
Modeling PCDH19-CE: From 2D Stem Cell Model to 3D Brain Organoids
 ,
,  , , , and
, , , and
Abstract
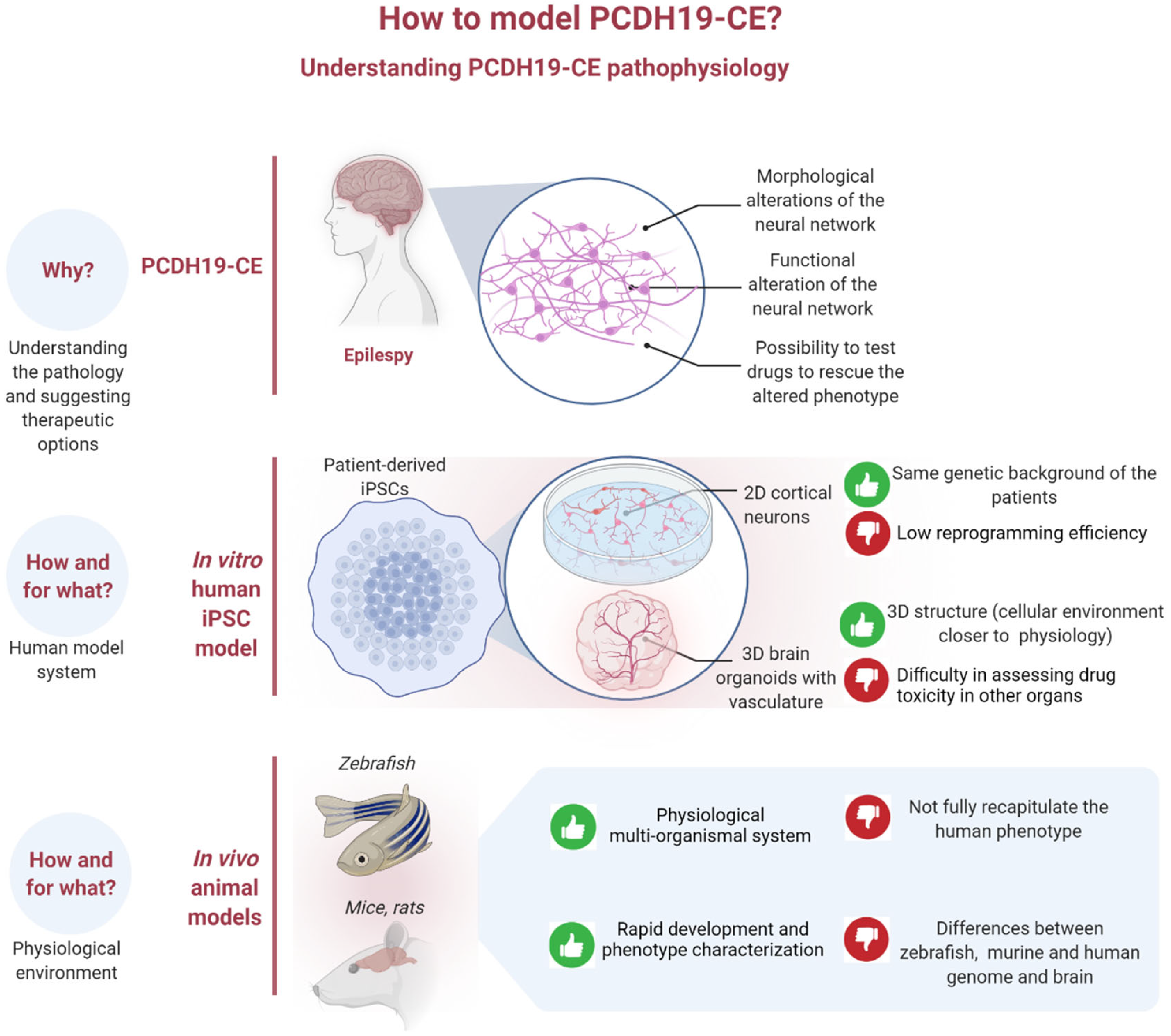
:1. Introduction
2. PCDH19-CE: Genetics
3. PCDH19 in Neurodevelopment
4. iPSCs as a Model System for Studying PCDH19-CE
5. From iPSCs to Brain Organoids
6. Blood-Brain Barrier and Brain Organoids
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dibbens, L.M.; Tarpey, P.S.; Hynes, K.; Bayly, M.A.; Scheffer, I.E.; Smith, R.; Bomar, J.; Sutton, E.; Vandeleur, L.; Shoubridge, C.; et al. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat. Genet. 2008, 40, 776–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juberg, R.C.; Hellman, C.D. A new familial form of convulsive disorder and mental retardation limited to females. J. Pediatr. 1971, 79, 726–732. [Google Scholar] [CrossRef]
- Depienne, C.; Bouteiller, D.; Keren, B.; Cheuret, E.; Poirier, K.; Trouillard, O.; Benyahia, B.; Quelin, C.; Carpentier, W.; Julia, S.; et al. Sporadic Infantile Epileptic Encephalopathy Caused by Mutations in PCDH19 Resembles Dravet Syndrome but Mainly Affects Females. PLoS Genet. 2009, 5, e1000381. [Google Scholar] [CrossRef]
- Duszyc, K.; Terczyńska, I.; Hoffman-Zacharska, D. Epilepsy and mental retardation restricted to females: X-linked epileptic infantile encephalopathy of unusual inheritance. J. Appl. Genet. 2015, 56, 49–56. [Google Scholar] [CrossRef]
- Trivisano, M.; Pietrafusa, N.; Terracciano, A.; Marini, C.; Mei, D.; Darra, F.; Accorsi, P.; Battaglia, D.; Caffi, L.; Canevini, M.P.; et al. Defining the electroclinical phenotype and outcome of PCDH19-related epilepsy: A multicenter study. Epilepsia 2018, 59, 2260–2271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheffer, I.E.; Turner, S.J.; Dibbens, L.; Bayly, M.; Friend, K.; Hodgson, B.; Burrows, L.; Shaw, M.; Wei, C.; Ullmann, R.; et al. Epilepsy and mental retardation limited to females: An under-recognized disorder. Brain 2008, 131 Pt 4, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Marini, C.; Mei, D.; Parmeggiani, L.; Norci, V.; Calado, E.; Ferrari, A.; Moreira, A.; Pisano, T.; Specchio, N.; Vigevano, F.; et al. Protocadherin 19 mutations in girls with infantile-onset epilepsy. Neurology 2010, 75, 646–653. [Google Scholar] [CrossRef]
- Specchio, N.; Marini, C.; Terracciano, A.; Mei, D.; Trivisano, M.; Sicca, F.; Fusco, L.; Cusmai, R.; Darra, F.; Bernardina, B.D.; et al. Spectrum of phenotypes in female patients with epilepsy due to protocadherin 19 mutations. Epilepsia 2011, 52, 1251–1257. [Google Scholar] [CrossRef]
- Piton, A.; Gauthier, J.; Hamdan, F.F.; Lafrenière, R.G.; Yang, Y.; Henrion, E.; Laurent, S.; Noreau, A.; Thibodeau, P.; Karemera, L.; et al. Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol. Psychiatry 2011, 16, 867–880. [Google Scholar] [CrossRef]
- Breuillard, D.; Leunen, D.; Chemaly, N.; Auclair, L.; Pinard, J.M.; Kaminska, A.; Desguerre, I.; Ouss, L.; Nabbout, R. Autism spectrum disorder phenotype and intellectual disability in females with epilepsy and PCDH-19 mutations. Epilepsy Behav. 2016, 60, 75–80. [Google Scholar] [CrossRef]
- Smith, L.; Singhal, N.; El Achkar, C.M.; Truglio, G.; Sheidley, B.R.; Sullivan, J.; Poduri, A. PCDH19 -related epilepsy is associated with a broad neurodevelopmental spectrum. Epilepsia 2018, 59, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlaskamp, D.R.M.; Bassett, A.S.; Sullivan, J.E.; Robblee, J.; Sadleir, L.G.; Scheffer, I.E.; Andrade, D.M. Schizophrenia is a later-onset feature of PCDH19 Girls Clustering Epilepsy. Epilepsia 2019, 60, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Depienne, C.; LeGuern, E. PCDH19-related infantile epileptic encephalopathy: An unusual X-linked inheritance disorder. Hum. Mutat. 2012, 33, 627–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depienne, C.; Trouillard, O.; Bouteiller, D.; Gourfinkel-An, I.; Poirier, K.; Rivier, F.; Berquin, P.; Nabbout, R.; Chaigne, D.; Steschenko, D.; et al. Mutations and deletions in PCDH19 account for various familial or isolated epilepsies in females. Hum. Mutat. 2011, 32, E1959–E1975. [Google Scholar] [CrossRef] [Green Version]
- Lotte, J.; Bast, T.; Borusiak, P.; Coppola, A.; Cross, H.; Dimova, P.; Fogarasi, A.; Graneß, I.; Guerrini, R.; Hjalgrim, H.; et al. Effectiveness of antiepileptic therapy in patients with PCDH19 mutations. Seizure 2016, 35, 106–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pederick, D.; Homan, C.; Jaehne, E.; Piltz, S.G.; Haines, B.P.; Baune, B.T.; Jolly, L.; Hughes, J.N.; Gecz, J.; Thomas, P.Q. Pcdh19 Loss-of-Function Increases Neuronal Migration In Vitro but is Dispensable for Brain Development in Mice. Sci. Rep. 2016, 6, 26765. [Google Scholar] [CrossRef]
- Pederick, D.T.; Richards, K.L.; Piltz, S.G.; Kumar, R.; Mincheva-Tasheva, S.; Mandelstam, S.A.; Dale, R.C.; Scheffer, I.E.; Gecz, J.; Petrou, S.; et al. Abnormal Cell Sorting Underlies the Unique X-Linked Inheritance of PCDH19 Epilepsy. Neuron 2018, 97, 59–66.e5. [Google Scholar] [CrossRef] [Green Version]
- Kurian, M.; Korff, C.M.; Ranza, E.; Bernasconi, A.; Lübbig, A.; Nangia, S.; Ramelli, G.P.; Wohlrab, G.; Nordli, D.R.; Bast, T. Focal cortical malformations in children with early infantile epilepsy and PCDH19 mutations: Case report. Dev. Med. Child Neurol. 2018, 60, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Lenge, M.; Marini, C.; Canale, E.; Napolitano, A.; De Masi, S.; Trivisano, M.; Mei, D.; Longo, D.; Rossi Espagnet, M.C.; Lucenteforte, E.; et al. Quantitative MRI-Based Analysis Identifies Developmental Limbic Abnormalities in PCDH19 Encephalopathy. Cereb. Cortex 2020, 30, 6039–6050. [Google Scholar] [CrossRef]
- Fujitani, M.; Zhang, S.; Fujiki, R.; Fujihara, Y.; Yamashita, T. A chromosome 16p13.11 microduplication causes hyperactivity through dysregulation of miR-484/protocadherin-19 signaling. Mol. Psychiatry 2016, 22, 364–374. [Google Scholar] [CrossRef]
- Hayashi, S.; Inoue, Y.; Hattori, S.; Kaneko, M.; Shioi, G.; Miyakawa, T.; Takeichi, M. Loss of X-linked Protocadherin-19 differentially affects the behavior of heterozygous female and hemizygous male mice. Sci. Rep. 2017, 7, 5801. [Google Scholar] [CrossRef] [PubMed]
- Homan, C.; Pederson, S.; To, T.-H.; Tan, C.; Piltz, S.; Corbett, M.A.; Wolvetang, E.; Thomas, P.Q.; Jolly, L.A.; Gecz, J. PCDH19 regulation of neural progenitor cell differentiation suggests asynchrony of neurogenesis as a mechanism contributing to PCDH19 Girls Clustering Epilepsy. Neurobiol. Dis. 2018, 116, 106–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassani, S.; Cwetsch, A.; Gerosa, L.; Serratto, G.M.; Folci, A.; Hall, I.F.; Mazzanti, M.; Cancedda, L.; Passafaro, M. The female epilepsy protein PCDH19 is a new GABAAR-binding partner that regulates GABAergic transmission as well as migration and morphological maturation of hippocampal neurons. Hum. Mol. Genet. 2018, 27, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.; Guo, M.; Martins-Taylor, K.; Wang, X.; Zhang, Z.; Park, J.W.; Zhan, S.; Kronenberg, M.S.; Lichtler, A.; Liu, H.-X.; et al. Specification of Region-Specific Neurons Including Forebrain Glutamatergic Neurons from Human Induced Pluripotent Stem Cells. PLoS ONE 2010, 5, e11853. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [Green Version]
- Kolc, K.L.; Sadleir, L.G.; Scheffer, I.; Ivancevic, A.; Roberts, R.; Pham, D.; Gecz, J. A systematic review and meta-analysis of 271 PCDH19-variant individuals identifies psychiatric comorbidities, and association of seizure onset and disease severity. Mol. Psychiatry 2019, 24, 241–251. [Google Scholar] [CrossRef]
- Van Harssel, J.J.T.; Weckhuysen, S.; Van Kempen, M.J.A.; Hardies, K.; Verbeek, N.E.; de Kovel, C.; Gunning, W.B.; Van Daalen, E.; De Jonge, M.V.; Jansen, A.; et al. Clinical and genetic aspects of PCDH19-related epilepsy syndromes and the possible role of PCDH19 mutations in males with autism spectrum disorders. Neurogenetics 2013, 14, 23–34. [Google Scholar] [CrossRef]
- Johnson, W.G. Metabolic interference and the +/- heterozygote. A hypothetical form of simple inheritance which is neither dominant nor recessive. Am. J. Hum. Genet. 1980, 32, 374–386. [Google Scholar]
- Wieland, I.; Jakubiczka, S.; Muschke, P.; Cohen, M.; Thiele, H.; Gerlach, K.L.; Adams, R.H.; Wieacker, P. Mutations of the Ephrin-B1 Gene Cause Craniofrontonasal Syndrome. Am. J. Hum. Genet. 2004, 74, 1209–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thiffault, I.; Farrow, E.; Smith, L.; Lowry, J.; Zellmer, L.; Black, B.; Abdelmoity, A.; Miller, N.; Soden, S.; Saunders, C. PCDH19-related epileptic encephalopathy in a male mosaic for a truncating variant. Am. J. Med. Genet. Part A 2016, 170, 1585–1589. [Google Scholar] [CrossRef] [PubMed]
- Terracciano, A.; Trivisano, M.; Cusmai, R.; De Palma, L.; Fusco, L.; Compagnucci, C.; Bertini, E.; Vigevano, F.; Specchio, N. PCDH19-related epilepsy in two mosaic male patients. Epilepsia 2016, 57, e51–e55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Lange, I.M.; Rump, P.; Neuteboom, R.F.; Augustijn, P.B.; Hodges, K.; Kistemaker, A.I.; Brouwer, O.F.; Mancini, G.M.S.; Newman, H.A.; Vos, Y.J.; et al. Male patients affected by mosaic PCDH19 mutations: Five new cases. Neurogenetics 2017, 18, 147–153. [Google Scholar] [CrossRef]
- Romasko, E.J.; DeChene, E.T.; Balciuniene, J.; Akgumus, G.T.; Helbig, I.; Tarpinian, J.M.; Keena, B.A.; Vogiatzi, M.G.; Zackai, E.H.; Izumi, K.; et al. PCDH19 -related epilepsy in a male with Klinefelter syndrome: Additional evidence supporting PCDH19 cellular interference disease mechanism. Epilepsy Res. 2018, 145, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Niazi, R.; Fanning, E.A.; Depienne, C.; Sarmady, M.; Tayoun, A.N.A. A mutation update for the PCDH19 gene causing early-onset epilepsy in females with an unusual expression pattern. Hum. Mutat. 2019, 40, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Wolverton, T.; Lalande, M. Identification and Characterization of Three Members of a Novel Subclass of Protocadherins. Genomics 2001, 76, 66–72. [Google Scholar] [CrossRef]
- Redies, C.; Vanhalst, K.; Van Roy, F. Delta-Protocadherins: Unique structures and functions. Cell Mol. Life Sci. 2005, 62, 2840–2852. [Google Scholar] [CrossRef]
- Hirano, S.; Takeichi, M. Cadherins in Brain Morphogenesis and Wiring. Physiol. Rev. 2012, 92, 597–634. [Google Scholar] [CrossRef]
- Hayashi, S.; Inoue, Y.; Kiyonari, H.; Abe, T.; Misaki, K.; Moriguchi, H.; Tanaka, Y.; Takeichi, M. Protocadherin-17 Mediates Collective Axon Extension by Recruiting Actin Regulator Complexes to Interaxonal Contacts. Dev. Cell 2014, 30, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Emond, M.R.; Duy, P.Q.; Hao, L.T.; Beattie, C.E.; Jontes, J.D. Protocadherin-18b interacts with Nap1 to control motor axon growth and arborization in zebrafish. Mol. Biol. Cell 2014, 25, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Tsai, N.-P.; Wilkerson, J.; Guo, W.; Maksimova, M.A.; DeMartino, G.N.; Cowan, C.W.; Huber, K.M. Multiple Autism-Linked Genes Mediate Synapse Elimination via Proteasomal Degradation of a Synaptic Scaffold PSD-95. Cell 2012, 151, 1581–1594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshina, N.; Tanimura, A.; Yamasaki, M.; Inoue, T.; Fukabori, R.; Kuroda, T.; Yokoyama, K.; Tezuka, T.; Sagara, H.; Hirano, S.; et al. Protocadherin 17 Regulates Presynaptic Assembly in Topographic Corticobasal Ganglia Circuits. Neuron 2013, 78, 839–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, H.; Hoshina, N.; Zhang, C.; Mattheisen, M.; Cao, H.; Wang, Y.; Wu, D.-D.; Bergen, S.; Landén, M.; Hultman, C.M.; et al. The protocadherin 17 gene affects cognition, personality, amygdala structure and function, synapse development and risk of major mood disorders. Mol. Psychiatry 2018, 23, 400–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pancho, A.; Aerts, T.; Mitsogiannis, M.; Seuntjens, E. Protocadherins at the Crossroad of Signaling Pathways. Front. Mol. Neurosci. 2020, 30, 117. [Google Scholar] [CrossRef] [PubMed]
- Cooper, S.R.; Emond, M.R.; Duy, P.Q.; Liebau, B.G.; Wolman, M.A.; Jontes, J.D. Protocadherins control the modular assembly of neuronal columns in the zebrafish optic tectum. J. Cell Biol. 2015, 211, 807–814. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Emond, M.R.; Jontes, J.D. Protocadherin-19 and N-cadherin interact to control cell movements during anterior neurulation. J. Cell Biol. 2010, 191, 1029–1041. [Google Scholar] [CrossRef] [Green Version]
- Emond, M.R.; Biswas, S.; Blevins, C.J.; Jontes, J.D. A complex of Protocadherin-19 and N-cadherin mediates a novel mechanism of cell adhesion. J. Cell Biol. 2011, 195, 1115–1121. [Google Scholar] [CrossRef] [Green Version]
- Emond, M.R.; Biswas, S.; Jontes, J.D. Protocadherin-19 is essential for early steps in brain morphogenesis. Dev. Biol. 2009, 334, 72–83. [Google Scholar] [CrossRef] [Green Version]
- Hoshina, N.; Johnson-Venkatesh, E.M.; Hoshina, M.; Umemori, H. Female-specific synaptic dysfunction and cognitive impairment in a mouse model of PCDH19 disorder. Science 2021, 372, eaaz3893. [Google Scholar] [CrossRef]
- Lv, X.; Ren, S.-Q.; Zhang, X.-J.; Shen, Z.; Ghosh, T.; Xianyu, A.; Gao, P.; Li, Z.; Lin, S.; Yu, Y.; et al. TBR2 coordinates neurogenesis expansion and precise microcircuit organization via Protocadherin 19 in the mammalian cortex. Nat. Commun. 2019, 10, 3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, S.G.; Chance, P.F.; Zou, C.-H.; Spinner, N.B.; Golden, J.A.; Smietana, S. Epilepsy and mental retardation limited to females: An X-linked dominant disorder with male sparing. Nat. Genet. 1997, 17, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Higurashi, N.; Nakamura, M.; Sugai, M.; Ohfu, M.; Sakauchi, M.; Sugawara, Y.; Nakamura, K.; Kato, M.; Usui, D.; Mogami, Y.; et al. PCDH19-related female-limited epilepsy: Further details regarding early clinical features and therapeutic efficacy. Epilepsy Res. 2013, 106, 191–199. [Google Scholar] [CrossRef]
- Lim, J.; Ryu, J.; Kang, S.; Noh, H.J.; Kim, C.H. Autism-like behaviors in male mice with a Pcdh19 deletion. Mol. Brain 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Mincheva-Tasheva, S.; Nieto Guil, A.F.; Homan, C.C.; Gecz, J.; Thomas, P.Q. Disrupted Excitatory Synaptic Contacts and Altered Neuronal Network Activity Underpins the Neurological Phenotype in PCDH19-Clustering Epilepsy (PCDH19-CE). Mol. Neurobiol. 2021, 58, 2005–2018. [Google Scholar] [CrossRef] [PubMed]
- Auvin, S.; Pineda, E.; Shin, D.; Gressens, P.; Mazarati, A. Novel Animal Models of Pediatric Epilepsy. Neurotherapeutics 2012, 9, 245–261. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Parent, J.M. Using Patient-Derived Induced Pluripotent Stem Cells to Model and Treat Epilepsies. Curr. Neurol. Neurosci. Rep. 2015, 15, 71. [Google Scholar] [CrossRef] [Green Version]
- Gagliano, O.; Luni, C.; Qin, W.; Bertin, E.; Torchio, E.; Galvanin, S.; Urciuolo, A.; Elvassore, N. Microfluidic reprogramming to pluripotency of human somatic cells. Nat. Protoc. 2019, 14, 722–737. [Google Scholar] [CrossRef]
- Borghi, R.; Magliocca, V.; Petrini, S.; Conti, L.; Moreno, S.; Bertini, E.; Tartaglia, M.; Compagnucci, C. Dissecting the Role of PCDH19 in Clustering Epilepsy by Exploiting Patient-Specific Models of Neurogenesis. J. Clin. Med. 2021, 10, 2754. [Google Scholar] [CrossRef]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef]
- Compagnucci, C.; Petrini, S.; Higuraschi, N.; Trivisano, M.; Specchio, N.; Hirose, S.; Bertini, E.; Terracciano, A. Characterizing PCDH19 in human induced pluripotent stem cells (iPSCs) and iPSC-derived developing neurons: Emerging role of a protein involved in controlling polarity during neurogenesis. Oncotarget 2015, 6, 26804–26813. [Google Scholar] [CrossRef] [PubMed]
- Compagnucci, C.; Nizzardo, M.; Corti, S.; Zanni, G.; Bertini, E. In vitro neurogenesis: Development and functional implications of iPSC technology. Cell Mol Life Sci. 2014, 71, 1623–1639. [Google Scholar] [CrossRef] [PubMed]
- Emond, M.R.; Biswas, S.; Morrow, M.L.; Jontes, J.D. Proximity-dependent Proteomics Reveals Extensive Interactions of Protocadherin-19 with Regulators of Rho GTPases and the Microtubule Cytoskeleton. Neuroscience 2021, 452, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Eiraku, M.; Watanabe, K.; Matsuo-Takasaki, M.; Kawada, M.; Yonemura, S.; Matsumura, M.; Wataya, T.; Nishiyama, A.; Muguruma, K.; Sasai, Y. Self-Organized Formation of Polarized Cortical Tissues from ESCs and Its Active Manipulation by Extrinsic Signals. Cell Stem Cell 2008, 3, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Mariani, J.; Simonini, M.V.; Palejev, D.; Tomasini, L.; Coppola, G.; Szekely, A.M.; Horvath, T.L.; Vaccarino, F.M. Modeling human cortical development in vitro using induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12770–12775. [Google Scholar] [CrossRef] [Green Version]
- Sasai, Y. Next-Generation Regenerative Medicine: Organogenesis from Stem Cells in 3D Culture. Cell Stem Cell 2013, 12, 520–530. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Kirwan, P.; Smith, J.; Robinson, H.P.C.; Livesey, F.J. Human cerebral cortex development from pluripotent stem cells to functional excitatory synapses. Nat. Neurosci. 2012, 15, 477–486. [Google Scholar] [CrossRef] [Green Version]
- Otani, T.; Marchetto, M.C.; Gage, F.H.; Simons, B.; Livesey, F.J. 2D and 3D Stem Cell Models of Primate Cortical Development Identify Species-Specific Differences in Progenitor Behavior Contributing to Brain Size. Cell Stem Cell 2016, 18, 467–480. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, H.; Ozaki, Y.; Ashida, T.; Matsubara, T.; Oishi, N.; Kihara, S.; Takahashi, J. Self-Organized Synchronous Calcium Transients in a Cultured Human Neural Network Derived from Cerebral Organoids. Stem Cell Rep. 2019, 13, 458–473. [Google Scholar] [CrossRef] [Green Version]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [Green Version]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Min Yang, S.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Buth, J.E.; Vishlaghi, N.; de la Torre-Ubieta, L.; Taxidis, J.; Khakh, B.S.; Coppola, G.; Pearson, C.A.; Yamauchi, K.; Gong, D.; et al. Self-Organized Cerebral Organoids with Human-Specific Features Predict Effective Drugs to Combat Zika Virus Infection. Cell Rep. 2017, 21, 517–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giandomenico, S.L.; Mierau, S.B.; Gibbons, G.M.; Wenger, L.M.D.; Masullo, L.; Sit, T.; Sutcliffe, M.; Boulanger, J.; Tripodi, M.; Derivery, E.; et al. Cerebral organoids at the air–liquid interface generate diverse nerve tracts with functional output. Nat. Neurosci. 2019, 22, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, C.A.; Gao, R.; Negraes, P.D.; Gu, J.; Buchanan, J.; Preissl, S.; Wang, A.; Wu, W.; Haddad, G.G.; Chaim, I.A.; et al. Complex Oscillatory Waves Emerging from Cortical Organoids Model Early Human Brain Network Development. Cell Stem Cell 2019, 25, 558–569.e7. [Google Scholar] [CrossRef]
- Fair, S.R.; Julian, D.; Hartlaub, A.M.; Pusuluri, S.T.; Malik, G.; Summerfied, T.L.; Zhao, G.; Hester, A.B.; Ackerman, W.E.; Hollingsworth, E.W.; et al. Electrophysiological Maturation of Cerebral Organoids Correlates with Dynamic Morphological and Cellular Development. Stem Cell Rep. 2020, 15, 855–868. [Google Scholar] [CrossRef]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Mariani, J.; Coppola, G.; Zhang, P.; Abyzov, A.; Provini, L.; Tomasini, L.; Amenduni, M.; Szekely, A.; Palejev, D.; Wilson, M.; et al. FOXG1-Dependent Dysregulation of GABA/Glutamate Neuron Differentiation in Autism Spectrum Disorders. Cell 2015, 162, 375–390. [Google Scholar] [CrossRef] [Green Version]
- Iefremova, V.; Manikakis, G.; Krefft, O.; Jabali, A.; Weynans, K.; Wilkens, R.; Marsoner, F.; Brändl, B.; Müller, F.-J.; Koch, P.; et al. An Organoid-Based Model of Cortical Development Identifies Non-Cell-Autonomous Defects in Wnt Signaling Contributing to Miller-Dieker Syndrome. Cell Rep. 2017, 19, 50–59. [Google Scholar] [CrossRef] [Green Version]
- Bershteyn, M.; Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Nene, A.; Wynshaw-Boris, A.; Kriegstein, A.R. Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia. Cell Stem Cell 2017, 20, 435–449.e4. [Google Scholar] [CrossRef] [Green Version]
- Mellios, N.; Feldman, D.A.; Sheridan, S.D.; Ip, J.P.K.; Kwok, S.; Amoah, S.K.; Rosen, B.; Rodriguez, B.A.; Crawford, B.; Swaminathan, R.; et al. MeCP2-regulated miRNAs control early human neurogenesis through differential effects on ERK and AKT signaling. Mol. Psychiatry 2018, 23, 1051–1065. [Google Scholar] [CrossRef] [Green Version]
- Xu, R.; Brawner, A.T.; Li, S.; Liu, J.-J.; Kim, H.; Xue, H.; Pang, Z.P.; Kim, W.-Y.; Hart, R.P.; Liu, Y.; et al. OLIG2 Drives Abnormal Neurodevelopmental Phenotypes in Human iPSC-Based Organoid and Chimeric Mouse Models of Down Syndrome. Cell Stem Cell 2019, 24, 908–926.e8. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Muffat, J.; Omer, A.; Bosch, I.; Lancaster, M.; Sur, M.; Gehrke, L.; Knoblich, J.; Jaenisch, R. Induction of Expansion and Folding in Human Cerebral Organoids. Cell Stem Cell 2017, 20, 385–396.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Ma, L.; Yang, M.; Shao, Q.; Xu, J.; Lu, Z.; Zhao, Z.; Chen, R.; Chai, Y.; Chen, J.-F. Cerebral organoid and mouse models reveal a RAB39b–PI3K–mTOR pathway-dependent dysregulation of cortical development leading to macrocephaly/autism phenotypes. Genes Dev. 2020, 34, 580–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Sun, L.; Fang, A.; Li, P.; Wu, Q.; Wang, X. Recapitulating cortical development with organoid culture in vitro and modeling abnormal spindle-like (ASPM related primary) microcephaly disease. Protein Cell 2017, 8, 823–833. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, S.-L.; Yang, M.; Herrlinger, S.; Shao, Q.; Collar, J.L.; Fierro, E.; Shi, Y.; Liu, A.; Lu, H.; et al. Modeling microcephaly with cerebral organoids reveals a WDR62–CEP170–KIF2A pathway promoting cilium disassembly in neural progenitors. Nat. Commun. 2019, 10, 2612. [Google Scholar] [CrossRef] [Green Version]
- Fish, J.L.; Kosodo, Y.; Enard, W.; Pääbo, S.; Huttner, W.B. Aspm specifically maintains symmetric proliferative divisions of neuroepithelial cells. Proc. Natl. Acad. Sci. USA 2006, 103, 10438–10443. [Google Scholar] [CrossRef] [Green Version]
- Fong, K.-W.; Choi, Y.-K.; Rattner, J.B.; Qi, R.Z. CDK5RAP2 Is a Pericentriolar Protein That Functions in Centrosomal Attachment of the γ-Tubulin Ring Complex. Mol. Biol. Cell 2008, 19, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Lippmann, E.S.; Azarin, S.M.; Kay, J.E.; Nessler, R.A.; Wilson, H.K.; Al-Ahmad, A.; Palecek, S.P.; Shusta, E.V. Derivation of blood-brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 2012, 30, 783–791. [Google Scholar] [CrossRef]
- Wang, Y.I.; Abaci, H.E.; Shuler, M.L. Microfluidic blood–brain barrier model provides in vivo-like barrier properties for drug permeability screening. Biotechnol. Bioeng. 2017, 114, 184–194. [Google Scholar] [CrossRef]
- Ribecco-Lutkiewicz, M.; Sodja, C.; Haukenfrers, J.; Haqqani, A.S.; Ly, D.; Zachar, P.; Baumann, E.; Ball, M.; Huang, J.; Rukhlova, M.; et al. A novel human induced pluripotent stem cell blood-brain barrier model: Applicability to study antibody-triggered receptor-mediated transcytosis. Sci. Rep. 2018, 8, 1873. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muldoon, L.L.; Alvarez, J.I.; Begley, D.J.; Boado, R.J.; Del Zoppo, G.J.; Doolittle, N.D.; Engelhardt, B.; Hallenbeck, J.M.; Lonser, R.R.; Ohlfest, J.R.; et al. Immunologic Privilege in the Central Nervous System and the Blood–Brain Barrier. J. Cereb. Blood Flow Metab. 2013, 33, 13–21. [Google Scholar] [CrossRef] [Green Version]
- Löscher, W.; Potschka, H. Role of multidrug transporters in pharmacoresistance to antiepileptic drugs. J. Pharmacol. Exp. Ther. 2002, 301, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarowski, A.; Czornyj, L.; Lubienieki, F.; Girardi, E.; Vazquez, S.; D’Giano, C. ABC Transporters during Epilepsy and Mechanisms Underlying Multidrug Resistance in Refractory Epilepsy. Epilepsia 2007, 48 (Suppl. S5), 140–149. [Google Scholar] [CrossRef] [PubMed]
- Leandro, K.; Bicker, J.; Alves, G.; Falcão, A.; Fortuna, A. ABC transporters in drug-resistant epilepsy: Mechanisms of upregulation and therapeutic approaches. Pharmacol. Res. 2019, 144, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, S.; Lawler, S.E.; Qu, Y.; Fadzen, C.M.; Wolfe, J.M.; Regan, M.S.; Pentelute, B.L.; Agar, N.Y.R.; Cho, C.-F. Blood–brain-barrier organoids for investigating the permeability of CNS therapeutics. Nat. Protoc. 2018, 13, 2827–2843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Research Council Canada. Available online: https://nrc.canada.ca/en/stories/novel-blood-brain-barrier-model-opens-door-advances-medical-biopharmaceutical-research (accessed on 7 March 2019).
- Ham, O.; Jin, Y.B.; Kim, J.; Lee, M.-O. Blood vessel formation in cerebral organoids formed from human embryonic stem cells. Biochem. Biophys. Res. Commun. 2020, 521, 84–90. [Google Scholar] [CrossRef]
- Cakir, B.; Xiang, Y.; Tanaka, Y.; Kural, M.H.; Parent, M.; Kang, Y.-J.; Chapeton, K.; Patterson, B.; Yuan, Y.; He, C.-S.; et al. Engineering of human brain organoids with a functional vascular-like system. Nat. Methods 2019, 16, 1169–1175. [Google Scholar] [CrossRef]
- Ahn, Y.; An, J.-H.; Yang, H.-J.; Gil Lee, D.; Kim, J.; Koh, H.; Park, Y.-H.; Song, B.-S.; Sim, B.-W.; Lee, H.J.; et al. Human Blood Vessel Organoids Penetrate Human Cerebral Organoids and Form a Vessel-Like System. Cells 2021, 10, 2036. [Google Scholar] [CrossRef]

{kind=link}
| Understanding the role of PCDH19 using in vivo and in vitro models | Model | Reference |
|---|---|---|
| Role in cell division (probable involvement in mitotic spindle orientation) | Human iPSCs | Compagnucci et al., 2015 [58]; |
| PCDH19/PCDH19 localizes at the neuron-neuron cell contact sites | Human iPSCs | Compagnucci et al., 2015 [58] |
| Mouse | Hayashi et al., 2017 [21] | |
| pcdh19 is necessary for early stages of neurulation of the zebrafish embryo (disruption of convergent cell movements and impaired brain morphogenesis in PCDH19 KO embryos) | Zebrafish | Emond et al., 2009 [49] |
| pcdh19 cis-interacts with ncad, which acts as a cofactor to enforce the adhesive properties of PCDH19 | Zebrafish | Biswas et al., 2010 [47] |
| HEK293 cells | Emond et al.,2011 [48] | |
| pcdh19 interacts with nedd1, an important protein for spindle assembly during development | HEK293 cells | Emond et al., 2021 [59] |
| Understanding the PCDH19-dependent neurological alterations in in vivo and in vitro models | Model | Reference |
| Mismatching between PCDH19 and Ncad interactions results in hippocampal presynaptic dysfunction and cognitive impairments | Mouse | Hoshina et al., 2021 [50] |
| Abnormal cell sorting and segregation of PCDH19+ and PCDH19- cortical NPCs and their progeny in heterozygous mice | Mouse | Pederick et al., 2018 [17]; Hayashi et al., 2017 [21] |
| Impaired migration and altered localization of cortical neurons | Zebrafish | Cooper et al., 2015 [46] |
| Mouse | Pederick et al., 2016 [16]; Lv et al., 2019 [51] | |
| Rat | Bassani et al., 2018 [23] | |
| Altered neuronal morphology | Zebrafish | Cooper et al., 2015 [46] |
| Rat | Bassani et al., 2018 [23] | |
| Mouse | Mincheva-Tasheva et al., 2021 [55] | |
| Impaired synaptic connectivity of neurons | Mouse | Lv et al., 2019 [51]; Mincheva-Tasheva et al., 2021 [55] |
| Decreased fear response and slight hyperactivity | Mouse | Hayashi et al., 2017 [21] |
| Increased numbers of neurons | Zebrafish | Cooper et al., 2015 [46] |
| Mouse | Homan et al., 2018 [22] | |
| Human iPSCs | Borghi et al., 2021 [60] | |
| Accelerated neural differentiation | Mouse | Homan et al., 2018 [22]; |
| Human iPSCs | Borghi et al., 2021 [60] | |
| Loss of apico-basal polarity of NPC | Human iPSCs | Homan et al., 2018 [22] |
| Reduced radial glia proliferation and increased radial glia differentiation | Mouse | Fujitani et al., 2017 [20] |
| Altered mitotic spindle and increased asymmetric cell division in progenitor cells | Human iPSCs | Borghi et al., 2021 [60] |
| Smaller size of patient-derived cerebral organoids compared to control ones | Human cerebral organoids | Borghi et al., 2021 [60] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borghi, R.; Magliocca, V.; Trivisano, M.; Specchio, N.; Tartaglia, M.; Bertini, E.; Compagnucci, C. Modeling PCDH19-CE: From 2D Stem Cell Model to 3D Brain Organoids. Int. J. Mol. Sci. 2022, 23, 3506. https://doi.org/10.3390/ijms23073506
Borghi R, Magliocca V, Trivisano M, Specchio N, Tartaglia M, Bertini E, Compagnucci C. Modeling PCDH19-CE: From 2D Stem Cell Model to 3D Brain Organoids. International Journal of Molecular Sciences. 2022; 23(7):3506. https://doi.org/10.3390/ijms23073506
Chicago/Turabian StyleBorghi, Rossella, Valentina Magliocca, Marina Trivisano, Nicola Specchio, Marco Tartaglia, Enrico Bertini, and Claudia Compagnucci. 2022. "Modeling PCDH19-CE: From 2D Stem Cell Model to 3D Brain Organoids" International Journal of Molecular Sciences 23, no. 7: 3506. https://doi.org/10.3390/ijms23073506
APA StyleBorghi, R., Magliocca, V., Trivisano, M., Specchio, N., Tartaglia, M., Bertini, E., & Compagnucci, C. (2022). Modeling PCDH19-CE: From 2D Stem Cell Model to 3D Brain Organoids. International Journal of Molecular Sciences, 23(7), 3506. https://doi.org/10.3390/ijms23073506