.; Beltrame, J. Histological regional analysis of the aortic root and thoracic ascending aorta: A complete analysis of aneurysms from root to arch. J. Cardiothorac. Surg. 2021, 16, 255. [Google Scholar] [CrossRef]
- Benetos, A.; Lacolley, P.; Safar, M.E. Prevention of aortic fibrosis by spironolactone in spontaneously hypertensive rats. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1152–1156. [Google Scholar] [CrossRef]
- Lacolley, P.; Safar, M.E.; Lucet, B.; Ledudal, K.; Labat, C.; Benetos, A. Prevention of aortic and cardiac fibrosis by spironolactone in old normotensive rats. J. Am. Coll. Cardiol. 2001, 37, 662–667. [Google Scholar] [CrossRef]
- Lacolley, P.; Labat, C.; Pujol, A.; Delcayre, C.; Benetos, A.; Safar, M. Increased carotid wall elastic modulus and fibronectin in aldosterone-salt-treated rats: Effects of eplerenone. Circulation 2002, 106, 2848–2853. [Google Scholar] [CrossRef]
- Iglarz, M.; Touyz, R.M.; Viel, E.C.; Amiri, F.; Schiffrin, E.L. Involvement of oxidative stress in the profibrotic action of aldosterone. Am. J. Hypertens. 2004, 17, 597–603. [Google Scholar] [CrossRef]
- Park, J.B.; Schiffrin, E.L. Cardiac and vascular fibrosis and hypertrophy in aldosterone-infused rats: Role of endothelin-1. Am. J. Hypertens. 2002, 15, 164–169. [Google Scholar] [CrossRef]
- Harvey, A.P.; Montezano, A.C.; Hood, K.Y.; Lopes, R.A.; Rios, F.; Ceravolo, G.; Graham, D.; Touyz, R.M. Vascular dysfunction and fibrosis in stroke-prone spontaneously hypertensive rats: The aldosterone-mineralocorticoid receptor-Nox1 axis. Life Sci. 2017, 179, 110–119. [Google Scholar] [CrossRef] [PubMed]
- De las Heras, N.; Ruiz-Ortega, M.; Miana, M.; Rupérez, M.; Sanz-Rosa, D.; Aragoncillo, P.; Mezzano, S.; Cachofeiro, V.; Egido, J.; Lahera, V. Interactions between aldosterone and connective tissue growth factor in vascular and renal damage in spontaneously hypertensive rats. J. Hypertens. 2007, 25, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Gekle, M.; Mildenberger, S.; Freudinger, R.; Grossmann, C. Altered collagen homeostasis in human aortic smooth muscle cells (HAoSMCs) induced by aldosterone. Pflug. Arch. 2007, 454, 403–413. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).