Abstract
Non-melanoma skin cancers are cutaneous malignancies representing the most common form of cancer in the United States. They are comprised predominantly of basal cell carcinomas and squamous cell carcinomas (cSCC). The incidence of cSCC is increasing, resulting in substantial morbidity and ever higher treatment costs; currently in excess of one billion dollars, per annum. Here, we review research defining the molecular basis and development of cSCC that aims to provide new insights into pathogenesis and drive the development of novel, cost and morbidity saving therapies.
1. Introduction
Cutaneous squamous cell carcinoma (cSCC) is the second most common cancer among Caucasians, with estimates of approximately 1.1 million new cases annually in the US [1,2]. While outcomes are favorable for most patients, and the vast majority of cSCCs are cured with complete excision, an estimated 3–7% of patients develop metastases that lead to significant morbidity and mortality [2,3,4]. Thus, early diagnosis and treatment of cSCCs, and their premalignant precursors, is crucial to minimize morbidity and conserve healthcare resources.
Most cSCCs arise in a progressive fashion from premalignant/noninvasive precursor lesions [5]. The earliest clinically detectable precursor lesion is actinic keratosis (AK). AKs can be distinguished from surrounding keratinocytes by the presence of hyperplasia and hyperkeratosis clinically, and histologically by the presence of basal keratinocyte dysplasia and overlying parakeratosis [6]. AKs share some histologic findings with other, less common lesions that may themselves be precursors to keratinocytic malignancy, such as acanthosis, which is associated with human papillomavirus infection or arsenic poisoning. The most well studied and broadly accepted precursor lesion in the field cancerization theory, however, is the AK, which may persist as premalignant lesions, or even regress spontaneously [7]. A small percentage of AKs, incidentally, acquire additional genetic and epigenetic changes and progress to cutaneous squamous cell carcinoma in situ (SCCIS) and ultimately cSCC, both of which are clinically larger lesions, similarly characterized histologically by the presence of parakeratosis, with more pronounced dysplasia wherein the full thickness of the epidermis is replaced by malignant keratinocytes that are either bounded by the basement membrane in the case of SCCIS, or invade the dermis and surrounding structures in the case of cSCC [5]. A small subset of cSCC may acquire additional genetic and epigenetic features that lead to metastatic disease [8,9].
The evolution of malignant properties that underlies this progression is a current focus of research and encompasses the concept of field cancerization [10,11], which proposes that precancerous lesions such as AKs and SCCIS arise from mutated, subclinical clones of keratinocytes within a clinically unremarkable epidermis. In most instances, UV-exposure initiates the mutagenic process in skin, causing mutations in individual keratinocytes that, provided they confer a survival advantage, are selected over time. This selection results in the presence of multiple, mutated keratinocyte subclones within clinically normal appearing skin. Additional genetic and epigenetic changes in these clones can promote neoplastic selection to produce AKs, which can then progress to SCCIS and beyond, ultimately resulting in polyclonal cSCCs that are comprised of multiple, competing keratinocyte clones.
Molecularly, cSCC arises from the accumulation of genetic and epigenetic alterations in keratinocytes over time that permit development of an invasive tumor [9]. The accumulation of genetic alterations, and development of malignant and premalignant lesions, is accelerated when intrinsic defenses are compromised. Examples include patients on chronic immunosuppression, or those with a heritable predisposition to cancer, such as the disease states of Xeroderma Pigmentosum or Bloom syndrome [12,13]. DNA mutations causing qualitative changes in gene expression can occur due to defects in DNA replication, repair, or recombination mechanisms. Endogenous mutagens result in spontaneous alterations of DNA, including free radical damage due to reactive oxygen species, deamination, or depurination [14,15]. Exogenous mutagens include sunlight (UVB and UVA), smoking, and dietary components. Epigenetic alterations, leading to quantitative changes in gene expression, can also facilitate inappropriate transcriptional activation and silencing of genes [16]. Both genetic and epigenetic alterations can increase the overall mutation rate, enhance proliferation, and decrease cell death.
Advances in our understanding of the epidemiologic risk factors and molecular mechanisms driving tumorigenesis have resulted in new therapeutic options of varying efficacy for patients with locally advanced or metastatic disease [11]. The prevention of such disease all together rests upon the development of field treatments that largely target early-stage precursor lesions and subclinical disease. Currently, most field treatment protocols are hampered by poor compliance due to prolonged and repeated treatment periods, as well as clinical toxicity; their efficacy is difficult to evaluate in the absence of universally accepted endpoints and objective measurements of field disease [10]. These circumstances motivate research to identify new molecular targets for field treatment strategies in the early stages of disease to prevent progression to invasive, late-stage disease, with its inherent risk of metastases. In this review, we discuss recent findings on the genetics, epigenetics, and biology of cSCC development. The therapeutic implications of these data will also be discussed.
2. Genetic Alterations in cSCC
2.1. Mechanisms of Genetic Alterations in cSCC
Mutations in DNA can occur through a variety of mechanisms. DNA damage due to exogenous factors including UV light, chemicals, and ionizing radiation can lead to mutations if not repaired [15]. Mutations can also occur through endogenous factors such as mitotic errors, errors in DNA repair, or genome editing, as well as reactive oxygen species [14]. For cSCC to arise, the mutations need to occur in long-lived cells in the epidermal compartment which reside in the basal cell layer. DNA mutations that alter/destabilize the double helix structure, such as UV-induced pyrimidine dimers, are typically corrected through DNA repair processes such as nucleotide excision repair (NER). The importance of this evolutionarily-conserved mechanism is demonstrated by patients with Xeroderma Pigmentosum (XP) [17]. Loss of any one effector protein of the NER pathway leads to early development of premalignant lesions and malignant skin cancers [17,18,19]. While numerous mechanisms contribute to mutagenesis, epidemiological and clinical data support the concept that cumulative lifetime exposure to ultraviolet radiation (UV) is the primary carcinogen responsible for cSCC [20].
UV irradiation is capable of initiating and promoting the progression of all stages of squamous carcinogenesis [18,21]. Both UVB and UVA radiation promote skin cancer by altering keratinocyte signaling, inducing oxidative stress, and producing DNA mutations [22]. UV exposure leads to ATP consumption due to the activation of DNA repair systems, which when overactivated (e.g., PARPs) is detrimental for the cell [23] (PMID: 34638427). UVB radiation possesses sufficient photonic energy to promote structural rearrangements (DNA damage) resulting in cyclobutane pyrimidine dimers (CPD) or (6-4) photoproducts [23,24]. If the damaged DNA strand is not repaired prior to replication, the daughter strand acquires the change in base (e.g., C ≥ T) and a mutation has occurred [25]. This process generates a “UVB signature” characterized by high rates of C > T transitions and CC > TT double base changes [21,26]. UVA and UVB can produce cellular oxidative stress, leading to formation of reactive oxygen species (ROS); these compounds can promote 8-oxo-deoxyguanine formation, which, if not repaired prior to DNA replication, will lead to a G > A mutation. The location of these mutations and the resulting changes in the gene products and genome structure determines the fate of mutated keratinocytes.
2.2. Genomic Characterization of cSCC
Genomic analyses of cSCCs have revealed much about the molecular mechanisms that drive the transition from benign to malignant keratinocytes. cSCCs have the highest tumor mutation burden among malignancies harboring, on average, ≈50 mutations per megabase pair (Mbp) of DNA [15,26,27,28]. It is hypothesized that most of these mutations are “passengers”, offering little-to-no growth advantage nor impact on tumor progression, while a subset represent “driver mutations” that promote tumorigenesis by regulating cell fate, growth, survival, or genomic maintenance [9]. Whole exome and targeted sequencing analyses of unremarkable skin, AKs, SCCIS, and cSCCs provide evidence that numerous driver pathways act preferentially at specific stages to promote tumorigenesis [20,29,30,31,32,33,34]. Some of the mutated driver genes include NOTCH1-3, TP53, FAT1, PIK3CA, CDKN2A, HRAS, KMT2C, KNSTRN, EGFR, CARD11, MYC, MLL2, MAP3K9, PTEN, SF3B1, VPS41, and WHSCI [20,26,31,32,33,34,35,36]. However, our understanding of the molecular mechanisms and genetic drivers of cSCCs are complicated by the relatively high (~5.8 mutations per Mb) burden of somatic mutations in normal sun-exposed skin [26,37]. This mutation rate is similar to that observed in many cancers [15], indicating that normal sun exposed skin is a patchwork of precancerous cells harboring mutations in known driver pathways. Some of the most frequently cited genes mutated in normal appearing skin include known drivers such as TP53, NOTCH1, NOTCH2, and FAT1 [34,37]. These findings have important implications for interpreting and understanding squamous carcinogenesis and suggest that certain mutations are somehow tolerated in vivo. Here, we will discuss the genetic alterations that have been repeatedly shown to play a role in cSCC tumorigenesis.
2.2.1. TP53
TP53 is the most commonly mutated gene in human cancers and plays a critical role in cSCC tumorigenesis [38]. The TP53 tumor suppressor gene encodes a 53-kDa phosphoprotein that functions to regulate cell cycle arrest, senescence, DNA-repair, and apoptosis in response to cellular stress [39,40,41,42]. p53 transmits stress-inducing signals to different anti-proliferative cellular responses primarily by regulating transcription [43]. In an inactive state, p53 is bound to Mdm2, which antagonizes the transcriptional activity of p53, induces its ubiquitylation, and promotes nuclear export of p53 [43,44]. Traditionally, p53 activation in response to cellular stress (e.g., UV-induced DNA damage) occurs through stabilization of p53, sequence specific DNA binding, and transcriptional activation of target genes [45]. Stabilization of p53 occurs primarily through disruption of Mdm2 binding, which can occur through post-translational modification of the amino terminus of p53 or through direct regulation of Mdm2 [46,47]. One such regulator is p14ARF, a tumor suppressor transcribed from the CDKN2A gene, which directly binds to and inhibits Mdm2 to promote p53 activation [47,48,49]. Once stabilized, p53 binds to target genes (e.g., CDKN1A [50], BAX [51], and DDB2 [52]) in a sequence-specific manner [53], activating or repressing gene expression [43]. p53 also transcriptionally regulates Notch genes in keratinocytes through a c-Jun dependent mechanism [54], and decreased p53 transcriptional activity correlates with lower levels of Notch-gene transcripts and protein [55].
Disruption of p53 activity can occur directly through inherited or acquired mutations in TP53 or indirectly through dysregulation of p53 signaling. For example, CDKN2A mutations occur frequently in AK and cSCC and certain mutations have been shown to disrupt p53-dependent functions (discussed below). Mutations in TP53 most frequently involve somatic deletions or missense mutations that alter p53 function [56]. Missense mutations in TP53 most frequently involve 6 “hotspot” amino acids within the DNA binding domain [41,57,58,59]. Multiple mechanisms of action have been ascribed to various TP53 alterations. Deletions and some mutations simply result in loss of function (LOF) of this important tumor suppressor. Other mutations inhibit p53 function by causing mutant p53 to act as a dominant negative inhibitor over WT p53 proteins [60]. Still, other missense mutations appear to confer gain of function (GOF) characteristics by altering the ability of mutant p53 to bind canonical p53 DNA binding regions and conferring the ability to bind alternative genes [61] and transcription factors (e.g., p63) [59]. GOF TP53 mutations have been observed in subsets of patients with missense germline TP53 mutations that have detectable mutant p53 protein expression and a significantly earlier cancer onset than patients with germline TP53 mutations, resulting in a loss of p53 protein expression [59,62]. GOF TP53 mutations have also been associated with an increased incidence of metastatic dissemination and drug resistance [63,64,65]. These findings indicate that malfunction of p53 is an important cause of carcinogenesis with implications for understanding cutaneous squamous cell carcinogenesis.
Analysis of mutations in TP53 has established a clear connection between UV exposure, DNA damage, and skin carcinogenesis [41]. Early sequencing studies of human cSCC identified an abundance of point mutations in TP53 resulting from UVB-induced C > T or CC > TT base substitutions [21]. In human sequencing studies, TP53 mutations have been identified in approximately 60% of actinic keratoses and 50–90% of cSCC [36,66]. Studies in mice have shown that inactivating mutations in TP53 promote AK and carcinoma in in situ formation [67,68]. In human cSCC, loss of p53 function has been shown to promote survival of mutant clones and the acquisition of additional mutations. Using whole exome sequencing, it was found that loss of the second TP53 allele in human cSCCs strongly increases the likelihood of additional DNA mutations and chromosomal aberrations [69]. GOF mutations have also recently been described to promote HNSCC tumorigenesis, invasion, and metastasis through transcription-independent control of the p53-AMPK-FOXO3a-FOXM1 cascade [70].
While the role of TP53 mutations in cSCC and its progression are clearly established, our understanding of the stage at which these mutations contribute most to cSCC carcinogenesis continues to evolve. Sequencing studies of grossly unremarkable sun-exposed skin identified clonal TP53 mutations in seemingly normal skin [15,37,71]. AKs are collections of atypical keratinocytes, localized to the basal layer of the interfollicular epidermis and have the potential to progress to higher grade lesions via lateral and superficial expansion [72]. Recent sequencing studies of AKs, confirmed by microscopic examination, found TP53 to be significantly mutated in AKs [33]. Another recent study, using laser capture microdissection of SCCIS and adjacent sun-exposed epithelium without microscopic evidence of dysplasia similarly found TP53 to be mutated in SCCIS, but did not identify TP53 mutations in microscopically normal skin [34]. These more recent studies that used microscopically confirmed lesional and non-lesional skin suggest that TP53 mutations arise as early mutations in cSCC carcinogenesis and are enriched at the AK/SCCIS stage, compared to UV-exposed, histologically unremarkable skin [73]. Quantifying the mutational burden of TP53 in “normal” skin is likely fraught with selection bias, as large samples of grossly normal skin likely contain microscopic collections of abnormal keratinocytes which may have gone on to form clinically discernable AKs capable of progression or regression.
Though TP53 alterations clearly contribute to cSCC, they are not in and of themselves sufficient to establish or maintain the malignancy. Up to 60% of cSCCs are believed to arise from AKs, approximately 30% of AKs will spontaneously regress in one year, and the estimated annual rate of malignant transformation to cSCC per AK is 0.025–16%; with most studies estimating it at <1% [74,75]. Therefore, while mutation in TP53 appears to occur early in the progression from normal to precancerous skin, and such mutations are selected for during malignant progression, additional mutations are needed to promote progression of precancerous lesion to cSCC. Further study of oncogenic TP53 mutations in the early stages of UV-induced skin cancer is warranted.
2.2.2. NOTCH
LOF mutations in NOTCH1, NOTCH2 and NOTCH3 have been found to occur commonly in cSCC and in sun exposed skin, including skin that shows no microscopic evidence of dysplasia [27,33,34,69,76,77]. NOTCH1–3 genes encode Notch proteins that function in keratinocytes to regulate cell fate determination, stem cell potential, lineage commitment, proliferation, and survival [78]. Activation of Notch signal transduction occurs through binding of ligands expressed on neighboring keratinocytes. Notch1 receptors are expressed in all layers of the epidermis, while Notch2 is primarily expressed in the basal layer. The ligands expressed in the epidermis include Jagged-1 and Jagged-2, which are expressed predominately in the suprabasal layers and Delta-like 1, which is expressed throughout the basal layer [79]. Ligand receptor interaction between two neighboring cells results in proteolytic cleavage of the Notch receptor, nuclear translocation of the cleaved cytoplasmic domain, and subsequent binding to a downstream transcription factor CBF1, which thereby activates transcription [80].
Notch signaling plays a critical role in the development and maintenance of the epidermis [81]. In the suprabasal layers of the epidermis, increased Notch1 activation either directly or indirectly, through IRF6-dependent mechanisms, acts to suppress P63 expression [82,83]. The shift from predominantly p63 to Notch signaling promotes cell cycle arrest and terminal differentiation. In addition, Notch signaling acts to promote keratinocyte differentiation through activation of caspase-3 and PKC-δ [84]. NOTCH1 is a direct target gene of p53 and is downregulated in keratinocytes with TP53 mutations [85].
Given its critical regulatory role in keratinocyte differentiation, it is not surprising that defects in Notch signaling are implicated in the pathogenesis of cSCC. Genetic loss of NOTCH promotes increased susceptibility to both chemically-induced and spontaneous cSCCs in mice, implicating Notch as a tumor suppressor [55,78,86,87,88]. Consistent with these findings, UV-signature LOF mutations in NOTCH1 and NOTCH2 have been found in approximately 75% of human cSCCs [27,77]. Recent studies indicate that UV-signature LOF NOTCH mutations also appear in clinically and microscopically non-dysplastic skin, suggesting that loss of Notch signaling may be one of the earliest mediators of transition from normal keratinocytes to a precancerous state [15,34,37,73].
Decreased Notch signaling may promote expansion of mutant keratinocytes in the epidermis via inhibition of UV-induced apoptosis of epidermal keratinocytes [89]. As a downstream effector of p53, NOTCH1 is further implicated as a significant regulator in UV-induced skin cancer [55,85]. Notch and TP53 alterations are not always mutually exclusive in cSCC and precancerous lesions, but the independent and coexistent alterations that have been reported vary, and further research into the relationship these alterations have with each other, and the progression of disease, is needed.
2.2.3. RAS
Mutant Ras has been identified to act as a driver oncogene in an estimated one third of human cancers [90]. The canonical members (HRAS, NRAS, KRAS4A, and KRAS4B) and noncanonical members of the Ras superfamily are small guanosine triphosphatases (GTPases) that play a central role in transmission of mitogenic signals [90,91]. Ras GTPases cycle between GDP-bound inactive and GTP-bound active states [92]. In an active state, Ras-GTP binds to and activates a spectrum of downstream effectors to regulate signaling networks that control gene expression, proliferation, differentiation, and survival.
In cSCCs, reported rates of amplifications and activating mutations in Ras genes range widely from 3–30% [93,94,95]. HRAS mutations are more common than NRAS or KRAS mutations; characteristically occurring in codons 12, 13, and 61, at UV-sensitive CC sites [96,97]. In one study, 13/21 cSCCs (60%) that developed in patients on vemurafenib had Ras mutations; with 11/13 occurring in HRAS, exhibiting a significantly higher frequency of Ras mutations than is seen in cSCC outside treatment with B-Raf inhibitors [93,94,95,98]. This phenomenon is thought to occur because B-Raf inhibition leads to paradoxical activation of MAPK signaling. Driving accelerated growth of subclones containing HRAS mutations. Such activating Ras mutations have demonstrated oncogenic capacity in mouse models of chemically-induced keratinocyte papillomatosis [99]. However, in human patients with UV-induced DNA damage, GOF Ras alterations are exceedingly rare in AKs/SCCIS, and are encountered predominantly in a small subset of late-stage cSCCs [33,34,73]. As such, Ras genes are not generally regarded as oncogenes driving the early stages of cSCC development [9].
2.2.4. CDKN2A
CDKN2A is inactivated in many human cancers and in families with hereditary melanoma and pancreatic cancer through homozygous deletion, methylation, or point mutation [100]. CDKN2A encodes two distinct tumor-suppressor proteins, p16INK4a and p14ARF that indirectly control the activities of p53 and retinoblastoma protein (RB) [47]. These two distinct proteins are derived from alternative transcription of the first exon of the CDKN2A gene [101]. INK4a and ARF, so-called because it is transcribed from an alternative reading frame (ARF), encode p16INK4a and p14ARF, respectively. P16INK4a protein directly inhibits the cyclin D-dependent kinases, CDK4 and CDK6, maintaining RB in its active, hypophosphorylated, anti-proliferative state [44]. While p14ARF acts primarily as a negative regulator of Mdm2 [48]. Blocking its interaction with p53 by isolating Mdm2 within the nucleolus, and by inhibiting its E3 ubiquitin protein ligase activity [102,103]. Thus, both proteins are involved in modulating responses to hyperproliferative signals via RB and p53 transcriptional programs.
Mutations in the CDKN2A locus have been reported to occur in 24–45% of sporadic cSCCs [20,27,104,105]. About half these mutations affect both p16INKa and p14ARF [101]. A similarly high frequency of CDKN2A mutations has also been observed in small studies of metastatic cSCCs [106]. Moreover, previous comparative studies of AKs and cSCCs suggest that deletion of p16INKa increases the incidence of progression from AK to cSCC [107]. Combined with evidence that CDKN2A mutations are rare and are not found to be under positive selection in normal skin, altered function of p16INKa and/or p14ARF likely play an important role in the development of precancerous lesions, as well as the progression from precancerous to cancerous lesions [33,73].
2.2.5. PIK3CA
A recent study of AKs and cSCCs found PIK3CA alterations, copy number amplifications, and/or GOF hotspot mutations in approximately half the sequenced samples in their cohort [33]. If these rates of alteration are confirmed in larger cohorts, they could be actionable therapeutic targets in cSCC. Similar alterations have been found in over one third of squamous cell carcinomas of the head and neck; where they have been shown to drive oncogenesis, along with increased sensitivity to targeted inhibitors, as well as show improved survival with nonsteroidal anti-inflammatory drugs [108,109]. Similar findings have been reported in colorectal and breast cancers [110]. This potential cross-cancer target could represent the next generation of application(s) for topical use tyrosine kinase inhibitors. An area of current research characterized by the December 2020 Food and Drug Administration approval of topical tirbanibulin, a Src kinase inhibitor with downstream activity against the phosphatidylinositol 3-kinase (PI3K) and other growth regulatory pathways, for the treatment of AKs.
2.2.6. KNSTRN
KNSTRN encodes a kinetochore-associated protein that modulates anaphase onset and chromosome segregation during mitosis [35,111]. KNSTRN was the third most frequently mutated gene in a cohort of cSCC samples using a combination of single nucleotide variant (SNV) determinations from whole exome sequencing and targeted sequencing [35]. Over half of these mutations were mapped to aserine-to-phenylalanine mutations with C > T transition characteristic of UVB-induced mutagenesis. Subsequent mutational analyses identified KNSTRN mutations in the same functional domain but in a smaller proportion of cSCC samples [26]. KNSTRN mutations disrupt chromatid cohesion and correlate with increased aneuploidy in primary tumors and tumorigenesis in vivo [35]. The frequency of KNSTRN mutations has also been shown to correlate with progression of dysplasia from unremarkable epidermis to cSCC, suggesting that KNSTRN may play a role in early tumorigenesis of cSCC, though future studies will be needed [112].
2.2.7. FAT1
FAT1 encodes a tumor suppressor-related member of the FAT protocadherin family that is frequently mutated in numerous types of human cancers including cutaneous, head and neck, and oral SCCs [20,26,76,113,114,115]. However, FAT1 mutations, including missense, deletion, and truncations have also been identified in a substantial subset of clinically unremarkable, sun-exposed skin samples [26,37]. The underlying functions of protocadherin proteins remain incompletely understood, especially in the skin. But two mechanisms that promote tumorigenesis have been reported; one in which inactivated FAT1 acts as a tumor suppressor, resulting in aberrant Wnt/β-catenin signaling in multiple cancer types, and another in which loss of FAT1 leads to increased CDK6 expression via activation of the Hippo pathway [116,117]. Future studies will need to investigate the underlying mechanism that controls FAT1 activity in normal, precancerous, and cancerous lesions.
2.2.8. CARD11
Caspase recruitment domain 11 (CARD11) encodes a protein that acts as a scaffold for nuclear factor kappa B (NF-κB) activity [29,118]. Activating mutations in CARD11 have been described in at least 2 independent studies suggesting a role for NF-κB signaling in carcinogenesis [29,119]. The NF-κB family of proteins consists of five transcription factors p65, RelB, c-Rel, NF-κB1, and NF-κB2 which form homo- and heterodimers that bind specific target genes to regulate transcription. In the epidermis, NF-κB plays a critical role in keratinocyte apoptosis, proliferation, and differentiation, but a more controversial role in tumorigenesis [120]. Findings from several studies point to a role of NF-κB in growth inhibition, as blockade of NF-κB activation induces spontaneous cSCC development in murine experimental models in part due to resulting CDK4 upregulation [8,121,122]. However, in one study, mutations in the CARD11 gene were found in 38% of 111 cSCCs with many of the mutations leading to constitutive NF-κB activity [119]. As has been seen with respect to NOTCH1 and TP53, CARD11 mutations have also been identified in sun-exposed and peri-tumoral skin, implicating aberrant NF-κB signaling in early steps of carcinogenesis. Thus, while changes in NF-κB-dependent gene expression have been shown to occur in cSCCs, additional studies are needed to clarify whether NF-κB plays a direct or indirect role in the progression to AK and/or cSCC.
2.2.9. SRCASM
SRCASM (Src activating and signaling molecule, previously known as TOM1L) encodes a tumor suppressor platform molecule containing a VHS and GAT domain and multiple conserved tyrosine phosphorylation sites that are phosphorylated by activated Src-family tyrosine kinases (SFKs) [123]. When phosphorylated, Srcasm engages SFKs and downregulates their activity through a lysosomal-dependent mechanism. Functionally, this acts to limit keratinocyte proliferation and promote keratinocyte differentiation by dampening the activation of PDK1/Akt/mTOR, MEK/ERK, and STAT3 [55,123,124,125]. In support of a role for Srcasm in cSCC tumorigenesis, human SCCIS and SCCs have been shown to have decreased Srcasm levels and increased SFK activity relative to non-lesional tissue [55]. Furthermore, overexpression of Srcasm completely inhibits development of tumors in a murine model of cSCC in which constitutive activation of SFK protein Fyn in mice leads to spontaneous formation of precancerous lesions resembling AKs and cSCCs. Thus, Srcasm may act as an important tumor suppressor in human cSCCs and targeting SFK activity through modulation of Srcasm function may provide an important therapeutic strategy.
2.2.10. TP63
While rarely mutated in human cancers, TP63 is frequently overexpressed in lung, head and neck, and skin SCCs [126,127]. A homologue of the TP53 gene, TP63 encodes the p63 protein which plays a critical role in epithelial development and homeostasis [128,129]. TP63−/− mice die shortly after birth, in part due to a complete lack of epidermis and epidermal structures [129]. TP63 is transcribed from two different promoters giving rise to distinct transcription factors with (TAp63) and without (ΔNp63) the N-terminal p53-homologous transactivation domain [128]. These factors are also subject to alternative splicing resulting in α, β, and γ isoforms with different COOH-termini [130]. In the epidermis, ΔNp63α is the predominate isoform expressed in developmentally mature keratinocytes of the basal layer where it plays a critical role in maintaining proliferation potential and lineage specification [127,131].
Diverse transcriptional and post-transcriptional mechanisms regulate ΔNp63 activity. ΔNp63 can act as a dominant negative suppressor of the TAp63 isoform, thereby regulating p53 and TAp63 dependent gene expression [128,132]. A balance between the tumor suppressive functions of the TAp63 isoform and the oncogenic functions of the ΔNp63 isoform may play a critical role in proliferation and differentiation of stem cells and tumor cells [133]. ΔNp63α also acts to induce or repress a wide range of transcriptional targets through direct target gene binding, recruitment of epigenetic modulators/chromatin remodeling factors and non-coding RNA [134,135,136,137]. Crosstalk between p63 and Notch signaling plays a critical role in the balance between proliferation and terminal differentiation [127]. p63 functions as a selective modulator of Notch1 target gene expression (e.g., HES1) and also acts as a negative regulator of NOTCH1 expression via p53 [83,138]. This serves to limit Notch signaling in the basal epidermis, which, together with p63′s suppression of the cyclin dependent kinase (CDK) inhibitor, p21WAF1/Cip1, maintains proliferation and inhibits terminal differentiation in the basal epidermis [83,127,139].
Consistent with a role in squamous cell carcinogenesis, forced overexpression of ΔNp63α in the basal layer of stratified epithelia is sufficient to induce mild epidermal hyperplasia and predisposes to papilloma formation in a murine chemical model of cSCC [140]. Genetic ablation of p63 in established murine SCC tumors results in regression via reduced cell proliferation and increased apoptosis driven by reduced expression of the p63 target gene FGFR2 [141]. ΔNp63α has also been shown to drive proliferation in SCC through suppression of TGFB2 expression and RHOA activity [142]. While TP63 amplification was reported to be present in a modest proportion of SCC, including 24% of metastatic cSCCs, additional mechanisms account for dysregulated ΔNp63 mRNA and protein expression in SCCs [29]. Nucleoporins (NUP62) have been shown to be important in the translocation of ΔNp63α to the nucleus in a ROCK-phosphorylation dependent manner [143]. Elevated levels of NUP62 have been demonstrated in cSCC, while reduction of NUP62 expression has been shown to prevent proliferation of SCC cells. Elevated ΔNp63 expression may also result from increased expression of Syntaxin binding protein 4 (Stxbp4), which acts to suppress the anaphase-promoting complex/cyclosome (APC/C) mediated ubiquitination and proteolysis of ΔNp63 [133,144]. Epigenetic dysregulation of the iASPP-P63 feedback loop at the microRNA level has also been shown promote proliferation and block epithelial-mesenchymal transition in cSCC [145]. Thus, through numerous regulatory pathways, TP63 plays a key role in cSCC tumorigenesis.
2.2.11. EGFR
Epidermal Growth Factor Receptor (EGFR) signaling is dysregulated in many human cancers, including cSCC. The EGFR is a transmembrane glycoprotein with numerous ligands (e.g., EGF, transforming growth factor alpha) that, upon binding, initiate homo- and hetero-dimerization, transphosphorylation, and subsequent activation of downstream pathways including Ras-Raf-MEK-ERK, PI3K, and JAK/STAT signaling [146]. Depending on the ligands present, receptor expression, and differentiation state, EGFR activation can regulate these signaling networks to influence keratinocyte proliferation, differentiation, and survival [147]. Normally, high EGFR activity acts in the basal epidermis to maintain self-renewal and suppress differentiation. In the upper layers, EGFR is downregulated to promote keratinocyte differentiation. Importantly, EGFR signaling acts to downregulate both TP53 and NOTCH1 transcription through a c-Jun dependent mechanism inhibiting keratinocyte differentiation and apoptosis [54].
Dysregulation of EGFR signaling can arise through EGFR mutations, receptor/ligand overexpression, and alterations in trafficking and signaling pathways [146]. As opposed to pulmonary SCC, EGFR is infrequently mutated in cSCC with reports ranging from 1–20% [148,149]. EGFR alterations were identified in only 6/122 (4.9%) recurrent or metastatic cSCCs, most of which (4/6), were amplifications [31]. Importantly, EGFR signaling is dysregulated in a much higher proportion of tumors than those predicted by genetic mutation alone. In one cohort of 94 cSCCs, EGFR was overexpressed, aberrantly localized and amplified in 35%, 53% and 7% of tumors, respectively [150]. Overexpression of EGFR may result from a combination of increased mRNA synthesis and decreased degradation. For example, TP53 mutations have previously been shown to positively regulate EGFR expression levels [151]. TP53 mutations in AK/SCCIS and cSCC may therefore promote enhanced EGFR signaling. Epigenetic regulation of EGFR expression may also play an important role in its overexpression. Long noncoding RNA (lncRNA) metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) promotes EGFR expression in cSCC through a c-Myc and kinectin 1 dependent mechanism [152]. A recent study identified microRNA-27a (miR-27a) as a negative regulator of EGFR signaling whose expression was significantly downregulated in human cSCCs [153]. Collectively, these findings indicate that EGFR signaling is dysregulated through multiple mechanisms during cSCC tumorigenesis.
2.3. Transcriptional Characterization of cSCC
Numerous studies have used transcriptional analyses of AKs and cSCCs to define how changes in gene expression regulate tumorigenesis [115,154,155,156,157,158,159,160,161,162]. However, findings from these studies have been limited by confounding variables including high inter-patient/sample variability, lack of histologic confirmation of skin tissue, and high annotation error rates.
A recent, well-designed study used combined next-generation sequencing analyses of patient-matched, histologically validated “normal,” skin (NS), AK and cSCC samples and a UVR-driven mouse model to identify transcriptional drivers involved in cSCC development [26]. Whole exome, RNA-seq, and miRNA-seq experiments were performed on multiple samples from 9 patients. Whole exome sequencing (WES) identified many of the commonly reported significantly mutated genes (SMG) including TP53, NOTCH1–2, FAT1, MLL2, and KNSTRN, but interestingly found that AKs contained the greatest proportion of SMGs, suggesting that AKs have acquired the mutational events necessary for cSCC formation. Consistent with these findings, global gene expression patterns revealed that dysregulated gene expression occurs at the earliest transition from NS to AK versus the subsequent transition to cSCC. Using a transcription factor (TF) binding motif analysis, the authors then identified several putative TFs responsible for altered gene expression some of which included ETS2, SP1, FREAC2 (FOXF2), AP1, NFAT, TCF3, LEF1, E2F MYC, and NFY. ETS2 is a pro-oncogenic TF downstream of the ERK MAP kinase pathway with targets that were broadly upregulated [163]. TCF3 and LEF1 are both downstream of the B-catenin/Wnt signaling pathway whose activity was upregulated across most samples. NFAT/AP1 target genes were downregulated early, which the authors proposed may reflect inhibition of keratinocyte differentiation programs that may be modulated by NOTCH signaling. While these findings suggest that these TFs may serve as key transcriptional regulations that drive cSCC development, their role in tumorigenesis remains to be functionally validated.
3. Beyond the Genome: Epigenetic Regulations in cSCC
Epigenetic alterations represent additional hallmarks in a cell’s transformation into cancer. These alterations result in a loss of gene expression by transcriptional silencing, via epigenetic promoter hypermethylation of CpG islands. Epigenetic alterations are estimated to be about 10 times more common across all cancer types than genetic mutations [164]. Epigenetic regulation in cells is achieved through a variety of mechanisms including DNA methylation, histone modifications (methylation, acetylation, phosphorylation, ubiquitination, and sumoylation), chromatin remodeling, and microRNAs. Epigenetic dysregulation can result from alterations to any of these systems, such as overactivation of methyltransferases or histone methyltransferases (PMID: 23455543). Affording a wide range of potential therapeutic targets holding great promise for cancer prevention, detection, and therapy [165]. The nascent field of epigenetic alterations in cSCC continues to garner increasing attention.
3.1. Aberrant DNA Methylation in cSCC
Genomic DNA methylation alterations, such as global DNA hypomethylation and gene specific hyper- or hypomethylation, are associated with SCC. Examples include CDKN2A (p14ARF and p16INK4A) promoter methylation, found in 40% of cSCC, and hypermethylation of the FOXE1 promoter [166,167]. FRZB, an antagonist of Wnt signaling, was found to be differentially methylated in metastatic cSCC as compared to primary cSCCs [168]. E-cadherin is among the genes most frequently hypermethylated in SCC [169]. This adhesion molecule promotes the structural integrity of the epidermis, loss of expression secondary to methylation may facilitate malignant invasion. Promoter hypermethylation of E-cadherin in keratinocytes coincided with the acquisition of a metastatic phenotype in a chemical carcinogenesis mouse model of cSCC [170]. This same study identified a number of novel methylation targets, such as insulin-like growth factor binding protein-3, in malignant murine keratinocytes, which correlated with hypermethylation patterns seen in human primary cSCCs.
3.2. Histone Modifications in Cutaneous SCC
Posttranslational covalent modifications of histones include acetylation, methylation, phosphorylation, ADP-ribosylation, ubiquitination, sumoylation, arginine deamination, and proline isomerization, occurring primarily at the amino-terminal histone tails. The location and type of modification impact the effect on gene transcription. Synergistic cooperation between different histone modifications acts to either repress or activate transcription. The overarching epigenetic state of the cell is ultimately defined by the specific combination of histone modifications present across the chromatin. This “histone code” represents an epigenetic readout reflecting the transcriptional state of a DNA region [171].
Histone acetyltransferase p300 (coded by EP300) has been shown to be essential for cell-cycle withdrawal of terminally differentiating keratinocytes, via several transcriptional regulatory mechanisms [172,173]. Similarly, histone methyltransferase (EZH2), a key component of the Polycomb repressive complex 2, is another epigenetic regulator of squamous differentiation and importantly, represents a druggable target [174]. The frequently mutated EZH2 regulates self-renewing keratinocyte populations through repression of INK4A-INK4B, preventing the recruitment of AP1 transcriptional factors to genes involved in terminal differentiation [175,176]. Sustained EZH2 activity is required for survival of keratinocyte cancer stem cell populations [177]. Increased EZH2 expression has been associated with malignant progression in both cutaneous and bronchial epithelium [178,179]. In HNSCC cells, EZH2 promotes enhanced malignant progression by mediating the effects that increased levels of the long non-coding RNA HOTAIR have on E-cadherin expression [180].
3.3. Long Noncoding RNA Dysregulation in cSCC
Stretches of DNA between genes, called non-coding DNA, also harbor genetic variants that are associated with diseases such as cSCC. Long noncoding RNAs (lncRNAs) are untranslated RNA transcripts >200 nucleotides in length that can regulate gene expression. Several lncRNAs have been shown to play an important role in keratinocyte differentiation and cSCC pathogenesis, including TINCR, PICSAR, LINC00520, LINC00319, MALAT1, LINC01048, GAS5, and HOTAIR [181]. TINCR is upregulated during keratinocyte differentiation and downregulate in cSCC, suggesting a pro-differentiation/antineoplastic role in keratinocytes [182]. PICSAR is expressed preferentially in cSCC tumor cells and promotes ERK1/2 activation by downregulating dual specificity phosphatase 6 (DUSP6) promoting cell migration through regulation of integrin expression [183]. LINC00520 expression is decreased in cSCC A431 cell lines and overexpression was shown to decrease the activity of EGFR and PI3K mRNA and protein in vitro [184]. LINC00319 expression, which promotes proliferation and migration by regulating miR-1207-5p mediated regulation of cyclin-dependent kinase 3, is significantly increased in cSCC [185]. MALAT1 promotes cSCC oncogenesis through regulation of EGFR activity [152]. A MALAT1-c-Myc complex binds the kinectin 1 (KTN1) promoter, enhancing its transactivation to positively regulate EGFR protein expression. LINC01048 expression, which increases binding of TAF15 to the Yes-Associated Protein 1 (YAP1) promoter, is associated with increased mortality in cSCC [181,186]. YAP1 encodes a key coactivator of Hippo signaling, which promotes RAS activation along with downstream AKT and ERK signaling [187]. GAS5 is a tumor suppressor with decreased expression in cSCC and other cancers [188]. In cSCC cell lines, HOTAIR is overexpressed and acts with miR-326 to regulate PRAF2 expression [189]. The network of lncRNA interactions that regulate cSCC pathogenesis is complex. Further validation and study of lncRNAs may present novel therapeutic opportunities in cSCC.
3.4. MicroRNA Dysregulation in cSCC
MicroRNAs (miRs) are short non-coding RNAs of 19–25 nucleotides that regulate gene expression post-transcriptionally. MiRs are involved in many biological processes and their dysregulation in cancer is believed to be important to tumorigenesis [190]. Nearly 20 oncogenic miRs have been identified (e.g., miR-21, miR-135b), along with tumor suppressor miRs (e.g., miR-124, miR-125b) in cSCC [191].
miR-21 is highly expressed in human cSCCs and HNSCCs. Where it inhibits PTEN and GHRL3 expression, leading to enhanced PI3K/AKT/mTOR activity [192]. Downregulation of miR-203, an antagonist of p63, and expression of miR-21, is associated with metastasis in HNSCC [193]. miR-124 and miR-214 are both downregulated in cSCC and may drive overexpression of ERK1/2 which promotes cellular proliferation in cSCC [194]. miR-27a targets and inhibits EGFR, the downregulation of miR-27a correlates with upregulation of EGFR in cSCCs [153]. Overexpression of miR-365 inhibits expression of the negative regulator of HIF1-alpha, HOXA9, which functions as a tumor suppressor in human cSCCs [195,196].
miRs, including miR-125b and miR-135b, have also been associated with tumor cell invasion. The downregulation of miR-125b in early cSCC promotes tumor growth and motility through a network of pro-tumorigenic genes including matrix metalloproteinases MMP13, MMP7, and MAP2K7 [197]. miR-135b is overexpressed in cSCC and promotes cancer cell motility and invasiveness [198].
TAp63 regulated miRs, miR-30c-2* and miR-497, act to inhibit tumor cell proliferation and promote apoptosis. Introduction of miR-30c-2* or miR-497 mimics massively inhibited growth of cSCC tumor xenografts in vivo [199], demonstrating that miR dysregulation in cSCC may contain opportunities for novel therapeutic development.
3.5. UV-Induced Epigenetic Regulation in cSCC
The primary environmental risk factor for BCC, SCC, and melanoma is ultraviolet radiation, which, like arsenic, acts at the molecular level, in part, through epigenetic mechanisms. Analysis of the epigenetic patterns of the tumor suppressor P16INK4a in chronically UVA-irradiated HaCaT human keratinocytes revealed a striking reduction of the permissive histone mark H3K4me3 in the promoter (4–9 fold reduction for 10 and 15 weeks, respectively), which has often been found deregulated in skin cancers [200]. This alteration, together with severe promoter hypermethylation, strongly impaired the transcription of P16INK4a (20–40 fold for 10 weeks and 15 weeks, respectively). Analysis of tumor cells also revealed downregulation of P16INK4a transcription, attributed to enrichment of the heterochromatin histone mark H3K9me3, the repressive mark H3K27me3 and promoter hypermethylation. Less pronounced UVA-induced epigenetic alterations were identified in other genes KLF4 and NANOG (stem cell fate determination), hTERT (telomere maintenance), and P21WAFI/CIPI (tumor suppressor), demonstrating that UVA can alter skin cancer associated transcriptomes by means of epigenetic DNA and histone alterations.
3.6. Epigenetic Regulation in cSCC Metastasis
Epigenetic alterations have also been shown to contribute to the final stages of cSCC progression metastasis. The epigenetic profiles of metastatic cSCCs diverge from those of their corresponding primary lesions, including significantly increased hypermethylation at FRZB [168]. Epigenetic profiling may prove to be a prognostic biomarker in nonmelanoma skin cancers. Super enhancer and transcriptional profiling of stem cell populations isolated from keratinocyte-derived SCCs suggest the Ets2 transcription factor to be a key regulator of epigenetic changes associated with malignant behavior [201].
3.7. Discovery of Novel Epigenetic Biomarkers in cSCC
Our understanding of the importance of global genomic DNA methylation in the molecular pathogenesis of neoplasia is growing. DNA methylation events may represent a specific and early marker of tumorigenesis that can be easily detected by minimally invasive PCR based methods. Promoter DNA methylation gene panels exist for HNSCC screening, risk of recurrence, and assessment of margin status during surgery. No correlation between global histone modifications and prognosis has yet been described in cSCC though such correlations have been identified in esophageal SCC. Scientists are now generating extensive maps of histone modifications and DNA methylation across mammalian cell types utilizing high throughput sequencing technologies totaling over 400 currently known marks [202]. Deciphering the specific functions of each of these marks in regulating gene expression will enable research into many aspects of human health and disease.
3.8. CRISPR/Cas9 Tools of Epigenomic Editing
Utilizing the CRISPR/Cas9 bacterial antiviral system for scientific research has been transformative in biomedicine [203]. Major limitations to editing human DNA with the CRISPR/Cas9 system are off target effects, however. The engineered Cas9-KRAB fusion protein activates specific genes by targeting specific histones for modifications such as the acetylation-one type epigenetic mark. Repression mediated by Cas9-KRAB via modification of the epigenome at specific positions has been shown to specifically disrupt gene expression [204,205]. This novel system allows for direct functional analysis of site-specific epigenetic modifications and modulation of target genes, offering a powerful novel platform for research and therapeutics in cSCC.
4. Metabolic Reprogramming in cSCC
Metabolic reprogramming is a hallmark of many cancers, including skin cancer [206,207]. Notably, melanoma metabolism has been intensively studied, and its metabolic signaling pathways have been shown to be involved in cancer cell survival, invasion, metastasis, and resistance to BRAF inhibitor therapy or PD-1 blockade immunotherapy [208]. While our understanding of the role and mechanisms of metabolic reprogramming in non-melanoma skin cancer is somewhat limited, recent studies have highlighted its importance in cSCC carcinogenesis including the effects of the DNA damage response, the potential role of commonly mutated oncogenes and tumor suppressors, and the role of deletions in mitochondrial DNA (mtDNA).
The Warburg effect is a metabolic feature of many cancer cells in which cells use glycolytic pathways despite the presence of adequate oxygen supply [209]. This reprogramming from oxidative phosphorylation (OXPHOS) to glycolysis is thought to allow cancer cells to produce ATP at higher rates/levels than OXPHOS while also producing substrates for anabolic metabolism (e.g., lactate). This appears to hold true for cSCC as recent studies have identified that, relative to peritumoral skin, hyperplasia and AKs, human cSCCs express higher levels of glycolytic enzymes, and lower levels of enzymes involved in lipid biosynthesis and the TCA cycle [210]. Murine models of cSCC utilizing oncogenic Ras mutation coupled with loss of p53 in hair follicular stem cells (HFSC) similarly find that aerobic glycolysis is enhanced in cSCCs [211]. Targeting this metabolic shift may provide a path to selectively target neoplastic cells. Indeed, selective inhibition of GLUT1 in squamous cell lung cancer using WZB117, a small molecular GLUT1 inhibitor, reduced tumor growth of HCC1588 and HCC2814 cSCCs by 40% while adenocarcinoma H1299 tumor growth remained unaffected [212]. However, the mechanisms and the functional role of metabolic reprograming in cSCC initiation and progression remain controversial.
Many of the commonly mutated oncogenes and tumor suppressors implicated in cSCC carcinogenesis play an important role in regulating energy metabolism. For instance, p53 influences energy metabolism by enhancing OXPHOS and inhibiting glycolysis through several mechanisms [207]. RAS mutations in cancer cell lines are associated with metabolic reprogramming through regulation of glycolytic enzyme gene expression and glutamine metabolism [213,214,215]. CDKN2A mutations and deficiency of p16INKa have been shown to regulate gluconeogenesis [216]. Furthermore, beyond somatic mutations, dysregulation of tumor suppressor and oncogene activity may influence cSCC carcinogenesis. In a recent study, Homeobox A9 (HOXA9), a direct target of onco-miR-365, was significantly downregulated in both cSCC primary tumors and cell lines. HOXA9 acts as a tumor suppressor and inhibits glycolysis in cSCC in vitro and in vivo by negatively regulating HIF-1α and its downstream glycolytic regulators, HK2, GLUT1 and PDK1 [196]. While the specific role of commonly mutated or dysregulated oncogenes and tumor suppressors in metabolic reprogramming of cSCC remains to be determined, these findings demonstrate multiple mechanisms through which metabolism may be altered in cSCC.
In addition to somatic mutations in oncogenes and tumor suppressors, mutations in mtDNA, triggered by genomic instability or UV exposure, are implicated in metabolic reprogramming and cSCC pathogenesis [217,218,219]. mtDNA encodes 13 essential genes for mitochondrial OXPHOS along with 2 ribosomal RNAs and 22 transfer RNAs [220]. Mutations in these critical genes have been shown to result in mitochondrial dysfunction, altered ATP production, and are associated with carcinogenesis [220]. For example, mtDNA mutations associated with genomic instability from knockdown of the tumor suppressor XPC lead to decreased OXPHOS and increased glycolysis through an AKT, NAPDH oxidase-1 (NOX1), and reactive oxygen species (ROS) dependent mechanism [221,222]. Moreover, several common mutations in mtDNA have been identified in sun-exposed skin including a 260 bp mtDNA tandem duplication in the regulatory site of mitochondrial DNA (D-loop) [217], a 4977 bp deletion [217,219], and a 3895 bp deletion [223]. Additional studies are needed to determine the specific role that each mtDNA mutation plays in keratinocyte metabolism and cSCC carcinogenesis.
While cumulatively this work sheds light on the mechanisms controlling metabolic reprogramming in cSCC, how metabolic changes regulate carcinogenesis/tumorigenesis remains controversial. Some contend that cSCC may not require increased glycolytic activity for transformation [211]. Aerobic glycolysis culminates in NADH-dependent fermentation of pyruvate to lactate by lactase dehydrogenase (Ldh) and loss of Ldha activity dramatically reduces glycolysis [211]. Surprisingly, deletion of Ldha in a genetic (Ras and p53-null driven) and a chemically induced DMBA/TPA model of cSCC failed to cause any changes in tumorigenesis, including tumor number, time to formation, proliferation, volume, gene expression, and immune response. While Ldha-null tumors took up and used more glutamine, suggesting a possible compensatory mechanism, the results suggest that cSCC does not require increased glycolytic activity or reprogramming to generate cancers. Investigations into whether metabolic changes occur during the initial phase of carcinogenesis identified that UVB exposed keratinocytes undergo a metabolic shift in response to DNA damage leading to a downregulation of glycolysis, TCA cycle, and fatty acid β oxidation, and an upregulation of the electron transport chain (ETC) before overt tumor formation [210]. Overactivation of the ETC results from upregulation dihydroorotate dehydrogenase (DHODH) which acts to coordinate nucleotide biosynthesis. Notably, Leflunomide, a non-specific inhibitor of DHODH, blocks UVB-induced tumorigenic transformation of keratinocytes, raising the possibility of targeted metabolic therapy for the prevention of UVB-induced cSCCs [224]. These studies provide compelling evidence that metabolic reprogramming is robust in cSCC, but its exact role and the underlying mechanisms controlling these processes remain to be elucidated.
5. Current Status of Molecular Targeted Therapies in cSCC
Targeted therapies for basal cell carcinoma and melanoma were derived via elucidation of the molecular mechanisms driving their tumorigenesis. Mutations in the hedgehog pathway led to the application of smoothened inhibitors for the treatment of basal cell carcinoma and BRAF mutations have been successfully targeted with BRAF inhibitors in melanoma. Enhanced understanding of cSCC tumorigenesis and mechanisms driving formation of its precursor lesions may similarly drive the development of novel treatment strategies. Identification of key driver genes in cSCC has been hampered, in part, by the high mutational burden of cSCCs. However, EGFR inhibitors and tyrosine kinase inhibitors have shown varying degrees of efficacy in clinical trials and are promising areas of ongoing research alongside the continued development of other novel targeted therapies, including immune checkpoint inhibitors, which represent the only FDA approved targeted therapies in cSCC at this time.
5.1. EGFR Inhibitors
EGFR inhibitors, including small molecule inhibitors (e.g., gefitnib and erlotinib) and monoclonal antibodies (e.g., cetuximab and panitumumab) are well studied. The relatively high frequency of tumors with dysregulated EGFR signaling and the established safety/efficacy of these therapies in other malignancies makes them attractive for use in cSCC. EGFR small molecule inhibitors act by blocking the tyrosine kinase ATP binding site, thereby inhibiting downstream signal transduction, whereas monoclonal antibodies bind to the extracellular domain of EGFR inhibiting phosphorylation and activation [225]. In phase II trials for patients with incurable, recurrent, or metastatic cSCC, oral gefitinib and erlotinib had limited efficacy with overall response rates (ORR = partial response plus complete response) ranging from 10–16% [226,227]. Cetuximab, a chimeric mouse-human anti-EGFR IgG1 monoclonal antibody, has demonstrated improved ORR of 28% in the initial phase II trial [228] and as high as 42–53% in recent retrospective analyses [229]. Similarly, phase II studies with panitumumab, a high affinity human anti-EGFR IgG2 monoclonal antibody, demonstrated an ORR of 31% [230]. While the response rates to date have been somewhat limited, the development of predictive biomarkers to identify susceptible subpopulations and/or combination therapy with other targeted therapies and platinum-based chemotherapy may further improve response rates [231,232]. A limitation to the use of EGFR inhibitors is their propensity to cause skin reaction side effects: acneiform rash, pruritus, desquamation, hypertrichosis, and/or nail disorders often requiring treatment that can be seen in as many as 80% of treated patients, in the case of cetuximab [233]. The major challenge of systemic EGFR targeted therapies is that low response rates have been seen alongside expected, and notable, side effect profiles, arguing against the use of these agents in unselected cSCC patient populations, on balance. One relatively unexplored avenue is the application of topical EGFR inhibitors.
5.2. Immune Checkpoint Inhibitors
Immune checkpoint blockade has been employed across many malignancies including cSCC. Cemiplimab is a high-affinity, human hinge-stabilized IgG4 monoclonal antibody to the PD-1 receptor that acts to enhance responses of human primary T cells [234]. Following promising early results and aggregated data analysis of 108 patients with advanced (75 metastatic/33 locally advanced) cSCC, from a phase I/II clinical trial (R2810-ONC-1423) and phase II clinical trial (R2810-ONC-1540), which found a combined ORR of 47% (95% CI: 38, 57), with 4% complete and 44% partial response rates including durable responses of 6 months or more in 61% of responders, the FDA approved cemiplimab as the first targeted immunotherapy in cSCC on Sept 28, 2018 [235,236]. On 24 June 2020, the FDA approved pembrolizumab, an IgG4-kappa humanized monoclonal antibody that also targets the PD-1 receptor, following KEYNOTE-629 (NCT03284424); a multicenter, multi-cohort, non-randomized, open-label trial in which ORR was 34% (95% CI: 24, 44) and median response duration was not reached (range: 2.7, 13.1+ months) [237]. Additional phase II trials of pembrolizumab as a first-line single-drug therapy in unresectable cSCC have shown similar results [238]. Some evidence exists to suggest that cemiplimab may be superior to other systemic therapies in cSCC [239]. However, these represent indirect treatment comparisons and many trials with agents such as nivolumab (NCT04204837), as single agents and in combination with other systemic therapies, are ongoing, which may provide substantial benefit as has been suggested in isolated case reports to date [240,241]. One potentially significant limitation of immunotherapy in advanced cSCC is that advanced cSCC is most prevalent among solid organ transplant patients, where the use of such agents may increase the risk of allograft rejection [233].
5.3. SRC Family Kinase (SFK) Inhibitors
Both preclinical murine data and early phase clinical trials suggest targeting SFKs may be a promising avenue for the treatment of AKs and SCCs [242,243]. Dasatinib is a multi-kinase inhibitor that potently inhibits SFK activity, along with other kinases including BCR/ABL, c-KIT, ephrin family kinases, and PDGFR [244]. Studies in mice demonstrated that a topical formulation of dasatanib induced similar rates of regression in cSCCs versus topical 5-Fluorouracil (5-FU) without the epidermal ulceration or severe toxicity/death that has been seen with topical 5-FU use in mice [242]. Tirbanibulin (KX2-391) is a well-characterized SFK inhibitor used as a systemic agent in multiple cancer clinical trials, that also disrupts actin polymerization and microtubule formation at higher doses and has been FDA approved for topical use in AK following results of two multi-center trials in which tirbanibulin was shown to be superior to placebo at 2 months but showed high rates (47%) of local recurrence among complete responders at one year [245,246].
5.4. PI3K/mTOR Inhibitors
The PI3K and mammalian target of rapamycin (mTOR) pathways act as key regulators of a broad range of cellular functions, including cell growth, metabolism, survival, and differentiation [247]. Upregulation of PI3K/mTOR signaling is a frequent finding in SCC, especially in HNSCC, and there are several clinical trials underway to investigate PI3K/mTOR inhibitors for HNSCC [248,249]. In cSCC, the number of studies investigating PI3K/mTOR inhibitors is limited. One study found systemic BEZ-235, a PI3K/mTOR inhibitor, by oral gavage to be effective in inhibiting the formation of papillomas and cSCCs in a chemically induced mouse model of cSCC, but to be ineffective in treating established lesions in preclinical models derived from human cSCC cell lines [250]. A subsequent study using the K14-Fyn Y528 transgenic mouse model of cSCC, however, showed topical application of BEZ-235 in mice yielded similar results to dasatinib; efficient regression of cSCC with less inflammation, no ulceration, and no mortality compared to 5-FU [242]. LY3023414, an orally bioavailable PI3K/AKT/mTOR inhibitor, has been shown to have cytotoxic activity in vivo in cSCC tumor xenograft models [251]. A clinical trial of a topical mTOR/PI3K inhibitor called CLL442 in patients with cSCCIS found twice daily topical application was safe and well tolerated with no severe adverse events. However, the primary endpoint of lesion reduction or complete lesion clearance was not met (NCT03333694). Given the promising preclinical data and mounting evidence of the efficacy of PI3K/mTOR inhibitors in other cancer types, additional studies are needed to investigate PI3K/mTOR inhibitors in cSCC.
5.5. Epigenetic Modulators
Cancer prevention, detection, and therapy would likely benefit from controlled manipulation of epigenetic alterations. The perturbation of epigenetic mechanisms that silence tumor suppressor genes and activate oncogenes via altered CpG island methylation patterns, histone modifications, and dysregulation of DNA binding proteins, holds promise. Epigenetic drugs, including two DNA methyltransferase enzyme (DNMT) inhibitors and a deacetylase (HDACs) inhibitor, have been approved by the FDA for cancer treatment. [252]. Various HDAC inhibitors including FK228, SAHA and MS-275, are in phase III clinical trials. Vorinostat, a broad inhibitor of histone deacetylases, belongs to the hydroxymate structural group [253]. Other drugs in this group include Givinostat, Abexinostat, Panobinostat, Belinostat, Remetinostat and Trichostatin A. Vorinostat is currently used as a second-line or concurrent agent in the management of persistent, progressive or recurrent cutaneous T cell lymphoma [254]. Vorinostat has demonstrated activity in human cSCC cell lines and xenograft models, putatively via inhibition of AKT/mTOR signaling and inhibition of EMT by E-cadherin upregulation [255]. A phase II trial of topical Remetinostat was initiated in cSCC (NCT03875859). However, it was terminated after enrolling only 4 patients during the COVID-19 pandemic. Trials of epigenetic therapies using vorinostat in combination with EGFR inhibitors and capecitabine, romidepsin, and 5-azacytidine are ongoing in HNSCC. Mcl-1 levels have been shown to be a key factor in the response to vorinostat, and FBXW7 mutation is a biomarker for sensitivity to HDAC inhibition [256]. There is also evidence for synergy between treatment with vorinostat and Bcl-2-targeted therapeutics [249]. Epigenetics as biomarkers and therapeutic targets warrant clinical evaluation in cSCC, an important part of advancing these technologies with the development of more specific and effective inhibitors to reduce side-effects, given the wide range of genes and organs epigenetics influence throughout the body. MiRNAs regulate multiple target genes simultaneously and may therefore represent promising therapeutic targets. Development of microRNA therapeutics has accelerated recently, and multiple agents are in preclinical trials.
6. Conclusions
The development, migration, differentiation, and regeneration of the body’s largest organ requires coordination of multiple mechanisms to perform the core skin functions of temperature regulation, protection from a wide range of insults including UV radiation, trauma, pathogens, microorganisms, and toxins, achieved via immunologic surveillance, sensory perception, control of fluid loss, and maintenance of general homeostasis. Similarly, dysregulation of these mechanisms is found in cutaneous tumors, including cSCC. A diagram summarizing the mutations and biological changes occurring at the different stages of UV-induced tumor progression is shown in Figure 1.
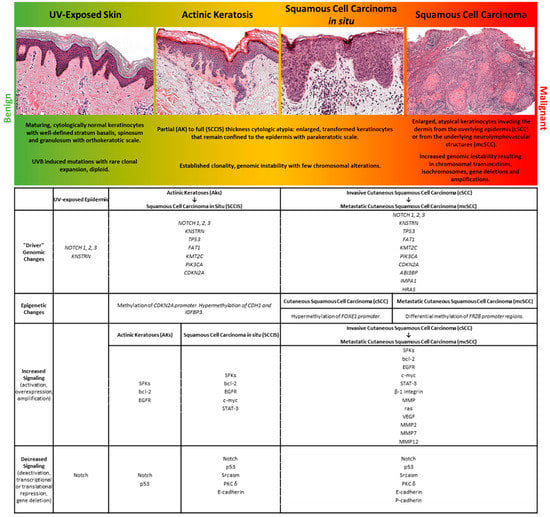
Figure 1.
A representation of stages within the spectrum of human cutaneous neoplasia is shown with select histologic and molecular features.
Through multi-“omic” studies that dissect the cellular microenvironment of cSCC, we have begun to identify and temporally organize the molecular alterations that underlie the field effect induced by UV irradiation that leads to cSCC [32,34,257]. The progression of oligo-cellular clones of UV irradiated keratinocytes to AKs, SCCIS, and cSCC is mirrored by an accumulation of expression profile changes across a wide array of cell signaling pathways driven by genetic and epigenetic alterations. The sheer number of intracellular and intercellular phenotypic alterations, and an even larger number of mechanisms driving them, is daunting for researchers attempting to understand the biology of this disease. However, this simultaneously offers abundant opportunities for therapeutic intervention and prevention of cSCC. Future studies on the early stages of cSCC tumor development designed to assess the interfacing mechanisms—genetic, epigenetic, metabolomic, and immunologic—that influence the development and progression of lesions to cSCC will offer new opportunities for tailored and combinatorial therapeutic approaches.
Author Contributions
Writing manuscripts drafts—M.L.H., C.T.B., A.S.M., Y.X., C.J.Y., C.A., B.C.C. and J.T.S.; revising manuscript drafts: M.L.H., C.T.B., A.S.M., Y.X., C.J.Y., C.A., B.C.C. and J.T.S., editing final version of manuscript: M.L.H., C.T.B. and J.T.S. Financial support for project—J.T.S. All authors have read and agreed to the published version of the manuscript.
Funding
MLH was supported by grant T32-AR007465 and through the Waine C. Johnson Research Fellowship in Dermatopathology. Funding also provided by NIH RO1 ES028114 to JTS.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Karia, P.S.; Han, J.; Schmults, C.D. Cutaneous squamous cell carcinoma: Estimated incidence of disease, nodal metastasis, and deaths from disease in the United States, 2012. J. Am. Acad. Dermatol. 2013, 68, 957–966. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Dermatol. 2015, 151, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Schmults, C.D.; Karia, P.S.; Carter, J.B.; Han, J.; Qureshi, A.A. Factors predictive of recurrence and death from cutaneous squamous cell carcinoma: A 10-year, single-institution cohort study. JAMA Dermatol. 2013, 149, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.R.; Zens, M.S.; Celaya, M.O.; Riddle, B.L.; Karagas, M.R.; Peacock, J.L. Survival after squamous cell and basal cell carcinoma of the skin: A retrospective cohort analysis. Int. J. Cancer 2015, 137, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Criscione, V.D.; Weinstock, M.A.; Naylor, M.F.; Luque, C.; Eide, M.J.; Bingham, S.F.; Department of Veteran Affairs Topical Tretinoin Chemoprevention Trial Group. Actinic keratoses: Natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer 2009, 115, 2523–2530. [Google Scholar] [CrossRef]
- Figueras Nart, I.; Cerio, R.; Dirschka, T.; Dreno, B.; Lear, J.T.; Pellacani, G.; Peris, K.; Ruiz de Casas, A. Defining the actinic keratosis field: A literature review and discussion. J. Eur. Acad. Dermatol. Venereol. JEADV 2018, 32, 544–563. [Google Scholar] [CrossRef]
- Sartini, D.; Campagna, R.; Lucarini, G.; Pompei, V.; Salvolini, E.; Mattioli-Belmonte, M.; Molinelli, E.; Brisigotti, V.; Campanati, A.; Bacchetti, T.; et al. Differential immunohistochemical expression of paraoxonase-2 in actinic keratosis and squamous cell carcinoma. Hum. Cell 2021, 34, 1929–1931. [Google Scholar] [CrossRef]
- Ratushny, V.; Gober, M.D.; Hick, R.; Ridky, T.W.; Seykora, J.T. From keratinocyte to cancer: The pathogenesis and modeling of cutaneous squamous cell carcinoma. J. Clin. Investig. 2012, 122, 464–472. [Google Scholar] [CrossRef]
- Harwood, C.A.; Proby, C.M.; Arron, S.T. Genomics of SCC: Tumor Formation, Progression, and Future Therapeutic Implications for High-Risk Cutaneous Squamous Cell Carcinoma. In High-Risk Cutaneous Squamous Cell Carcinoma: A Practical Guide for Patient Management; Schmults, C.D., Ed.; Springer: Berlin/Heidelberg, Germany, 2016; pp. 67–102. [Google Scholar] [CrossRef]
- Cornejo, C.M.; Jambusaria-Pahlajani, A.; Willenbrink, T.J.; Schmults, C.D.; Arron, S.T.; Ruiz, E.S. Field cancerization: Treatment. J. Am. Acad. Dermatol. 2020, 83, 719–730. [Google Scholar] [CrossRef]
- Ishitsuka, Y.; Hanaoka, Y.; Tanemura, A.; Fujimoto, M. Cutaneous Squamous Cell Carcinoma in the Age of Immunotherapy. Cancers 2021, 13, 1148. [Google Scholar] [CrossRef]
- Kim, C.; Cheng, J.; Colegio, O.R. Cutaneous squamous cell carcinomas in solid organ transplant recipients: Emerging strategies for surveillance, staging, and treatment. Semin. Oncol. 2016, 43, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Schierbeck, J.; Vestergaard, T.; Bygum, A. Skin Cancer Associated Genodermatoses: A Literature Review. Acta Derm. Venereol. 2019, 99, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Errol, C.F.; Graham, C.W.; Wolfram, S.; Richard, D.W.; Roger, A.S.; Tom, E. DNA Repair and Mutagenesis, 2nd ed.; American Society of Microbiology: Washington, DC, USA, 2006. [Google Scholar] [CrossRef]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, N.; Rabbani, S.A. DNA Methylation Readers and Cancer: Mechanistic and Therapeutic Applications. Front. Oncol. 2019, 9, 489. [Google Scholar] [CrossRef]
- Leibeling, D.; Laspe, P.; Emmert, S. Nucleotide excision repair and cancer. J. Mol. Histol. 2006, 37, 225–238. [Google Scholar] [CrossRef]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef]
- Kraemer, K.H.; DiGiovanna, J.J. Xeroderma Pigmentosum. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Mirzaa, G.M., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Inman, G.J.; Wang, J.; Nagano, A.; Alexandrov, L.B.; Purdie, K.J.; Taylor, R.G.; Sherwood, V.; Thomson, J.; Hogan, S.; Spender, L.C.; et al. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018, 9, 3667. [Google Scholar] [CrossRef]
- Brash, D.E.; Rudolph, J.A.; Simon, J.A.; Lin, A.; McKenna, G.J.; Baden, H.P.; Halperin, A.J.; Ponten, J. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 1991, 88, 10124–10128. [Google Scholar] [CrossRef]
- Molho-Pessach, V.; Lotem, M. Ultraviolet radiation and cutaneous carcinogenesis. Curr. Probl. Derm. 2007, 35, 14–27. [Google Scholar]
- Campagna, R.; Valentina, p.; Davide, S. Beyond Nicotinamide Metabolism: Potential Role of Nicotinamide N-Methyltransferase as a Biomarker in Skin Cancers. Cancers 2021, 13, 4943. [Google Scholar] [CrossRef]
- Setlow, R.B.; Carrier, W.L. Pyrimidine dimers in ultraviolet-irradiated DNA’s. J. Mol. Biol. 1966, 17, 237–254. [Google Scholar] [CrossRef]
- Mitchell, D.L.; Nairn, R.S. The biology of the (6-4) photoproduct. Photochem. Photobiol. 1989, 49, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E. UV signature mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Chitsazzadeh, V.; Coarfa, C.; Drummond, J.A.; Nguyen, T.; Joseph, A.; Chilukuri, S.; Charpiot, E.; Adelmann, C.H.; Ching, G.; Nguyen, T.N.; et al. Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nat. Commun. 2016, 7, 12601. [Google Scholar] [CrossRef]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; den Breems, N.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A.; et al. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Li, Y.Y.; Hanna, G.J.; Laga, A.C.; Haddad, R.I.; Lorch, J.H.; Hammerman, P.S. Genomic analysis of metastatic cutaneous squamous cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1447–1456. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef]
- Al-Rohil, R.N.; Tarasen, A.J.; Carlson, J.A.; Wang, K.; Johnson, A.; Yelensky, R.; Lipson, D.; Elvin, J.A.; Vergilio, J.A.; Ali, S.M.; et al. Evaluation of 122 advanced-stage cutaneous squamous cell carcinomas by comprehensive genomic profiling opens the door for new routes to targeted therapies. Cancer 2016, 122, 249–257. [Google Scholar] [CrossRef]
- Ji, A.L.; Rubin, A.J.; Thrane, K.; Jiang, S.; Reynolds, D.L.; Meyers, R.M.; Guo, M.G.; George, B.M.; Mollbrink, A.; Bergenstrahle, J.; et al. Multimodal Analysis of Composition and Spatial Architecture in Human Squamous Cell Carcinoma. Cell 2020, 182, 497–514.e22. [Google Scholar] [CrossRef]
- Thomson, J.; Bewicke-Copley, F.; Anene, C.A.; Gulati, A.; Nagano, A.; Purdie, K.; Inman, G.J.; Proby, C.M.; Leigh, I.M.; Harwood, C.A.; et al. The Genomic Landscape of Actinic Keratosis. J. Investig. Dermatol. 2021, 141, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Capell, B.C.; Parekh, V.; O’Day, C.; Atillasoy, C.; Bashir, H.M.; Yeh, C.; Shim, E.H.; Prouty, S.M.; Dentchev, T.; et al. Whole-Exome and Transcriptome Analysis of UV-Exposed Epidermis and Carcinoma In Situ Reveals Early Drivers of Carcinogenesis. J. Investig. Derm. 2021, 141, 295–307.e13. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Bhaduri, A.; Mah, A.; Johnson, W.L.; Ungewickell, A.; Aros, C.J.; Nguyen, C.B.; Rios, E.J.; Siprashvili, Z.; Straight, A.; et al. Recurrent point mutations in the kinetochore gene KNSTRN in cutaneous squamous cell carcinoma. Nat. Genet. 2014, 46, 1060–1062. [Google Scholar] [CrossRef] [PubMed]
- Kanellou, P.; Zaravinos, A.; Zioga, M.; Stratigos, A.; Baritaki, S.; Soufla, G.; Zoras, O.; Spandidos, D.A. Genomic instability, mutations and expression analysis of the tumour suppressor genes p14(ARF), p15(INK4b), p16(INK4a) and p53 in actinic keratosis. Cancer Lett. 2008, 264, 145–161. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef]
- Lane, D.P. Cancer. p53, guardian of the genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef]
- Benjamin, C.L.; Ananthaswamy, H.N. p53 and the pathogenesis of skin cancer. Toxicol. Appl. Pharmacol. 2007, 224, 241–248. [Google Scholar] [CrossRef]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Zilfou, J.T.; Lowe, S.W. Tumor suppressive functions of p53. Cold Spring Harb. Perspect. Biol. 2009, 1, a001883. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2001, 2, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Yee, K.S.; Vousden, K.H. Complicating the complexity of p53. Carcinogenesis 2005, 26, 1317–1322. [Google Scholar] [CrossRef] [PubMed]
- Meek, D.W.; Anderson, C.W. Posttranslational modification of p53: Cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009, 1, a000950. [Google Scholar] [CrossRef]
- Sherr, C.J. Divorcing ARF and p53: An unsettled case. Nat. Rev. Cancer 2006, 6, 663–673. [Google Scholar] [CrossRef]
- Pomerantz, J.; Schreiber-Agus, N.; Liegeois, N.J.; Silverman, A.; Alland, L.; Chin, L.; Potes, J.; Chen, K.; Orlow, I.; Lee, H.W.; et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998, 92, 713–723. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiong, Y.; Yarbrough, W.G. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998, 92, 725–734. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Miyashita, T.; Reed, J.C. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell 1995, 80, 293–299. [Google Scholar] [CrossRef]
- Tan, T.; Chu, G. p53 Binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice. Mol. Cell. Biol. 2002, 22, 3247–3254. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Kern, S.E.; Pietenpol, J.A.; Kinzler, K.W.; Vogelstein, B. Definition of a consensus binding site for p53. Nat. Genet. 1992, 1, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Kolev, V.; Mandinova, A.; Guinea-Viniegra, J.; Hu, B.; Lefort, K.; Lambertini, C.; Neel, V.; Dummer, R.; Wagner, E.F.; Dotto, G.P. EGFR signalling as a negative regulator of Notch1 gene transcription and function in proliferating keratinocytes and cancer. Nat. Cell Biol. 2008, 10, 902–911. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, W.; Marshall, C.; Griffin, T.; Hanson, M.; Hick, R.; Dentchev, T.; Williams, E.; Werth, A.; Miller, C.; et al. Srcasm inhibits Fyn-induced cutaneous carcinogenesis with modulation of Notch1 and p53. Cancer Res. 2009, 69, 9439–9447. [Google Scholar] [CrossRef]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Greenblatt, M.S.; Bennett, W.P.; Hollstein, M.; Harris, C.C. Mutations in the p53 tumor suppressor gene: Clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994, 54, 4855–4878. [Google Scholar] [PubMed]
- Hainaut, P.; Hollstein, M. p53 and human cancer: The first ten thousand mutations. Adv. Cancer Res. 2000, 77, 81–137. [Google Scholar]
- Muller, P.A.; Vousden, K.H. Mutant p53 in cancer: New functions and therapeutic opportunities. Cancer Cell 2014, 25, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Suh, Y.A.; Fuller, M.Y.; Jackson, J.G.; Xiong, S.; Terzian, T.; Quintas-Cardama, A.; Bankson, J.A.; El-Naggar, A.K.; Lozano, G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J. Clin. Investig. 2011, 121, 893–904. [Google Scholar] [CrossRef]
- Weisz, L.; Oren, M.; Rotter, V. Transcription regulation by mutant p53. Oncogene 2007, 26, 2202–2211. [Google Scholar] [CrossRef]
- Bougeard, G.; Sesboue, R.; Baert-Desurmont, S.; Vasseur, S.; Martin, C.; Tinat, J.; Brugieres, L.; Chompret, A.; de Paillerets, B.B.; Stoppa-Lyonnet, D.; et al. Molecular basis of the Li-Fraumeni syndrome: An update from the French LFS families. J. Med. Genet. 2008, 45, 535–538. [Google Scholar] [CrossRef]
- Alexandrova, E.M.; Mirza, S.A.; Xu, S.; Schulz-Heddergott, R.; Marchenko, N.D.; Moll, U.M. p53 loss-of-heterozygosity is a necessary prerequisite for mutant p53 stabilization and gain-of-function in vivo. Cell Death Dis. 2017, 8, e2661. [Google Scholar] [CrossRef] [PubMed]
- Amelio, I.; Mancini, M.; Petrova, V.; Cairns, R.A.; Vikhreva, P.; Nicolai, S.; Marini, A.; Antonov, A.A.; Le Quesne, J.; Baena Acevedo, J.D.; et al. p53 mutants cooperate with HIF-1 in transcriptional regulation of extracellular matrix components to promote tumor progression. Proc. Natl. Acad. Sci. USA 2018, 115, E10869–E10878. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Thoenen, E.; Iwakuma, T. The interplay between mutant p53 and the mevalonate pathway. Cell Death Differ. 2018, 25, 460–470. [Google Scholar] [CrossRef]
- Giglia-Mari, G.; Sarasin, A. TP53 mutations in human skin cancers. Hum. Mutat. 2003, 21, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.; Jonason, A.S.; Leffell, D.J.; Simon, J.A.; Sharma, H.W.; Kimmelman, J.; Remington, L.; Jacks, T.; Brash, D.E. Sunburn and p53 in the onset of skin cancer. Nature 1994, 372, 773–776. [Google Scholar] [CrossRef]
- Brash, D.E.; Ziegler, A.; Jonason, A.S.; Simon, J.A.; Kunala, S.; Leffell, D.J. Sunlight and sunburn in human skin cancer: p53, apoptosis, and tumor promotion. J. Investig. Derm. Symp. Proc. 1996, 1, 136–142. [Google Scholar]
- Durinck, S.; Ho, C.; Wang, N.J.; Liao, W.; Jakkula, L.R.; Collisson, E.A.; Pons, J.; Chan, S.W.; Lam, E.T.; Chu, C.; et al. Temporal dissection of tumorigenesis in primary cancers. Cancer Discov. 2011, 1, 137–143. [Google Scholar] [CrossRef]
- Tanaka, N.; Zhao, M.; Tang, L.; Patel, A.A.; Xi, Q.; Van, H.T.; Takahashi, H.; Osman, A.A.; Zhang, J.; Wang, J.; et al. Gain-of-function mutant p53 promotes the oncogenic potential of head and neck squamous cell carcinoma cells by targeting the transcription factors FOXO3a and FOXM1. Oncogene 2018, 37, 1279–1292. [Google Scholar] [CrossRef]
- Stahl, P.L.; Stranneheim, H.; Asplund, A.; Berglund, L.; Ponten, F.; Lundeberg, J. Sun-induced nonsynonymous p53 mutations are extensively accumulated and tolerated in normal appearing human skin. J. Investig. Dermatol. 2011, 131, 504–508. [Google Scholar] [CrossRef]
- Fernandez-Figueras, M.T.; Saenz-Sarda, X.; Vargas, P.; Thompson, C.T.; Carrato, C.; Puig, L.; Ferrandiz, C.; Ariza, A. The depth of follicular extension in actinic keratosis correlates with the depth of invasion in squamous cell carcinoma: Implication for clinical treatment. J. Eur. Acad. Dermatol. Venereol. JEADV 2018, 32, 1657–1661. [Google Scholar] [CrossRef]
- Hedberg, M.; Seykora, J.T. Clarifying Progress on the Genomic Landscape of Actinic Keratosis. J. Investig. Dermatol. 2021, 141, 1622–1624. [Google Scholar] [CrossRef] [PubMed]
- Marks, R.; Rennie, G.; Selwood, T.S. Malignant transformation of solar keratoses to squamous cell carcinoma. Lancet 1988, 1, 795–797. [Google Scholar] [CrossRef]
- Gilchrest, B.A. Actinic Keratoses: Reconciling the Biology of Field Cancerization with Treatment Paradigms. J. Investig. Dermatol. 2021, 141, 727–731. [Google Scholar] [CrossRef]
- Quintana, R.M.; Dupuy, A.J.; Bravo, A.; Casanova, M.L.; Alameda, J.P.; Page, A.; Sanchez-Viera, M.; Ramirez, A.; Navarro, M. A transposon-based analysis of gene mutations related to skin cancer development. J. Investig. Dermatol. 2013, 133, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P. Notch tumor suppressor function. Oncogene 2008, 27, 5115. [Google Scholar] [CrossRef]
- Nowell, C.; Radtke, F. Cutaneous Notch signaling in health and disease. Cold Spring Harb. Perspect. Med. 2013, 3, a017772. [Google Scholar] [CrossRef]
- Okuyama, R.; Tagami, H.; Aiba, S. Notch signaling: Its role in epidermal homeostasis and in the pathogenesis of skin diseases. J. Dermatol. Sci. 2008, 49, 187–194. [Google Scholar] [CrossRef]
- Moretti, F.; Marinari, B.; Lo Iacono, N.; Botti, E.; Giunta, A.; Spallone, G.; Garaffo, G.; Vernersson-Lindahl, E.; Merlo, G.; Mills, A.A.; et al. A regulatory feedback loop involving p63 and IRF6 links the pathogenesis of 2 genetically different human ectodermal dysplasias. J. Clin. Investig. 2010, 120, 1570–1577. [Google Scholar] [CrossRef]
- Nguyen, B.C.; Lefort, K.; Mandinova, A.; Antonini, D.; Devgan, V.; Della Gatta, G.; Koster, M.I.; Zhang, Z.; Wang, J.; Tommasi di Vignano, A.; et al. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006, 20, 1028–1042. [Google Scholar] [CrossRef]
- Okuyama, R.; Nguyen, B.C.; Talora, C.; Ogawa, E.; Tommasi di Vignano, A.; Lioumi, M.; Chiorino, G.; Tagami, H.; Woo, M.; Dotto, G.P. High commitment of embryonic keratinocytes to terminal differentiation through a Notch1-caspase 3 regulatory mechanism. Dev. Cell 2004, 6, 551–562. [Google Scholar] [CrossRef]
- Lefort, K.; Mandinova, A.; Ostano, P.; Kolev, V.; Calpini, V.; Kolfschoten, I.; Devgan, V.; Lieb, J.; Raffoul, W.; Hohl, D.; et al. Notch1 is a p53 target gene involved in human keratinocyte tumor suppression through negative regulation of ROCK1/2 and MRCKalpha kinases. Genes Dev. 2007, 21, 562–577. [Google Scholar] [CrossRef] [PubMed]
- Demehri, S.; Turkoz, A.; Kopan, R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 2009, 16, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.C.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416–421. [Google Scholar] [CrossRef]
- Proweller, A.; Tu, L.; Lepore, J.J.; Cheng, L.; Lu, M.M.; Seykora, J.; Millar, S.E.; Pear, W.S.; Parmacek, M.S. Impaired notch signaling promotes de novo squamous cell carcinoma formation. Cancer Res. 2006, 66, 7438–7444. [Google Scholar] [CrossRef]
- Mandinova, A.; Lefort, K.; Tommasi di Vignano, A.; Stonely, W.; Ostano, P.; Chiorino, G.; Iwaki, H.; Nakanishi, J.; Dotto, G.P. The FoxO3a gene is a key negative target of canonical Notch signalling in the keratinocyte UVB response. EMBO J 2008, 27, 1243–1254. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Wennerberg, K.; Rossman, K.L.; Der, C.J. The Ras superfamily at a glance. J. Cell Sci. 2005, 118, 843–846. [Google Scholar] [CrossRef]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef]
- van der Schroeff, J.G.; Evers, L.M.; Boot, A.J.; Bos, J.L. Ras oncogene mutations in basal cell carcinomas and squamous cell carcinomas of human skin. J. Investig. Dermatol. 1990, 94, 423–425. [Google Scholar] [CrossRef]
- Spencer, J.M.; Kahn, S.M.; Jiang, W.; DeLeo, V.A.; Weinstein, I.B. Activated ras genes occur in human actinic keratoses, premalignant precursors to squamous cell carcinomas. Arch. Dermatol. 1995, 131, 796–800. [Google Scholar] [CrossRef] [PubMed]
- Pierceall, W.E.; Goldberg, L.H.; Tainsky, M.A.; Mukhopadhyay, T.; Ananthaswamy, H.N. Ras gene mutation and amplification in human nonmelanoma skin cancers. Mol. Carcinog. 1991, 4, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Ashford, B.G.; Clark, J.; Gupta, R.; Iyer, N.G.; Yu, B.; Ranson, M. Reviewing the genetic alterations in high-risk cutaneous squamous cell carcinoma: A search for prognostic markers and therapeutic targets. Head Neck 2017, 39, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Boukamp, P. Non-melanoma skin cancer: What drives tumor development and progression? Carcinogenesis 2005, 26, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef]
- Huang, P.Y.; Balmain, A. Modeling cutaneous squamous carcinoma development in the mouse. Cold Spring Harb. Perspect. Med. 2014, 4, a013623. [Google Scholar] [CrossRef]
- Serrano, M.; Lee, H.; Chin, L.; Cordon-Cardo, C.; Beach, D.; DePinho, R.A. Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996, 85, 27–37. [Google Scholar] [CrossRef]
- Quelle, D.E.; Zindy, F.; Ashmun, R.A.; Sherr, C.J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 1995, 83, 993–1000. [Google Scholar] [CrossRef]
- Honda, R.; Yasuda, H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J 1999, 18, 22–27. [Google Scholar] [CrossRef]
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26. [Google Scholar] [CrossRef]
- Soufir, N.; Moles, J.P.; Vilmer, C.; Moch, C.; Verola, O.; Rivet, J.; Tesniere, A.; Dubertret, L.; Basset-Seguin, N. P16 UV mutations in human skin epithelial tumors. Oncogene 1999, 18, 5477–5481. [Google Scholar] [CrossRef] [PubMed]
- Saridaki, Z.; Liloglou, T.; Zafiropoulos, A.; Koumantaki, E.; Zoras, O.; Spandidos, D.A. Mutational analysis of CDKN2A genes in patients with squamous cell carcinoma of the skin. Br. J. Dermatol. 2003, 148, 638–648. [Google Scholar] [CrossRef] [PubMed]
- Blokx, W.A.; Ruiter, D.J.; Verdijk, M.A.; de Wilde, P.C.; Willems, R.W.; de Jong, E.M.; Ligtenberg, M.J. INK4-ARF and p53 mutations in metastatic cutaneous squamous cell carcinoma: Case report and archival study on the use of Ink4a-ARF and p53 mutation analysis in identification of the corresponding primary tumor. Am. J. Surg. Pathol. 2005, 29, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Mortier, L.; Marchetti, P.; Delaporte, E.; Martin de Lassalle, E.; Thomas, P.; Piette, F.; Formstecher, P.; Polakowska, R.; Danze, P.M. Progression of actinic keratosis to squamous cell carcinoma of the skin correlates with deletion of the 9p21 region encoding the p16(INK4a) tumor suppressor. Cancer Lett. 2002, 176, 205–214. [Google Scholar] [CrossRef]
- Hedberg, M.L.; Peyser, N.D.; Bauman, J.E.; Gooding, W.E.; Li, H.; Bhola, N.E.; Zhu, T.R.; Zeng, Y.; Brand, T.M.; Kim, M.O.; et al. Use of nonsteroidal anti-inflammatory drugs predicts improved patient survival for PIK3CA-altered head and neck cancer. J. Exp. Med. 2019, 216, 419–427. [Google Scholar] [CrossRef]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef]
- Cai, Y.; Yousef, A.; Grandis, J.R.; Johnson, D.E. NSAID therapy for PIK3CA-Altered colorectal, breast, and head and neck cancer. Adv. Biol. Regul. 2020, 75, 100653. [Google Scholar] [CrossRef]
- Fang, L.; Seki, A.; Fang, G. SKAP associates with kinetochores and promotes the metaphase-to-anaphase transition. Cell Cycle 2009, 8, 2819–2827. [Google Scholar] [CrossRef]
- Schmitz, L.; Grinblat, B.; Novak, B.; Hoeh, A.K.; Handschke, K.; von Dobbeler, C.; Bierhoff, E.; Szeimies, R.M.; Gambichler, T.; Torezan, L.; et al. Somatic mutations in kinetochore gene KNSTRN are associated with basal proliferating actinic keratoses and cutaneous squamous cell carcinoma. J. Eur. Acad. Dermatol. Venereol. JEADV 2019, 33, 1535–1540. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef]
- Yilmaz, A.S.; Ozer, H.G.; Gillespie, J.L.; Allain, D.C.; Bernhardt, M.N.; Furlan, K.C.; Castro, L.T.; Peters, S.B.; Nagarajan, P.; Kang, S.Y.; et al. Differential mutation frequencies in metastatic cutaneous squamous cell carcinomas versus primary tumors. Cancer 2017, 123, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Pickering, C.R.; Zhang, J.; Yoo, S.Y.; Bengtsson, L.; Moorthy, S.; Neskey, D.M.; Zhao, M.; Ortega Alves, M.V.; Chang, K.; Drummond, J.; et al. Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov. 2013, 3, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Morris, L.G.; Kaufman, A.M.; Gong, Y.; Ramaswami, D.; Walsh, L.A.; Turcan, S.; Eng, S.; Kannan, K.; Zou, Y.; Peng, L.; et al. Recurrent somatic mutation of FAT1 in multiple human cancers leads to aberrant Wnt activation. Nat. Genet. 2013, 45, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Razavi, P.; Li, Q.; Toy, W.; Liu, B.; Ping, C.; Hsieh, W.; Sanchez-Vega, F.; Brown, D.N.; Da Cruz Paula, A.F.; et al. Loss of the FAT1 Tumor Suppressor Promotes Resistance to CDK4/6 Inhibitors via the Hippo Pathway. Cancer Cell 2018, 34, 893–905.e8. [Google Scholar] [CrossRef] [PubMed]
- Bertin, J.; Wang, L.; Guo, Y.; Jacobson, M.D.; Poyet, J.L.; Srinivasula, S.M.; Merriam, S.; DiStefano, P.S.; Alnemri, E.S. CARD11 and CARD14 are novel caspase recruitment domain (CARD)/membrane-associated guanylate kinase (MAGUK) family members that interact with BCL10 and activate NF-kappa B. J. Biol. Chem. 2001, 276, 11877–11882. [Google Scholar] [CrossRef]
- Watt, S.A.; Purdie, K.J.; den Breems, N.Y.; Dimon, M.; Arron, S.T.; McHugh, A.T.; Xue, D.J.; Dayal, J.H.; Proby, C.M.; Harwood, C.A.; et al. Novel CARD11 Mutations in Human Cutaneous Squamous Cell Carcinoma Lead to Aberrant NF-kappaB Regulation. Am. J. Pathol. 2015, 185, 2354–2363. [Google Scholar] [CrossRef]
- Sur, I.; Ulvmar, M.; Toftgard, R. The two-faced NF-kappaB in the skin. Int. Rev. Immunol. 2008, 27, 205–223. [Google Scholar] [CrossRef]
- van Hogerlinden, M.; Rozell, B.L.; Ahrlund-Richter, L.; Toftgard, R. Squamous cell carcinomas and increased apoptosis in skin with inhibited Rel/nuclear factor-kappaB signaling. Cancer Res. 1999, 59, 3299–3303. [Google Scholar]
- Zhang, J.Y.; Tao, S.; Kimmel, R.; Khavari, P.A. CDK4 regulation by TNFR1 and JNK is required for NF-kappaB-mediated epidermal growth control. J. Cell Biol. 2005, 168, 561–566. [Google Scholar] [CrossRef]
- Seykora, J.T.; Mei, L.; Dotto, G.P.; Stein, P.L. ‘Srcasm: A novel Src activating and signaling molecule. J. Biol. Chem. 2002, 277, 2812–2822. [Google Scholar] [CrossRef]
- Li, W.; Marshall, C.; Mei, L.; Gelfand, J.; Seykora, J.T. Srcasm corrects Fyn-induced epidermal hyperplasia by kinase down-regulation. J. Biol. Chem. 2007, 282, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Marshall, C.; Mei, L.; Dzubow, L.; Schmults, C.; Dans, M.; Seykora, J. Srcasm modulates EGF and Src-kinase signaling in keratinocytes. J. Biol. Chem. 2005, 280, 6036–6046. [Google Scholar] [CrossRef] [PubMed]
- Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [CrossRef] [PubMed]
- Gatti, V.; Fierro, C.; Annicchiarico-Petruzzelli, M.; Melino, G.; Peschiaroli, A. ΔNp63 in squamous cell carcinoma: Defining the oncogenic routes affecting epigenetic landscape and tumour microenvironment. Mol. Oncol. 2019, 13, 981–1001. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.; Kaghad, M.; Wang, Y.; Gillett, E.; Fleming, M.D.; Dotsch, V.; Andrews, N.C.; Caput, D.; McKeon, F. p63, a p53 homolog at 3q27-29, encodes multiple products with transactivating, death-inducing, and dominant-negative activities. Mol. Cell 1998, 2, 305–316. [Google Scholar] [CrossRef]
- Yang, A.; Schweitzer, R.; Sun, D.; Kaghad, M.; Walker, N.; Bronson, R.T.; Tabin, C.; Sharpe, A.; Caput, D.; Crum, C.; et al. p63 is essential for regenerative proliferation in limb, craniofacial and epithelial development. Nature 1999, 398, 714–718. [Google Scholar] [CrossRef]
- Vanbokhoven, H.; Melino, G.; Candi, E.; Declercq, W. p63, a story of mice and men. J. Investig. Dermatol. 2011, 131, 1196–1207. [Google Scholar] [CrossRef]
- Candi, E.; Cipollone, R.; Rivetti di Val Cervo, P.; Gonfloni, S.; Melino, G.; Knight, R. p63 in epithelial development. Cell. Mol. Life Sci. CMLS 2008, 65, 3126–3133. [Google Scholar] [CrossRef]
- Petitjean, A.; Ruptier, C.; Tribollet, V.; Hautefeuille, A.; Chardon, F.; Cavard, C.; Puisieux, A.; Hainaut, P.; Caron de Fromentel, C. Properties of the six isoforms of p63: p53-like regulation in response to genotoxic stress and cross talk with DeltaNp73. Carcinogenesis 2008, 29, 273–281. [Google Scholar] [CrossRef]
- Rokudai, S.; Li, Y.; Otaka, Y.; Fujieda, M.; Owens, D.M.; Christiano, A.M.; Nishiyama, M.; Prives, C. STXBP4 regulates APC/C-mediated p63 turnover and drives squamous cell carcinogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E4806–E4814. [Google Scholar] [CrossRef]
- Dohn, M.; Zhang, S.; Chen, X. p63alpha and DeltaNp63alpha can induce cell cycle arrest and apoptosis and differentially regulate p53 target genes. Oncogene 2001, 20, 3193–3205. [Google Scholar] [CrossRef] [PubMed]
- Mardaryev, A.N.; Gdula, M.R.; Yarker, J.L.; Emelianov, V.U.; Poterlowicz, K.; Sharov, A.A.; Sharova, T.Y.; Scarpa, J.A.; Joffe, B.; Solovei, I.; et al. p63 and Brg1 control developmentally regulated higher-order chromatin remodelling at the epidermal differentiation complex locus in epidermal progenitor cells. Development 2014, 141, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Mardaryev, A.N.; Liu, B.; Rapisarda, V.; Poterlowicz, K.; Malashchuk, I.; Rudolf, J.; Sharov, A.A.; Jahoda, C.A.; Fessing, M.Y.; Benitah, S.A.; et al. Cbx4 maintains the epithelial lineage identity and cell proliferation in the developing stratified epithelium. J. Cell Biol. 2016, 212, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Cao, Y.; Gong, X.; Li, H. Long noncoding RNAs in head and neck cancer. Oncotarget 2017, 8, 10726–10740. [Google Scholar] [CrossRef]
- Yugawa, T.; Narisawa-Saito, M.; Yoshimatsu, Y.; Haga, K.; Ohno, S.-i.; Egawa, N.; Fujita, M.; Kiyono, T. ΔNp63α Repression of the Notch1 Gene Supports the Proliferative Capacity of Normal Human Keratinocytes and Cervical Cancer Cells. Cancer Res. 2010, 70, 4034–4044. [Google Scholar] [CrossRef]
- Westfall, M.D.; Mays, D.J.; Sniezek, J.C.; Pietenpol, J.A. The Delta Np63 alpha phosphoprotein binds the p21 and 14-3-3 sigma promoters in vivo and has transcriptional repressor activity that is reduced by Hay-Wells syndrome-derived mutations. Mol. Cell. Biol. 2003, 23, 2264–2276. [Google Scholar] [CrossRef]
- Devos, M.; Gilbert, B.; Denecker, G.; Leurs, K.; Mc Guire, C.; Lemeire, K.; Hochepied, T.; Vuylsteke, M.; Lambert, J.; Van Den Broecke, C.; et al. Elevated DeltaNp63alpha Levels Facilitate Epidermal and Biliary Oncogenic Transformation. J. Investig. Dermatol. 2017, 137, 494–505. [Google Scholar] [CrossRef]
- Ramsey, M.R.; Wilson, C.; Ory, B.; Rothenberg, S.M.; Faquin, W.; Mills, A.A.; Ellisen, L.W. FGFR2 signaling underlies p63 oncogenic function in squamous cell carcinoma. J. Clin. Investig. 2013, 123, 3525–3538. [Google Scholar] [CrossRef]
- Abraham, C.G.; Ludwig, M.P.; Andrysik, Z.; Pandey, A.; Joshi, M.; Galbraith, M.D.; Sullivan, K.D.; Espinosa, J.M. ΔNp63α Suppresses TGFB2 Expression and RHOA Activity to Drive Cell Proliferation in Squamous Cell Carcinomas. Cell Rep. 2018, 24, 3224–3236. [Google Scholar] [CrossRef]
- Hazawa, M.; Lin, D.C.; Kobayashi, A.; Jiang, Y.Y.; Xu, L.; Dewi, F.R.P.; Mohamed, M.S.; Nakada, M.; Meguro-Horike, M.; Horike, S.I.; et al. ROCK-dependent phosphorylation of NUP62 regulates p63 nuclear transport and squamous cell carcinoma proliferation. EMBO Rep. 2018, 19, 73–88. [Google Scholar] [CrossRef]
- Li, Y.; Peart, M.J.; Prives, C. Stxbp4 regulates DeltaNp63 stability by suppression of RACK1-dependent degradation. Mol. Cell. Biol. 2009, 29, 3953–3963. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.J.; Patel, A.; Purdie, K.J.; Wang, J.; Rizvi, H.; Hufbauer, M.; Ostano, P.; Akgul, B.; Chiorino, G.; Harwood, C.A.; et al. Epigenetic Regulation of iASPP-p63 Feedback Loop in Cutaneous Squamous Cell Carcinoma. J. Investig. Dermatol. 2019, 139, 1658–1671.e8. [Google Scholar] [CrossRef] [PubMed]
- de Lima, P.O.; Joseph, S.; Panizza, B.; Simpson, F. Epidermal Growth Factor Receptor’s Function in Cutaneous Squamous Cell Carcinoma and Its Role as a Therapeutic Target in the Age of Immunotherapies. Curr. Treat. Options Oncol. 2020, 21, 9. [Google Scholar] [CrossRef] [PubMed]
- Jost, M.; Kari, C.; Rodeck, U. The EGF receptor—An essential regulator of multiple epidermal functions. Eur. J. Dermatol. EJD 2000, 10, 505–510. [Google Scholar] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef]
- Toll, A.; Salgado, R.; Yebenes, M.; Martin-Ezquerra, G.; Gilaberte, M.; Baro, T.; Sole, F.; Alameda, F.; Espinet, B.; Pujol, R.M. Epidermal growth factor receptor gene numerical aberrations are frequent events in actinic keratoses and invasive cutaneous squamous cell carcinomas. Exp. Dermatol. 2010, 19, 151–153. [Google Scholar] [CrossRef]
- Canueto, J.; Cardenoso, E.; Garcia, J.L.; Santos-Briz, A.; Castellanos-Martin, A.; Fernandez-Lopez, E.; Blanco Gomez, A.; Perez-Losada, J.; Roman-Curto, C. Epidermal growth factor receptor expression is associated with poor outcome in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2017, 176, 1279–1287. [Google Scholar] [CrossRef]
- Grandis, J.R.; Zeng, Q.; Drenning, S.D.; Tweardy, D.J. Normalization of EGFR mRNA levels following restoration of wild-type p53 in a head and neck squamous cell carcinoma cell line. Int. J. Oncol. 1998, 13, 375–378. [Google Scholar] [CrossRef]
- Zhang, Y.; Gao, L.; Ma, S.; Ma, J.; Wang, Y.; Li, S.; Hu, X.; Han, S.; Zhou, M.; Zhou, L.; et al. MALAT1-KTN1-EGFR regulatory axis promotes the development of cutaneous squamous cell carcinoma. Cell Death Differ. 2019, 26, 2061–2073. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, X.; Dai, Y.; Niu, X.; Zhou, M. miR-27a Downregulation Promotes Cutaneous Squamous Cell Carcinoma Progression via Targeting EGFR. Front. Oncol. 2019, 9, 1565. [Google Scholar] [CrossRef]
- Nindl, I.; Dang, C.; Forschner, T.; Kuban, R.J.; Meyer, T.; Sterry, W.; Stockfleth, E. Identification of differentially expressed genes in cutaneous squamous cell carcinoma by microarray expression profiling. Mol. Cancer 2006, 5, 30. [Google Scholar] [CrossRef] [PubMed]
- Dooley, T.P.; Reddy, S.P.; Wilborn, T.W.; Davis, R.L. Biomarkers of human cutaneous squamous cell carcinoma from tissues and cell lines identified by DNA microarrays and qRT-PCR. Biochem. Biophys. Res. Commun. 2003, 306, 1026–1036. [Google Scholar] [CrossRef]
- Hameetman, L.; Commandeur, S.; Bavinck, J.N.B.; Wisgerhof, H.C.; de Gruijl, F.R.; Willemze, R.; Mullenders, L.; Tensen, C.P.; Vrieling, H. Molecular profiling of cutaneous squamous cell carcinomas and actinic keratoses from organ transplant recipients. BMC Cancer 2013, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diez, I.; Hernandez-Munoz, I.; Hernandez-Ruiz, E.; Nonell, L.; Puigdecanet, E.; Bodalo-Torruella, M.; Andrades, E.; Pujol, R.M.; Toll, A. Transcriptome and cytogenetic profiling analysis of matched in situ/invasive cutaneous squamous cell carcinomas from immunocompetent patients. Genes Chromosomes Cancer 2019, 58, 164–174. [Google Scholar] [CrossRef]
- Mitsui, H.; Suarez-Farinas, M.; Gulati, N.; Shah, K.R.; Cannizzaro, M.V.; Coats, I.; Felsen, D.; Krueger, J.G.; Carucci, J.A. Gene expression profiling of the leading edge of cutaneous squamous cell carcinoma: IL-24-driven MMP-7. J. Investig. Dermatol. 2014, 134, 1418–1427. [Google Scholar] [CrossRef]
- Padilla, R.S.; Sebastian, S.; Jiang, Z.; Nindl, I.; Larson, R. Gene expression patterns of normal human skin, actinic keratosis, and squamous cell carcinoma: A spectrum of disease progression. Arch. Dermatol. 2010, 146, 288–293. [Google Scholar] [CrossRef]
- Kathpalia, V.P.; Mussak, E.N.; Chow, S.S.; Lam, P.H.; Skelley, N.; Time, M.; Markelewicz, R.J., Jr.; Kanduc, D.; Lomas, L.; Xiang, Z.; et al. Genome-wide transcriptional profiling in human squamous cell carcinoma of the skin identifies unique tumor-associated signatures. J. Dermatol. 2006, 33, 309–318. [Google Scholar] [CrossRef]
- Ra, S.H.; Li, X.; Binder, S. Molecular discrimination of cutaneous squamous cell carcinoma from actinic keratosis and normal skin. Mod. Pathol. 2011, 24, 963–973. [Google Scholar] [CrossRef]
- Hudson, L.G.; Gale, J.M.; Padilla, R.S.; Pickett, G.; Alexander, B.E.; Wang, J.; Kusewitt, D.F. Microarray analysis of cutaneous squamous cell carcinomas reveals enhanced expression of epidermal differentiation complex genes. Mol. Carcinog. 2010, 49, 619–629. [Google Scholar] [CrossRef]
- Kar, A.; Gutierrez-Hartmann, A. Molecular mechanisms of ETS transcription factor-mediated tumorigenesis. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 522–543. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Banno, K.; Kisu, I.; Yanokura, M.; Tsuji, K.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Yamagami, W.; Nomura, H.; Tominaga, E.; et al. Epimutation and cancer: A new carcinogenic mechanism of Lynch syndrome (Review). Int. J. Oncol. 2012, 41, 793–797. [Google Scholar] [CrossRef] [PubMed]
- Brown, V.L.; Harwood, C.A.; Crook, T.; Cronin, J.G.; Kelsell, D.P.; Proby, C.M. p16INK4a and p14ARF tumor suppressor genes are commonly inactivated in cutaneous squamous cell carcinoma. J. Investig. Dermatol. 2004, 122, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Venza, I.; Visalli, M.; Tripodo, B.; De Grazia, G.; Loddo, S.; Teti, D.; Venza, M. FOXE1 is a target for aberrant methylation in cutaneous squamous cell carcinoma. Br. J. Dermatol. 2010, 162, 1093–1097. [Google Scholar] [CrossRef] [PubMed]
- Darr, O.A.; Colacino, J.A.; Tang, A.L.; McHugh, J.B.; Bellile, E.L.; Bradford, C.R.; Prince, M.P.; Chepeha, D.B.; Rozek, L.S.; Moyer, J.S. Epigenetic alterations in metastatic cutaneous carcinoma. Head Neck 2015, 37, 994–1001. [Google Scholar] [CrossRef]
- Chiles, M.C.; Ai, L.; Zuo, C.; Fan, C.Y.; Smoller, B.R. E-cadherin promoter hypermethylation in preneoplastic and neoplastic skin lesions. Mod. Pathol. 2003, 16, 1014–1018. [Google Scholar] [CrossRef]
- Fraga, M.F.; Herranz, M.; Espada, J.; Ballestar, E.; Paz, M.F.; Ropero, S.; Erkek, E.; Bozdogan, O.; Peinado, H.; Niveleau, A.; et al. A mouse skin multistage carcinogenesis model reflects the aberrant DNA methylation patterns of human tumors. Cancer Res. 2004, 64, 5527–5534. [Google Scholar] [CrossRef]
- Nessa, A.; Rashid, M.H.U.; E-Ferdous, N.; Chowdhury, A. Screening for and management of high-grade cervical intraepithelial neoplasia in Bangladesh: A cross-sectional study comparing two protocols. J. Obstet. Gynaecol. Res. 2013, 39, 564–571. [Google Scholar] [CrossRef]
- Missero, C.; Calautti, E.; Eckner, R.; Chin, J.; Tsai, L.H.; Livingston, D.M.; Dotto, G.P. Involvement of the cell-cycle inhibitor Cip1/WAF1 and the E1A-associated p300 protein in terminal differentiation. Proc. Natl. Acad. Sci. USA 1995, 92, 5451–5455. [Google Scholar] [CrossRef]
- Wang, F.; Marshall, C.B.; Ikura, M. Transcriptional/epigenetic regulator CBP/p300 in tumorigenesis: Structural and functional versatility in target recognition. Cell. Mol. Life Sci. CMLS 2013, 70, 3989–4008. [Google Scholar] [CrossRef]
- McCabe, M.T.; Creasy, C.L. EZH2 as a potential target in cancer therapy. Epigenomics 2014, 6, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Hung, M.C. Regulation and Role of EZH2 in Cancer. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2014, 46, 209–222. [Google Scholar] [CrossRef] [PubMed]
- Ezhkova, E.; Pasolli, H.A.; Parker, J.S.; Stokes, N.; Su, I.H.; Hannon, G.; Tarakhovsky, A.; Fuchs, E. Ezh2 orchestrates gene expression for the stepwise differentiation of tissue-specific stem cells. Cell 2009, 136, 1122–1135. [Google Scholar] [CrossRef] [PubMed]
- Adhikary, G.; Grun, D.; Balasubramanian, S.; Kerr, C.; Huang, J.M.; Eckert, R.L. Survival of skin cancer stem cells requires the Ezh2 polycomb group protein. Carcinogenesis 2015, 36, 800–810. [Google Scholar] [CrossRef]
- Behrens, C.; Solis, L.M.; Lin, H.; Yuan, P.; Tang, X.; Kadara, H.; Riquelme, E.; Galindo, H.; Moran, C.A.; Kalhor, N.; et al. EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 6556–6565. [Google Scholar] [CrossRef]
- Xie, Q.; Wang, H.; Heilman, E.R.; Walsh, M.G.; Haseeb, M.A.; Gupta, R. Increased expression of enhancer of Zeste Homolog 2 (EZH2) differentiates squamous cell carcinoma from normal skin and actinic keratosis. Eur. J. Dermatol. EJD 2014, 24, 41–45. [Google Scholar] [CrossRef]
- Wu, C.; Chen, Z.; Kuchroo, V.K. Ezh2 lines up the chromatin in T regulatory cells. Immunity 2015, 42, 201–203. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, B.; Wen, X.; Hao, D.; Du, D.; He, G.; Jiang, X. The Roles of lncRNA in Cutaneous Squamous Cell Carcinoma. Front. Oncol. 2020, 10, 158. [Google Scholar] [CrossRef]
- Kretz, M. TINCR, staufen1, and cellular differentiation. RNA Biol. 2013, 10, 1597–1601. [Google Scholar] [CrossRef]
- Piipponen, M.; Nissinen, L.; Farshchian, M.; Riihila, P.; Kivisaari, A.; Kallajoki, M.; Peltonen, J.; Peltonen, S.; Kahari, V.M. Long Noncoding RNA PICSAR Promotes Growth of Cutaneous Squamous Cell Carcinoma by Regulating ERK1/2 Activity. J. Investig. Dermatol. 2016, 136, 1701–1710. [Google Scholar] [CrossRef]
- Mei, X.L.; Zhong, S. Long noncoding RNA LINC00520 prevents the progression of cutaneous squamous cell carcinoma through the inactivation of the PI3K/Akt signaling pathway by downregulating EGFR. Chin. Med. J. 2019, 132, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Liao, J.; Duan, X.; He, Y.; Liao, Y. Upregulation of LINC00319 indicates a poor prognosis and promotes cell proliferation and invasion in cutaneous squamous cell carcinoma. J. Cell. Biochem. 2018, 119, 10393–10405. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, Q.; Kuang, S.; Zhao, C.; Yang, L.; Zhang, Y.; Zhu, H.; Yang, R. USF1-induced upregulation of LINC01048 promotes cell proliferation and apoptosis in cutaneous squamous cell carcinoma by binding to TAF15 to transcriptionally activate YAP1. Cell Death Dis. 2019, 10, 296. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Li, C.; Luo, S.; Liu-Smith, F.; Yang, J.; Wang, X.; Wang, N.; Lai, B.; Lei, T.; Wang, Q.; et al. Yes-Associated Protein Contributes to the Development of Human Cutaneous Squamous Cell Carcinoma via Activation of RAS. J. Investig. Dermatol. 2016, 136, 1267–1277. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.H.; Chan, C.W.; Fang, J.Y.; Shih, Y.M.; Liu, Y.W.; Wang, T.V.; Chen, C.Y. 2-O-Methylmagnolol upregulates the long non-coding RNA, GAS5, and enhances apoptosis in skin cancer cells. Cell Death Dis. 2017, 8, e2638. [Google Scholar] [CrossRef]
- Yu, G.J.; Sun, Y.; Zhang, D.W.; Zhang, P. Long non-coding RNA HOTAIR functions as a competitive endogenous RNA to regulate PRAF2 expression by sponging miR-326 in cutaneous squamous cell carcinoma. Cancer Cell Int. 2019, 19, 270. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Croce, C.M. The role of MicroRNAs in human cancer. Signal Transduct. Target. Ther. 2016, 1, 15004. [Google Scholar] [CrossRef] [PubMed]
- García-Sancha, N.; Corchado-Cobos, R.; Pérez-Losada, J.; Cañueto, J. MicroRNA Dysregulation in Cutaneous Squamous Cell Carcinoma. Int. J. Mol. Sci. 2019, 20, 2181. [Google Scholar] [CrossRef]
- Darido, C.; Georgy, S.R.; Wilanowski, T.; Dworkin, S.; Auden, A.; Zhao, Q.; Rank, G.; Srivastava, S.; Finlay, M.J.; Papenfuss, A.T.; et al. Targeting of the tumor suppressor GRHL3 by a miR-21-dependent proto-oncogenic network results in PTEN loss and tumorigenesis. Cancer Cell 2011, 20, 635–648. [Google Scholar] [CrossRef]
- Benaich, N.; Woodhouse, S.; Goldie, S.J.; Mishra, A.; Quist, S.R.; Watt, F.M. Rewiring of an epithelial differentiation factor, miR-203, to inhibit human squamous cell carcinoma metastasis. Cell Rep. 2014, 9, 104–117. [Google Scholar] [CrossRef]
- Yamane, K.; Jinnin, M.; Etoh, T.; Kobayashi, Y.; Shimozono, N.; Fukushima, S.; Masuguchi, S.; Maruo, K.; Inoue, Y.; Ishihara, T.; et al. Down-regulation of miR-124/-214 in cutaneous squamous cell carcinoma mediates abnormal cell proliferation via the induction of ERK. J. Mol. Med. 2013, 91, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Liu, W.; Ma, S.; Cao, H.; Peng, X.; Guo, L.; Zhou, X.; Zheng, L.; Guo, L.; Wan, M.; et al. A novel onco-miR-365 induces cutaneous squamous cell carcinoma. Carcinogenesis 2013, 34, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Wang, Y.; Zhou, M.; Zhang, Y.; Wang, P.; Li, X.; Yang, J.; Wang, H.; Ding, Z. HOXA9 inhibits HIF-1alpha-mediated glycolysis through interacting with CRIP2 to repress cutaneous squamous cell carcinoma development. Nat. Commun. 2018, 9, 1480. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Zhang, L.; Meisgen, F.; Harada, M.; Heilborn, J.; Homey, B.; Grander, D.; Stahle, M.; Sonkoly, E.; Pivarcsi, A. MicroRNA-125b down-regulates matrix metallopeptidase 13 and inhibits cutaneous squamous cell carcinoma cell proliferation, migration, and invasion. J. Biol. Chem. 2012, 287, 29899–29908. [Google Scholar] [CrossRef]
- Olasz, E.B.; Seline, L.N.; Schock, A.M.; Duncan, N.E.; Lopez, A.; Lazar, J.; Flister, M.J.; Lu, Y.; Liu, P.; Sokumbi, O.; et al. MicroRNA-135b Regulates Leucine Zipper Tumor Suppressor 1 in Cutaneous Squamous Cell Carcinoma. PLoS ONE 2015, 10, e0125412. [Google Scholar] [CrossRef]
- Davis, A.J.; Tsinkevich, M.; Rodencal, J.; Abbas, H.A.; Su, X.H.; Gi, Y.J.; Fang, B.; Rajapakshe, K.; Coarfa, C.; Gunaratne, P.H.; et al. TAp63-regulated microRNAs suppress cutaneous squamous cell carcinoma through inhibition of a network of cell cycle genes. Cancer Res. 2020, 80, 2484–2497. [Google Scholar] [CrossRef]
- Chen, I.P.; Henning, S.; Faust, A.; Boukamp, P.; Volkmer, B.; Greinert, R. UVA-induced epigenetic regulation of P16(INK4a) in human epidermal keratinocytes and skin tumor derived cells. Photochem. Photobiol. Sci. Off. J. Eur. Photochem. Assoc. Eur. Soc. Photobiol. 2012, 11, 180–190. [Google Scholar] [CrossRef]
- Yang, H.; Schramek, D.; Adam, R.C.; Keyes, B.E.; Wang, P.; Zheng, D.; Fuchs, E. ETS family transcriptional regulators drive chromatin dynamics and malignancy in squamous cell carcinomas. eLife 2015, 4, e10870. [Google Scholar] [CrossRef]
- Huang, H.; Sabari, B.R.; Garcia, B.A.; Allis, C.D.; Zhao, Y. SnapShot: Histone modifications. Cell 2014, 159, 458. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Klann, T.S.; Black, J.B.; Chellappan, M.; Safi, A.; Song, L.; Hilton, I.B.; Crawford, G.E.; Reddy, T.E.; Gersbach, C.A. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol. 2017, 35, 561–568. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, M.; Kasraian, Z.; Rezvani, H.R. Energy metabolism in skin cancers: A therapeutic perspective. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 712–722. [Google Scholar] [CrossRef]
- Ratnikov, B.I.; Scott, D.A.; Osterman, A.L.; Smith, J.W.; Ronai, Z.A. Metabolic rewiring in melanoma. Oncogene 2017, 36, 147–157. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Hosseini, M.; Dousset, L.; Mahfouf, W.; Serrano-Sanchez, M.; Redonnet-Vernhet, I.; Mesli, S.; Kasraian, Z.; Obre, E.; Bonneu, M.; Claverol, S.; et al. Energy Metabolism Rewiring Precedes UVB-Induced Primary Skin Tumor Formation. Cell Rep. 2018, 23, 3621–3634. [Google Scholar] [CrossRef]
- Flores, A.; Sandoval-Gonzalez, S.; Takahashi, R.; Krall, A.; Sathe, L.; Wei, L.; Radu, C.; Joly, J.H.; Graham, N.A.; Christofk, H.R.; et al. Increased lactate dehydrogenase activity is dispensable in squamous carcinoma cells of origin. Nat. Commun. 2019, 10, 91. [Google Scholar] [CrossRef]
- Goodwin, J.; Neugent, M.L.; Lee, S.Y.; Choe, J.H.; Choi, H.; Jenkins, D.M.R.; Ruthenborg, R.J.; Robinson, M.W.; Jeong, J.Y.; Wake, M.; et al. The distinct metabolic phenotype of lung squamous cell carcinoma defines selective vulnerability to glycolytic inhibition. Nat. Commun. 2017, 8, 15503. [Google Scholar] [CrossRef]
- Zheng, W.; Tayyari, F.; Gowda, G.A.; Raftery, D.; McLamore, E.S.; Porterfield, D.M.; Donkin, S.S.; Bequette, B.; Teegarden, D. Altered glucose metabolism in Harvey-ras transformed MCF10A cells. Mol. Carcinog. 2015, 54, 111–120. [Google Scholar] [CrossRef]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.; Kimmelman, A.C.; Lyssiotis, C.A.; Hua, S.; Chu, G.C.; Fletcher-Sananikone, E.; Locasale, J.W.; Son, J.; Zhang, H.; Coloff, J.L.; et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 2012, 149, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Bantubungi, K.; Hannou, S.A.; Caron-Houde, S.; Vallez, E.; Baron, M.; Lucas, A.; Bouchaert, E.; Paumelle, R.; Tailleux, A.; Staels, B. Cdkn2a/p16Ink4a regulates fasting-induced hepatic gluconeogenesis through the PKA-CREB-PGC1alpha pathway. Diabetes 2014, 63, 3199–3209. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Krishnan, K.J.; Birch-Machin, M.A. The incidence of both tandem duplications and the common deletion in mtDNA from three distinct categories of sun-exposed human skin and in prolonged culture of fibroblasts. J. Investig. Dermatol. 2006, 126, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.M.; Murphy, G.; Ralph, N.; O’Gorman, S.M.; Murphy, J.E. Mitochondrial DNA deletion percentage in sun exposed and non sun exposed skin. J. Photochem. Photobiol. B Biol. 2016, 165, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.J.; Turner, R.; Nikaido, O.; Rees, J.L.; Birch-Machin, M.A. The spectrum of mitochondrial DNA deletions is a ubiquitous marker of ultraviolet radiation exposure in human skin. J. Investig. Dermatol. 2000, 115, 674–679. [Google Scholar] [CrossRef]
- Yusoff, A.A.M.; Abdullah, W.S.W.; Khair, S.; Radzak, S.M.A. A comprehensive overview of mitochondrial DNA 4977-bp deletion in cancer studies. Oncol. Rev. 2019, 13, 409. [Google Scholar] [CrossRef]
- Rezvani, H.R.; Kim, A.L.; Rossignol, R.; Ali, N.; Daly, M.; Mahfouf, W.; Bellance, N.; Taieb, A.; de Verneuil, H.; Mazurier, F.; et al. XPC silencing in normal human keratinocytes triggers metabolic alterations that drive the formation of squamous cell carcinomas. J. Clin. Investig. 2011, 121, 195–211. [Google Scholar] [CrossRef]
- Rezvani, H.R.; Rossignol, R.; Ali, N.; Benard, G.; Tang, X.; Yang, H.S.; Jouary, T.; de Verneuil, H.; Taieb, A.; Kim, A.L.; et al. XPC silencing in normal human keratinocytes triggers metabolic alterations through NOX-1 activation-mediated reactive oxygen species. Biochim. Biophys. Acta 2011, 1807, 609–619. [Google Scholar] [CrossRef]
- Harbottle, A.; Birch-Machin, M.A. Real-time PCR analysis of a 3895 bp mitochondrial DNA deletion in nonmelanoma skin cancer and its use as a quantitative marker for sunlight exposure in human skin. Br. J. Cancer 2006, 94, 1887–1893. [Google Scholar] [CrossRef]
- Hosseini, M.; Dousset, L.; Michon, P.; Mahfouf, W.; Muzotte, E.; Bergeron, V.; Bortolotto, D.; Rossignol, R.; Moisan, F.; Taieb, A.; et al. UVB-induced DHODH upregulation, which is driven by STAT3, is a promising target for chemoprevention and combination therapy of photocarcinogenesis. Oncogenesis 2019, 8, 52. [Google Scholar] [CrossRef] [PubMed]
- Uribe, P.; Gonzalez, S. Epidermal growth factor receptor (EGFR) and squamous cell carcinoma of the skin: Molecular bases for EGFR-targeted therapy. Pathol. Res. Pract. 2011, 207, 337–342. [Google Scholar] [CrossRef] [PubMed]
- William, W.N., Jr.; Feng, L.; Ferrarotto, R.; Ginsberg, L.; Kies, M.; Lippman, S.; Glisson, B.; Kim, E.S. Gefitinib for patients with incurable cutaneous squamous cell carcinoma: A single-arm phase II clinical trial. J. Am. Acad. Dermatol. 2017, 77, 1110–1113.e2. [Google Scholar] [CrossRef] [PubMed]
- Gold, K.A.; Kies, M.S.; William, W.N., Jr.; Johnson, F.M.; Lee, J.J.; Glisson, B.S. Erlotinib in the treatment of recurrent or metastatic cutaneous squamous cell carcinoma: A single-arm phase 2 clinical trial. Cancer 2018, 124, 2169–2173. [Google Scholar] [CrossRef] [PubMed]
- Maubec, E.; Petrow, P.; Scheer-Senyarich, I.; Duvillard, P.; Lacroix, L.; Gelly, J.; Certain, A.; Duval, X.; Crickx, B.; Buffard, V.; et al. Phase II study of cetuximab as first-line single-drug therapy in patients with unresectable squamous cell carcinoma of the skin. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 3419–3426. [Google Scholar] [CrossRef] [PubMed]
- Montaudie, H.; Viotti, J.; Combemale, P.; Dutriaux, C.; Dupin, N.; Robert, C.; Mortier, L.; Kaphan, R.; Duval-Modeste, A.B.; Dalle, S.; et al. Cetuximab is efficient and safe in patients with advanced cutaneous squamous cell carcinoma: A retrospective, multicentre study. Oncotarget 2020, 11, 378–385. [Google Scholar] [CrossRef]
- Foote, M.C.; McGrath, M.; Guminski, A.; Hughes, B.G.; Meakin, J.; Thomson, D.; Zarate, D.; Simpson, F.; Porceddu, S.V. Phase II study of single-agent panitumumab in patients with incurable cutaneous squamous cell carcinoma. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2014, 25, 2047–2052. [Google Scholar] [CrossRef]
- Galbiati, D.; Cavalieri, S.; Alfieri, S.; Resteghini, C.; Bergamini, C.; Orlandi, E.; Platini, F.; Locati, L.; Giacomelli, L.; Licitra, L.; et al. Activity of platinum and cetuximab in cutaneous squamous cell cancer not amenable to curative treatment. Drugs Context 2019, 8, 212611. [Google Scholar] [CrossRef]
- Reigneau, M.; Robert, C.; Routier, E.; Mamelle, G.; Moya-Plana, A.; Tomasic, G.; Mateus, C. Efficacy of neoadjuvant cetuximab alone or with platinum salt for the treatment of unresectable advanced nonmetastatic cutaneous squamous cell carcinomas. Br. J. Dermatol. 2015, 173, 527–534. [Google Scholar] [CrossRef]
- Corchado-Cobos, R.; Garcia-Sancha, N.; Gonzalez-Sarmiento, R.; Perez-Losada, J.; Canueto, J. Cutaneous Squamous Cell Carcinoma: From Biology to Therapy. Int. J. Mol. Sci. 2020, 21, 2956. [Google Scholar] [CrossRef]
- Burova, E.; Hermann, A.; Waite, J.; Potocky, T.; Lai, V.; Hong, S.; Liu, M.; Allbritton, O.; Woodruff, A.; Wu, Q.; et al. Characterization of the Anti-PD-1 Antibody REGN2810 and Its Antitumor Activity in Human PD-1 Knock-In Mice. Mol. Cancer Ther. 2017, 16, 861–870. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. N. Engl. J. Med. 2018, 379, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Migden, M.R.; Khushalani, N.I.; Chang, A.L.S.; Lewis, K.D.; Schmults, C.D.; Hernandez-Aya, L.; Meier, F.; Schadendorf, D.; Guminski, A.; Hauschild, A.; et al. Cemiplimab in locally advanced cutaneous squamous cell carcinoma: Results from an open-label, phase 2, single-arm trial. Lancet. Oncol. 2020, 21, 294–305. [Google Scholar] [CrossRef]
- Grob, J.J.; Gonzalez, R.; Basset-Seguin, N.; Vornicova, O.; Schachter, J.; Joshi, A.; Meyer, N.; Grange, F.; Piulats, J.M.; Bauman, J.R.; et al. Pembrolizumab Monotherapy for Recurrent or Metastatic Cutaneous Squamous Cell Carcinoma: A Single-Arm Phase II Trial (KEYNOTE-629). J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 2916–2925. [Google Scholar] [CrossRef]
- Maubec, E.; Boubaya, M.; Petrow, P.; Beylot-Barry, M.; Basset-Seguin, N.; Deschamps, L.; Grob, J.J.; Dreno, B.; Scheer-Senyarich, I.; Bloch-Queyrat, C.; et al. Phase II Study of Pembrolizumab As First-Line, Single-Drug Therapy for Patients With Unresectable Cutaneous Squamous Cell Carcinomas. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 3051–3061. [Google Scholar] [CrossRef]
- Keeping, S.; Xu, Y.; Chen, C.I.; Cope, S.; Mojebi, A.; Kuznik, A.; Konidaris, G.; Ayers, D.; Sasane, M.; Allen, R.; et al. Comparative efficacy of cemiplimab versus other systemic treatments for advanced cutaneous squamous cell carcinoma. Future Oncol. 2021, 17, 611–627. [Google Scholar] [CrossRef]
- Chen, A.; Ali, N.; Boasberg, P.; Ho, A.S. Clinical Remission of Cutaneous Squamous Cell Carcinoma of the Auricle with Cetuximab and Nivolumab. J. Clin. Med. 2018, 7, 10. [Google Scholar] [CrossRef]
- Sernicola, A.; Lampitelli, S.; Marraffa, F.; Maddalena, P.; Grassi, S.; Richetta, A.G.; Calvieri, S. Case Report: Cetuximab use in advanced cutaneous squamous cell carcinoma resistant to chemotherapy. F1000Research 2019, 8, 933. [Google Scholar] [CrossRef]
- Yang, X.; Daifallah, A.E.M.; Shankar, S.; Beer, J.; Marshall, C.; Dentchev, T.; Seykora, F.; D’Armas, S.; Hahn, J.; Lee, V.; et al. Topical kinase inhibitors induce regression of cutaneous squamous cell carcinoma. Exp. Dermatol. 2019, 28, 609–613. [Google Scholar] [CrossRef]
- Naing, A.; Cohen, R.; Dy, G.K.; Hong, D.S.; Dyster, L.; Hangauer, D.G.; Kwan, R.; Fetterly, G.; Kurzrock, R.; Adjei, A.A. A phase I trial of KX2-391, a novel non-ATP competitive substrate-pocket- directed SRC inhibitor, in patients with advanced malignancies. Investig. New Drugs 2013, 31, 967–973. [Google Scholar] [CrossRef]
- Olivieri, A.; Manzione, L. Dasatinib: A new step in molecular target therapy. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2007, 18 (Suppl. 6), vi42–vi46. [Google Scholar] [CrossRef] [PubMed]
- Smolinski, M.P.; Bu, Y.; Clements, J.; Gelman, I.H.; Hegab, T.; Cutler, D.L.; Fang, J.W.S.; Fetterly, G.; Kwan, R.; Barnett, A.; et al. Discovery of Novel Dual Mechanism of Action Src Signaling and Tubulin Polymerization Inhibitors (KX2-391 and KX2-361). J. Med. Chem. 2018, 61, 4704–4719. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Kempers, S.; Lain, E.; Schlesinger, T.; Tyring, S.; Forman, S.; Ablon, G.; Martin, G.; Wang, H.; Cutler, D.L.; et al. Phase 3 Trials of Tirbanibulin Ointment for Actinic Keratosis. N. Engl. J. Med. 2021, 384, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Yanai, H.; Kaji, T.; Kubo, T.; Ennishi, D.; Hirasawa, A.; Yoshino, T. Secretory carcinoma of the skin with lymph node metastases and recurrence in both lungs: A case report. J. Cutan. Pathol. 2021, 48, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.; Kang, H.; Mehra, R. Targeting phosphoinositide 3-kinase (PI3K) in head and neck squamous cell carcinoma (HNSCC). Cancers Head Neck 2018, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Darido, C.; Georgy, S.R.; Cullinane, C.; Partridge, D.D.; Walker, R.; Srivastava, S.; Roslan, S.; Carpinelli, M.R.; Dworkin, S.; Pearson, R.B.; et al. Stage-dependent therapeutic efficacy in PI3K/mTOR-driven squamous cell carcinoma of the skin. Cell Death Differ. 2018, 25, 1146–1159. [Google Scholar] [CrossRef]
- Zou, Y.; Ge, M.; Wang, X. Targeting PI3K-AKT-mTOR by LY3023414 inhibits human skin squamous cell carcinoma cell growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2017, 490, 385–392. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef]
- Chung, C. Current targeted therapies in lymphomas. Am. J. Health Syst. Pharm. 2019, 76, 1825–1834. [Google Scholar] [CrossRef] [PubMed]
- Kurundkar, D.; Srivastava, R.K.; Chaudhary, S.C.; Ballestas, M.E.; Kopelovich, L.; Elmets, C.A.; Athar, M. Vorinostat, an HDAC inhibitor attenuates epidermoid squamous cell carcinoma growth by dampening mTOR signaling pathway in a human xenograft murine model. Toxicol. Appl. Pharmacol. 2013, 266, 233–244. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Torres-Lockhart, K.; Forster, N.; Ramakrishnan, S.; Greninger, P.; Garnett, M.J.; McDermott, U.; Rothenberg, S.M.; Benes, C.H.; Ellisen, L.W. Mcl-1 and FBW7 control a dominant survival pathway underlying HDAC and Bcl-2 inhibitor synergy in squamous cell carcinoma. Cancer Discov. 2013, 3, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Slaughter, D.P.; Southwick, H.W.; Smejkal, W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer 1953, 6, 963–968. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).