
Combination Effect of Cilengitide with Erlotinib on TGF-β1-Induced Epithelial-to-Mesenchymal Transition in Human Non-Small Cell Lung Cancer Cells


Abstract
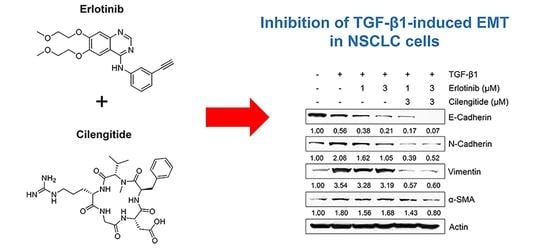
:
1. Introduction
2. Results
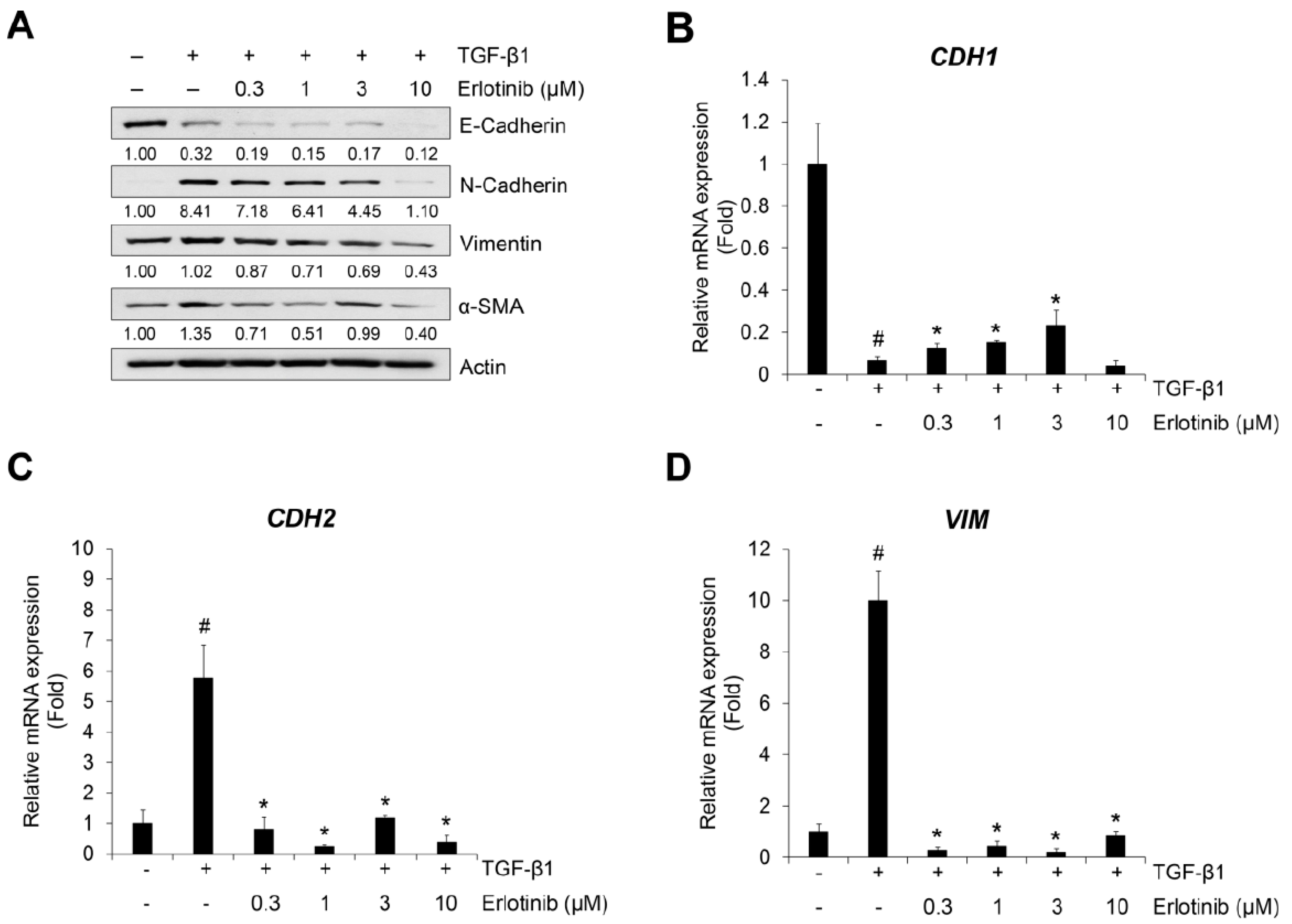
2.1. Erlotinib Inhibits Tgf-Β1–Induced Emt-Related Phenotype Changes
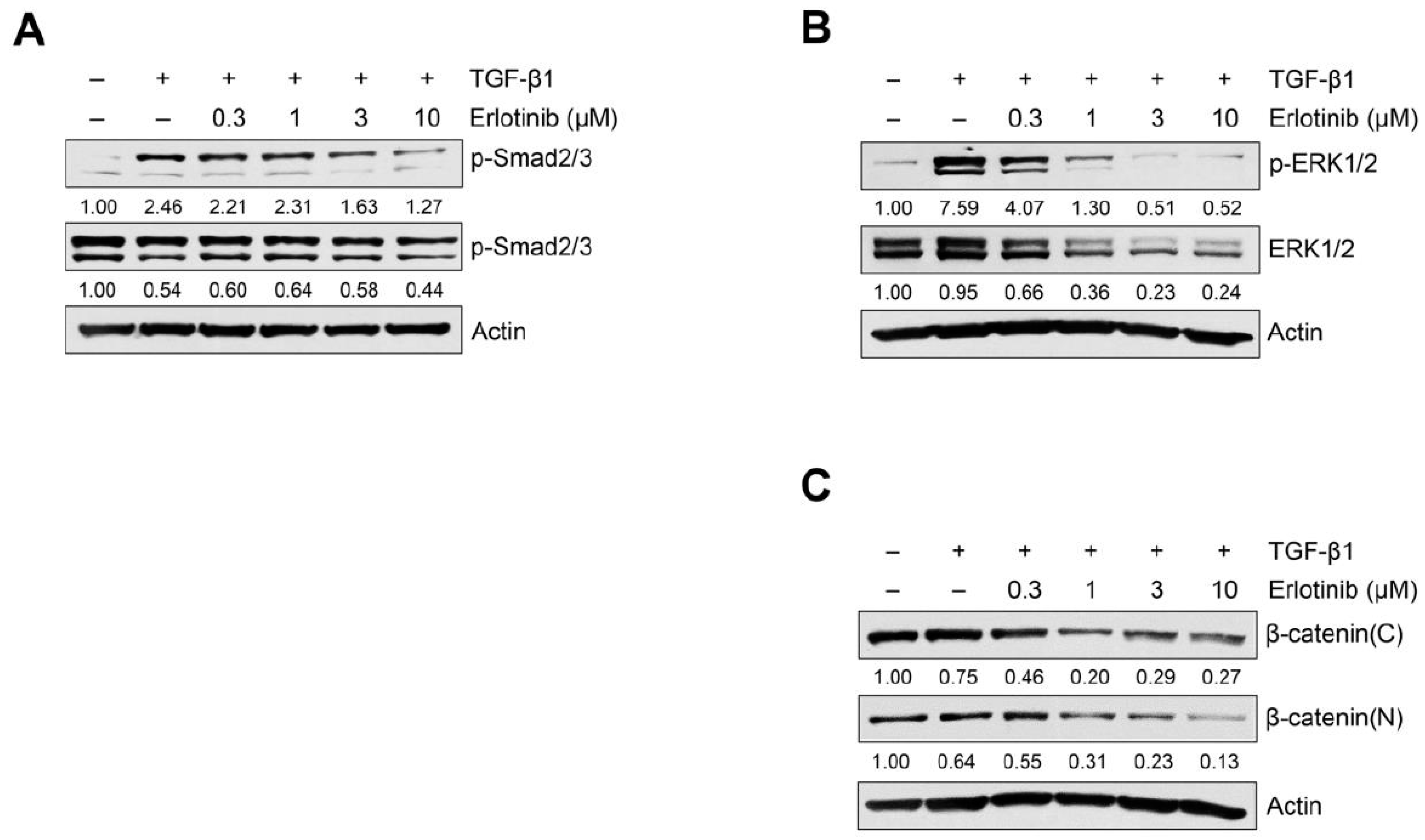
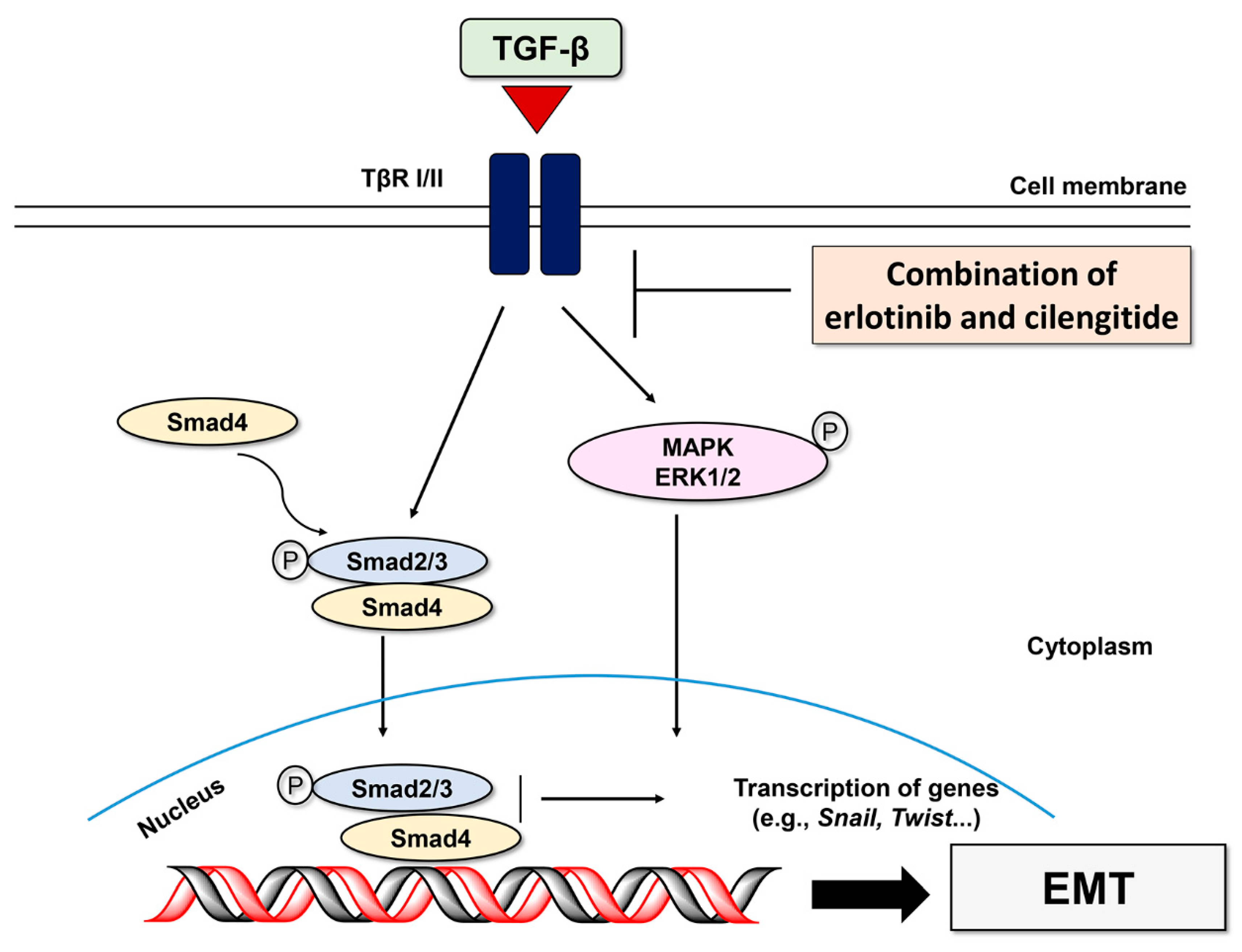
2.2. Erlotinib Inhibits Tgf-Β1–Induced Smad and Non-Smad Signaling
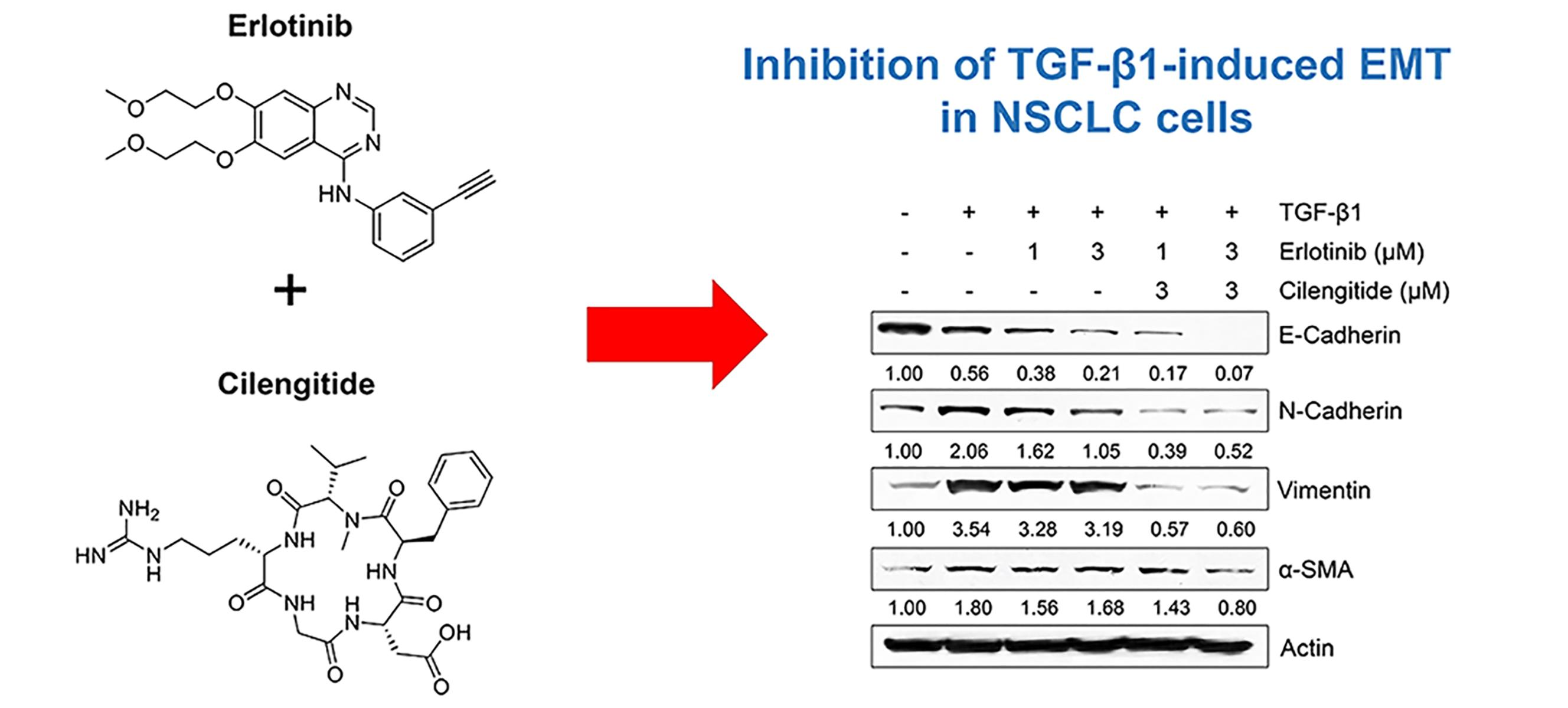
2.3. Combination Cilengitide with Erlotinib Exerts a Synergistic Inhibitory Effect on NSCLC Cell Viability and EMT Markers
2.4. The Combination of Cilengitide with Erlotinib Exerts a Synergistic Inhibitory Effect on Tgf-Β1–Mediated Invasion and MMP Secretion
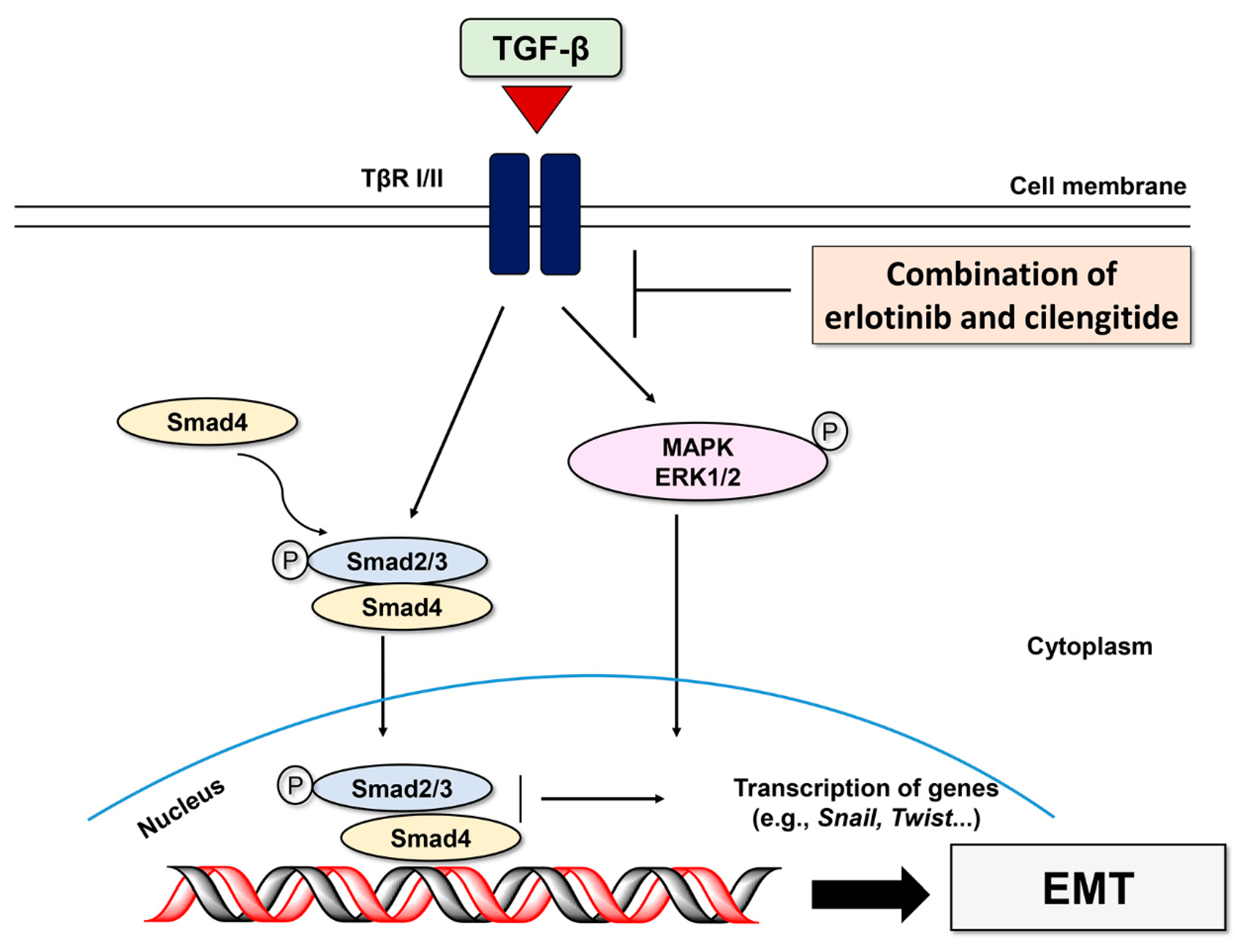
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture and Cell Viability Assay
4.3. Western Blot Analysis
4.4. Invasion Assay
4.5. Quantitative Real-Time PCR (qRT-PCR) Analysis
4.6. Gelatin Zymography
4.7. Analysis of Combined Drug Effects
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.; Ward, E.; Brawley, O.; Jemal, A. Cancer statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin. 2011, 61, 212–236. [Google Scholar] [CrossRef]
- Duma, N.; Santana-Davila, R.; Molina, J.R. Non-Small Cell Lung Cancer: Epidemiology, Screening, Diagnosis, and Treatment. Mayo Clin. Proc. 2019, 94, 1623–1640. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Drabsch, Y.; ten Dijke, P. TGF-β signalling and its role in cancer progression and metastasis. Cancer Metastasis Rev. 2012, 31, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; Mareel, M.M.; Van Roy, F.M.; Birchmeier, W. Dissecting tumor cell invasion: Epithelial cells acquire invasive properties after the loss of uvomorulin-mediated cell-cell adhesion. J. Cell Biol. 1989, 108, 2435–2447. [Google Scholar] [CrossRef] [PubMed]
- Barcellos-Hoff, M.H.; Akhurst, R.J. Transforming growth factor-beta in breast cancer: Too much, too late. Breast Cancer Res. 2009, 11, 202. [Google Scholar] [CrossRef]
- Buck, M.B.; Knabbe, C. TGF-beta signaling in breast cancer. Ann. N. Y. Acad. Sci. 2006, 1089, 119–126. [Google Scholar] [CrossRef]
- Massagué, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef]
- Massagué, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef]
- Peinado, H.; Portillo, F.; Cano, A. Transcriptional regulation of cadherins during development and carcinogenesis. Int. J. Dev. Biol. 2004, 48, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Mani, S.A.; Donaher, J.; Ramaswamy, L.S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galliher, A.J.; Neil, J.R.; Schiemann, W.P. Role of transforming growth factor-beta in cancer progression. Future Oncol. 2006, 2, 743–763. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.F.; Zhang, H.J.; Wang, H.B.; Zhu, J.; Zhou, W.Y.; Zhang, H.; Zhao, M.C.; Su, J.M.; Gao, W.; Zhang, L.; et al. Transforming growth factor-β1 induces epithelial-to-mesenchymal transition in human lung cancer cells via PI3K/Akt and MEK/Erk1/2 signaling pathways. Mol. Biol. Rep. 2012, 39, 3549–3556. [Google Scholar] [CrossRef] [PubMed]
- Medici, D.E.; Hay, D.; Olsen, B.R. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol. Biol. Cell 2008, 19, 4875–4887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, W.H.; Yang, J.C.; Mok, T.S.; Loong, H.H. Overview of current systemic management of EGFR-mutant NSCLC. Ann. Oncol. 2018, 29 (Suppl. 1), i3–i9. [Google Scholar] [CrossRef]
- Molina, J.R.; Yang, P.; Cassivi, S.D.; Schild, S.E.; Adjei, A.A. Non-small cell lung cancer: Epidemiology, risk factors, treatment, and survivorship. Mayo Clin. Proc. 2008, 83, 584–594. [Google Scholar] [CrossRef]
- Suda, K.; Tomizawa, K.; Fujii, M.; Murakami, H.; Osada, H.; Maehara, Y.; Yatabe, Y.; Sekido, Y.; Mitsudomi, T. Epithelial to mesenchymal transition in an epidermal growth factor receptor-mutant lung cancer cell line with acquired resistance to erlotinib. J. Thorac. Oncol. 2011, 6, 1152–1161. [Google Scholar] [CrossRef] [Green Version]
- Qu, Y.; Wu, X.; Yin, Y.; Yang, Y.; Ma, D.; Li, H. Antitumor activity of selective MEK1/2 inhibitor AZD6244 in combination with PI3K/mTOR inhibitor BEZ235 in gefitinib-resistant NSCLC xenograft models. J. Exp. Clin. Cancer Res. 2014, 33, 52. [Google Scholar] [CrossRef] [Green Version]
- La Monica, S.; Madeddu, D.; Tiseo, M.; Vivo, V.; Galetti, M.; Cretella, D.; Bonelli, M.; Fumarola, C.; Cavazzoni, A.; Falco, A.; et al. Combination of Gefitinib and Pemetrexed Prevents the Acquisition of TKI Resistance in NSCLC Cell Lines Carrying EGFR-Activating Mutation. J. Thorac. Oncol. 2016, 11, 1051–1063. [Google Scholar] [CrossRef] [Green Version]
- Han, S.Y.; Zhao, W.; Sun, H.; Zhou, N.; Zhou, F.; An, G.; Li, P.P. Marsdenia tenacissima extract enhances gefitinib efficacy in non-small cell lung cancer xenografts. Phytomedicine 2015, 22, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Tam, K.Y. Combination Strategies Using EGFR-TKi in NSCLC Therapy: Learning from the Gap between Pre-Clinical Results and Clinical Outcomes. Int. J. Biol. Sci. 2018, 14, 204–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.C.; Gadgeel, S.M.; Sequist, L.V.; Wu, C.L.; Papadimitrakopoulou, V.A.; Su, W.C.; Fiore, J.; Saraf, S.; Raftopoulos, H.; Patnaik, A. Pembrolizumab in Combination with Erlotinib or Gefitinib as First-Line Therapy for Advanced NSCLC with Sensitizing EGFR Mutation. J. Thorac. Oncol. 2019, 14, 553–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, H.; Fukuhara, T.; Furuya, N.; Watanabe, K.; Sugawara, S.; Iwasawa, S.; Tsunezuka, Y.; Yamaguchi, O.; Okada, M.; Yoshimori, K.; et al. Erlotinib plus bevacizumab versus erlotinib alone in patients with EGFR-positive advanced non-squamous non-small-cell lung cancer (NEJ026): Interim analysis of an open-label, randomised, multicentre, phase 3 trial. Lancet Oncol. 2019, 20, 625–635. [Google Scholar] [CrossRef]
- Yue, D.; Xu, S.; Wang, Q.; Li, X.; Shen, Y.; Zhao, H.; Chen, C.; Mao, W.; Liu, W.; Liu, J.; et al. Erlotinib versus vinorelbine plus cisplatin as adjuvant therapy in Chinese patients with stage IIIA EGFR mutation-positive non-small-cell lung cancer (EVAN): A randomised, open-label, phase 2 trial. Lancet Respir. Med. 2018, 6, 863–873. [Google Scholar] [CrossRef]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [Green Version]
- Plow, E.F.; Haas, T.A.; Zhang, L.; Loftus, J.; Smith, J.W. Ligand binding to integrins. J. Biol. Chem. 2000, 275, 21785–21788. [Google Scholar] [CrossRef] [Green Version]
- Heckmann, D.; Kessler, H. Design and chemical synthesis of integrin ligands. Methods Enzymol. 2007, 426, 463–503. [Google Scholar] [CrossRef]
- Cox, D.; Brennan, M.; Moran, N. Integrins as therapeutic targets: Lessons and opportunities. Nat. Rev. Drug Discov. 2010, 9, 804–820. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Barlesi, F.; Waller, C.F.; Bennouna, J.; Gridelli, C.; Goekkurt, E.; Vorhoeven, D.; Szczesna, A.; Feurer, M.; Milanowski, J.; et al. Cilengitide combined with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small-cell lung cancer (NSCLC) patients: Results of an open-label, randomized, controlled phase II study (CERTO). Ann. Oncol. 2015, 26, 1734–1740. [Google Scholar] [CrossRef]
- Heiduschka, G.; Lill, C.; Schneider, S.; Seemann, R.; Kornek, G.; Schmid, R.; Kotowski, U.; Thurnher, D. The effect of cilengitide in combination with irradiation and chemotherapy in head and neck squamous cell carcinoma cell lines. Strahlenther. Onkol. 2014, 190, 472–479. [Google Scholar] [CrossRef]
- Lautenschlaeger, T.; Perry, J.; Peereboom, D.; Li, B.; Ibrahim, A.; Huebner, A.; Meng, W.; White, J.; Chakravarti, A. In vitro study of combined cilengitide and radiation treatment in breast cancer cell lines. Radiat. Oncol. 2013, 8, 246. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Wan, X.; Xu, J.; Wang, H.; Chen, H.; Zeng, Q.; Zhang, W.; Zhao, B. Therapeutic options for advanced epidermal growth factor receptor (EGFR)-mutant non-small cell lung cancer: A Bayesian network secondary analysis. Aging 2020, 12, 7129–7162. [Google Scholar] [CrossRef]
- Haddad, T.; Qin, R.; Lupu, R.; Satele, D.; Eadens, M.; Goetz, M.P.; Erlichman, C.; Molina, J. A phase I study of cilengitide and paclitaxel in patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2017, 79, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Massabeau, C.; Khalifa, J.; Filleron, T.; Modesto, A.; Bigay-Gamé, L.; Plat, G.; Dierrichx, L.; Aziza, R.; Rouquetter, I.; Gomez-Roca, C.; et al. Continuous Infusion of Cilengitide Plus Chemoradiotherapy for Patients with Stage III Non-Small-cell Lung Cancer: A Phase I Study. Clin. Lung Cancer 2018, 19, e277–e285. [Google Scholar] [CrossRef]
- Jeong, J.; Kim, J. Cyclic RGD Pentapeptide Cilengitide Enhances Efficacy of Gefitinib on TGF-β1-Induced Epithelial-to-Mesenchymal Transition and Invasion in Human Non-Small Cell Lung Cancer Cells. Front. Pharmacol. 2021, 12, 639095. [Google Scholar] [CrossRef] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Extracellular matrix (ECM) stiffness and degradation as cancer drivers. J. Cell. Biochem. 2019, 120, 2782–2790. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428, Erratum in: J. Clin. Investig. 2010, 120, 1786. [Google Scholar] [CrossRef] [Green Version]
- Jakobsen, K.R.; Demuth, C.; Sorensen, B.S.; Nielsen, A.L. The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl. Lung Cancer Res. 2016, 5, 172–182. [Google Scholar] [CrossRef] [Green Version]
- Ong, Y.S.; Gao, L.; Kalesh, K.A.; Yu, Z.; Wang, J.; Liu, C.; Li, Y.; Sun, H.; Lee, S.S. Recent Advances in Synthesis and Identification of Cyclic Peptides for Bioapplications. Curr. Top Med. Chem. 2017, 17, 2302–2318. [Google Scholar] [CrossRef]
- Kulhari, H.; Pooja, D.; Kota, R.; Reddy, T.S.; Tabor, R.F.; Shukla, R.; Adams, D.J.; Sistla, R.; Bansal, V. Cyclic RGDfK Peptide Functionalized Polymeric Nanocarriers for Targeting Gemcitabine to Ovarian Cancer Cells. Mol. Pharm. 2016, 13, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, X. Integrin targeted delivery of chemotherapeutics. Theranostics 2011, 1, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, K.Y.; Kim, J. Cyclic pentapeptide cRGDfK enhances the inhibitory effect of sunitinib on TGF-β1-induced epithelial-to-mesenchymal transition in human non-small cell lung cancer cells. PLoS ONE 2020, 15, e0232917. [Google Scholar] [CrossRef]
- Wichmann, G.; Cedra, S.; Schlegel, D.; Kolb, M.; Wiegand, S.; Boehm, A.; Hofer, M.; Dietz, A. Cilengitide and Cetuximab Reduce Cytokine Production and Colony Formation of Head and Neck Squamous Cell Carcinoma Cells. Ex Vivo Anticancer Res. 2017, 37, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kim, J.; Moon, S.H.; Kim, B.T.; Chae, C.H.; Lee, J.Y.; Kim, S.H. A novel aminothiazole KY-05009 with potential to inhibit Traf2- and Nck-interacting kinase (TNIK) attenuates TGF-β1-mediated epithelial-to-mesenchymal transition in human lung adenocarcinoma A549 cells. PLoS ONE 2014, 9, e110180. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzym. Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Zhao, L.; Wientjes, M.G.; Au, J.L. Evaluation of combination chemotherapy: Integration of nonlinear regression, curve shift, isobologram, and combination index analyses. Clin. Cancer Res. 2004, 10, 7994–8004. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell line. | Erlotinib (μM) | Cilengitide (μM) | CI Value |
|---|---|---|---|
| A549 | 0.1 | 0.1 | 0.2655 |
| 0.3 | 0.3 | 0.5058 | |
| 1 | 1 | 0.6415 | |
| 3 | 3 | 0.2346 | |
| 10 | 10 | 0.0298 | |
| 30 | 30 | 0.0100 | |
| H1299 | 0.1 | 0.1 | 0.5450 |
| 0.3 | 0.3 | 0.7446 | |
| 1 | 1 | 1.7137 | |
| 3 | 3 | 0.4097 | |
| 10 | 10 | 0.0357 | |
| 30 | 30 | 0.0359 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeong, J.; Kim, J. Combination Effect of Cilengitide with Erlotinib on TGF-β1-Induced Epithelial-to-Mesenchymal Transition in Human Non-Small Cell Lung Cancer Cells. Int. J. Mol. Sci. 2022, 23, 3423. https://doi.org/10.3390/ijms23073423
Jeong J, Kim J. Combination Effect of Cilengitide with Erlotinib on TGF-β1-Induced Epithelial-to-Mesenchymal Transition in Human Non-Small Cell Lung Cancer Cells. International Journal of Molecular Sciences. 2022; 23(7):3423. https://doi.org/10.3390/ijms23073423
Chicago/Turabian StyleJeong, Jisu, and Jiyeon Kim. 2022. "Combination Effect of Cilengitide with Erlotinib on TGF-β1-Induced Epithelial-to-Mesenchymal Transition in Human Non-Small Cell Lung Cancer Cells" International Journal of Molecular Sciences 23, no. 7: 3423. https://doi.org/10.3390/ijms23073423
APA StyleJeong, J., & Kim, J. (2022). Combination Effect of Cilengitide with Erlotinib on TGF-β1-Induced Epithelial-to-Mesenchymal Transition in Human Non-Small Cell Lung Cancer Cells. International Journal of Molecular Sciences, 23(7), 3423. https://doi.org/10.3390/ijms23073423